
Abstract
A role for stress in the onset and relapse of schizophrenia is an ‘inescapable clinical fact’ according to a leading biological researcher [1]. According to the stressvulnerability model of psychotic disorders, ‘each of us is endowed with a degree of vulnerability that, under suitable circumstances, will express itself in an episode of schizophrenic illness’ [2], p.109], with one circumstance being excessive levels of stress. Although the essence of this model has been widely accepted as a model to explain the onset and maintenance of disorders such as schizophrenia, vulnerability markers have not been reliably identified and it is unclear how the psychological experience of stress influences brain biology and, ultimately, the expression of psychotic symptoms.
In recent years, it has been suggested that the Hypothalamic-Pituitary-Adrenal (HPA) axis, one of the primary biological systems that mediates the psychological experience of stress, may be impaired in some individuals with schizophrenia and other psychoses [3]. It is known that the hippocampus is an area of the brain that is intimately involved in regulation of the stress response as well as potentially playing a central role in the development and maintenance of schizophrenia and other psychoses [4, 5, 6, 7]. Thus, the HPA-axis and the hippocampus might mediate between the experience of stressful events and the onset of psychotic illnesses.
In this paper, we review evidence supporting a role for stress, HPA-axis dysfunction and hippocampal abnormality in the development and maintenance of schizophrenia and other psychoses and propose three aetiological models linking these elements. Strategies for further research investigating these models are discussed. This review will not focus on the vulnerability component of the stress-vulnerability model - a review of this area of research that has been published recently [8].
Stress and the HPA-axis
The HPA-axis is one of the primary systems moderating the physiological response to psychological and physiological stressors in mammals [9, 10]. Neural signals associated with a stressor are transduced into an endocrine response at the level of the hypothalamus (Fig. 1). The paraventricular nucleus in the hypothalamus is a complex integration centre that receives and coordinates neuroendocrine, autonomic, cognitive and emotional input and is responsible for initiating glucocorticoid secretion (for review, see [11]). Corticotropin releasing hormone (CRH) and, to a lesser extent, arginine vasopressin are then secreted from the paraventricular nucleus of the hypothalamus into the hypophyseal portal system where they reach the anterior pituitary gland and synergistically stimulate the release of adrenocorticotropin hormone. Adrenocorticotropin hormone is then transported in the bloodstream where it stimulates the release of glucocorticoids from the adrenal cortex. The release of glucocorticoids is ultimately inhibited through a negative feedback mechanism.
The HPA-axis. ACTH, adrenocorticotropin hormone; AVP, arginine vasopressin; CRF, corticotropin releasing hormone; GR, glucocorticoid receptors; MR, mineralocorticoid receptors; -ve, negative feedback.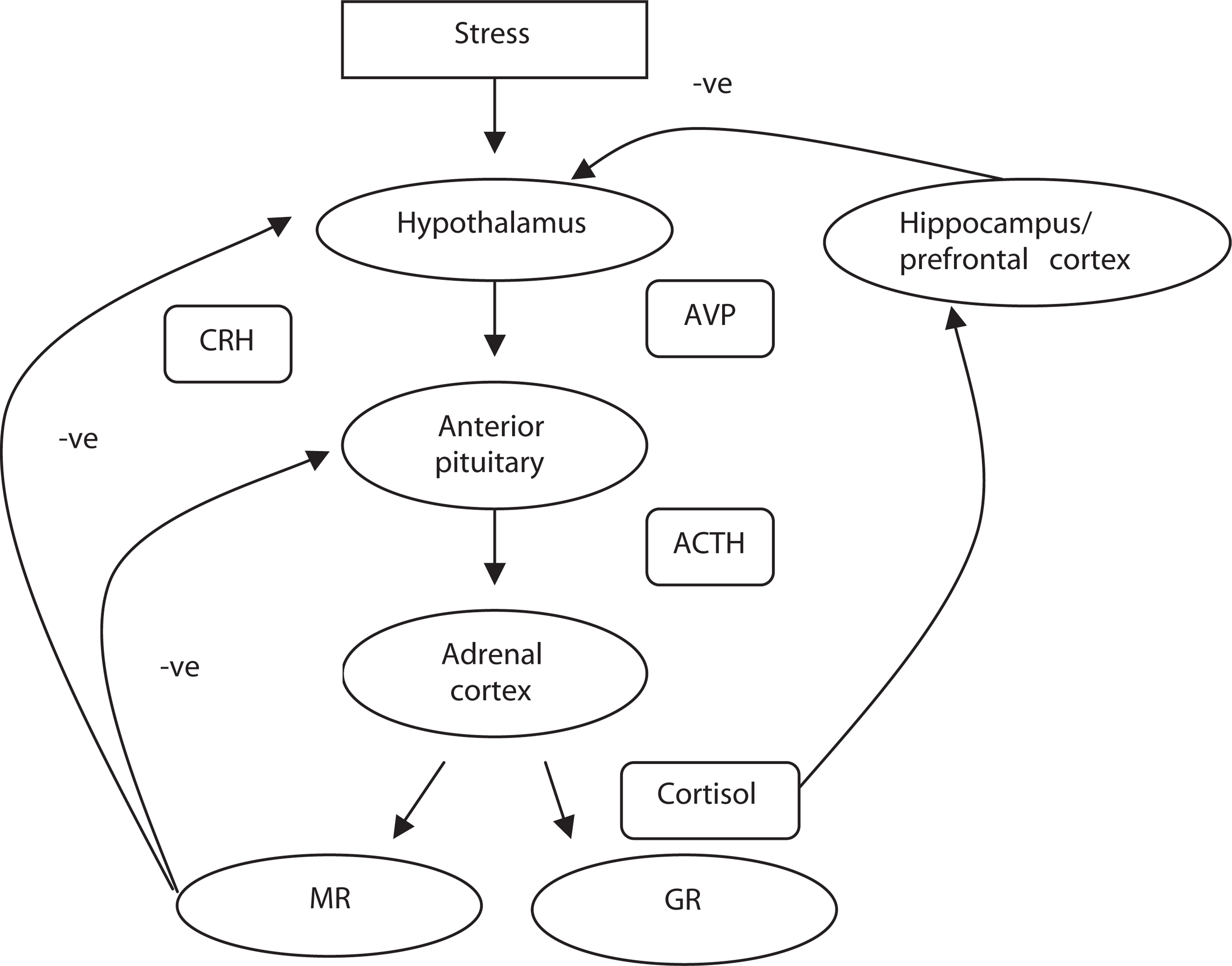
The glucocorticoid cortisol is a major mediator of the physiological stress response and impacts on many physiological systems to allow the body to react to a stressor. Acute, time-limited rises in cortisol levels are adaptive, but long-term elevation may have a negative effect on the central nervous system, and has been associated with ventricular dilation, cerebral atrophy, cognitive impairment [9] and possibly neurotoxicity [12]. Prolonged heightened cortisol levels also damage the hippocampus, thereby reducing negative feedback further. This vicious cycle of events is often referred to as the ‘glucocorticoid cascade’ [13].
Other glucocorticoids include corticosterone and cortisone. Corticosterone is released from the adrenal gland in response to stimulation by adrenocorticotropin hormone and assists in regulation of the conversion of amino acids into carbohydrates and glycogen by the liver, and helps to stimulate glycogen formation in the tissues. A recent post-mortem study has indicated that corticosterone appears to penetrate more easily into the human brain than cortisol [14], suggesting that that corticosterone might play a more prominent role in brain functioning than has previously been recognized. Most research to date has focused on cortisol, however. Unlike cortisol and corticosterone, cortisone is produced from cholesterol in the adrenal gland. The primary effect of cortisone is on carbohydrate metabolism.
Glucocorticoid receptors
Two types of receptors for glucocorticoids have been identified. Type I or mineralocorticoid receptors (MR) have a high affinity for natural glucocorticoids such as cortisol, and are densely localized in the hippocampus and septal neurones [15]. Type II receptors or glucocorticoid receptors (GR) have a low affinity for natural glucocorticoids but a higher affinity for synthetic steroids such as dexamethasone [16]. Glucocorticoid receptors are widely distributed throughout the mammalian brain including the limbic system, hypothalamus, pituitary cerebral cortex and brainstem monoaminergic nuclei [15]. Mineralocorticoid receptors are activated at low levels of circulating cortisol and are thought to be primarily involved in maintaining basal HPA activity. Glucocorticoid receptors, on the other hand, respond when glucocorticoid levels rise and MR become saturated, either as a result of the natural circadian rhythm or in response to stress. When this occurs, GR moderate glucocorticoid activity and are responsible for termination of the stress response via negative feedback inhibition, mostly at the level of the hypothalamus and pituitary. Additionally, HPA-axis activity is also regulated by limbic structures, including the hippocampus, amygdala, as well as the prefrontal cortex. The hippocampus contains high concentrations of both MR and GR, and has an important inhibitory role on both basal HPA-axis activity and termination of the stress response [17].
Dexamethasone suppression test
The HPA-axis responsiveness is commonly assessed using the dexamethasone suppression test (DST) [18]. In this test, a synthetic glucocorticoid, dexamethasone, is ingested. The normal response to the ingestion of dexamethasone is the inhibition of cortisol secretion through the initiation of GR-mediated feedback mechanisms. This occurs mainly in the hypothalamus and pituitary as dexamethasone is unable to penetrate the blood-brain barrier. A failure to suppress cortisol release following the administration of dexamethasone reflects a failure in the negative feedback mechanism. This response is seen in conjunction with major depressive disorder [19]. Hypersuppression of cortisol levels following ingestion of dexamethasone, as is typically seen in conjunction with posttraumatic stress disorder [20], indicates an overreactive negative feedback mechanism.
A variant of the DST is the combined dexamethasone/ CRH suppression test (DEX/CRH test), in which participants take an oral dose of dexamethasone in the evening. The following afternoon blood samples are taken at baseline and at regular intervals following intravenous infusion of CRH [21]. The combined test is more sensitive in detecting differences in HPA-axis activity between individuals with and without major depressive disorder than the original DST [22].
The relationship between the HPA-axis and the dopamine system
A synergistic relationship appears to exist between the HPA-axis and the dopaminergic system (for review, see [3, 23]). The rate of dopamine (DA) synthesis and release is known to increase in response to acute stress [24], and heightened HPA-axis activity also influences the rate of DA activity [25]. The magnitude of the DA response to stress differs throughout the brain [26]. In rats, for example, stress induces the biggest turnover of DA in the prefrontal cortex, a moderate rate of turnover in the nucleus accumbens and smallest in the neostriatum [27]. It is also thought that heightened DA activity in prefrontal areas reduces the DA response to stress in subcortical regions [26, 28]. Evidence also suggests that exposure to a chronic stressor may result in the downregulation of tonic activity of DA neurones in midbrain regions which could then down-regulate DA levels in the nucleus accumbens [27].
Stress, HPA-axis dysfunction and hippocampal damage
Animal studies
As mentioned earlier, the hippocampus plays a crucial role in the negative feedback loops that ultimately terminate the physiological stress response and is therefore particularly important in the regulation of glucocorticoid levels [9, 29]. Because of its high density of MR and GR, the hippocampus is the region of the brain that is most susceptible to alterations in glucocorticoid levels. Animal studies have demonstrated that prolonged, severe stress causes damage to hippocampal CA3 pyramidal neurones [30, 31, 32, 33, 34, 35], and suppresses long-term potentiation of neurones in the CA1 region [36, 37], and that neurogenesis in the dentate gyrus is reduced by stress and enhanced by adrenalectomy [38]. Prolonged exposure to elevated levels of corticosterone (the rodent equivalent of cortisol) has also been associated with hippocampal neuronal cell death, changes to cell morphology including atrophy of dendritic processes, and reduced neurogenesis in animals [38, 39, 40, 41, 42, 43] and stress-induced memory impairment has been reported in rats [44, 45]. Reducing corticosterone secretion in rats has been shown to prevent the degree of hippocampal cell loss that is normally associated with chronic stress [46, 47]. The effect of glucocorticoids on hippocampal plasticity is thought to be mediated, in part, by excitatory amino acids and N-methyl-D-aspartic acid (NMDA) receptors [48]. In rats, stress- and corticoster-one-induced hippocampal dendritic atrophy is blocked by the administration of phenytoin (which is known to interfere with excitatory amino acids release and actions) [49] and by treatment with an NMDA receptor antagonist, but not a non-NMDA glutamate receptor blocker [48]. Further understanding of the underlying mechanisms of stress-induced structural changes may provide the opportunity for pharmacological prevention and treatment of effects caused by stress.
Human studies
In humans, high plasma cortisol levels also appear to be associated with diminished hippocampal volumes and cognitive impairment. For example, individuals with acute Cushing's disease (marked by chronically high levels of cortisol) show reductions in hippocampal volume, which are correlated with poor performance on tests of verbal memory [50]. Reduced hippocampal volumes and deficits in aspects of memory have also been described in physically healthy elderly individuals with high cortisol levels [51, 52] and prolonged exposure to cortisol at plasma concentrations commensurate to that experienced during physical and psychological stress has been demonstrated to decrease memory performance [53]. Nonpsychiatric patients receiving corticosteroid therapy have also been shown to have reduced hippocampal volume and impaired cognition compared with control subjects [54]. Further evidence of alterations to the hippocampus in association with the experience of stress comes from many structural imaging studies that have shown reduced hippocampal brain volumes in anxiety disorders such as posttraumatic stress disorder [55, 56, 57] and obsessivecompulsive disorder [58, 59]. Finally, it has been shown that treatment of Cushing's Disease and subsequent reduction in cortisol level is associated with hippocampal volume increase [60].
Evidence of abnormal HPA-axis function in schizophrenia and other psychotic disorders
Schizophrenia
Results of studies that have evaluated HPA-axis functioning in association with schizophrenia by assessing plasma, urinary or salivary cortisol levels have been varied. Higher cortisol levels (plasma, salivary or urinary) and abnormal circadian cortisol rhythms have been reported in patients with schizophrenia compared with healthy control subjects in a number of studies [61, 62, 63, 64, 65, 66, 67, 68], but not in others [69, 70, 71, 72, 73, 74, 75]. In comparison with other psychiatric patient groups, one study reported that patients with schizophrenia have lower urinary cortisol levels than patients with bipolar disorder (manic phase) and major depressive disorder [76], but no differences in salivary cortisol levels were found between patients with schizophrenia and bipolar disorder in another study [67].
Three studies have reported a blunted cortisol response to psychosocial, psychological and physical stressors in schizophrenic patients [72, 77, 78]. A blunted cortisol response by individuals with schizophrenia compared with control participants has also been shown in a study of the response to acute metabolic stress induced by the injection of 2-deoxy-D-glucose (2-DG) [79]. A blunted cortisol response to stress may reflect an impaired ability to adapt to stress at the biological level. Once again, it should be noted that a number of studies have had different results. Three studies have failed to show any difference in cortisol responses to metabolic stress between patients and controls [70, 80, 81].
Most studies have reported higher levels of DST non-suppression of cortisol in patients with schizophrenia compared with healthy control subjects [61, 63, 64, 66, 68, 69, 71, 82, 83, 84, 85], but lower levels of non-suppression compared with depressed patients [82, 86, 87]. Consistent with these findings, a reduction in GR mRNA levels has been reported in various brain regions, including the hippocampus, prefrontal cortex and amygdala, in schizophrenia patients [88, 89], providing further evidence of impairment glucocorticoid negative feedback inhibition.
Other psychotic disorders
The HPA-axis activity also appears to be affected in individuals experiencing psychotic disorders other than schizophrenia. Individuals with major depressive disorder with psychotic features were found to have significantly higher plasma cortisol levels than individuals with non-psychotic major depressive disorder and healthy controls in a study by Belanoff et al. [90] and patients with schizoaffective disorder (manic subtype) had significantly higher plasma cortisol levels than controls in a study by Whalley [67]. The particularly high cortisol levels observed in patients with major depression disorder and psychotic features may reflect the combination of HPA pathology associated with depression, and the distress of experiencing psychosis and HPA activation associated with psychosis.
Mediating factors
The heterogeneous nature of psychotic disorders might account for some of the varying results that are reported in the studies which have been mentioned in the preceding paragraphs. For example, a number of studies have reported that individuals with schizophrenia who are also experiencing depression[84, 90, 91, 92, 93] or high levels of negative symptoms[61, 84, 94, 95, 96] are more likely to show non-suppression in response to the DST than psychotic individuals without either of those symptoms profiles. Although it is noted that not all studies support these findings[85, 97, 98, 99, 100], methodological differences between studies can explain some of this inconsistency.
Duration of illness may also be important in determining HPA-axis functioning in relation to psychotic disorders. Duration of psychotic symptoms has been negatively correlated with cortisol levels in one study [101], and hyperactivity of the HPA-axis has been reported more consistently in patients who are newly hospitalized or experiencing their first psychotic episode as opposed to chronically ill patients [102]. In particular, an enlarged pituitary volume, an indicator of HPA-axis activation, has been reported in subjects before the onset of psychosis and in first-episode patients, but not in chronic patients [102, 103]. Further investigation of the relationship between HPA-axis functioning and phase of psychotic illness are warranted.
Finally, treatment with neuroleptic medication might also account for some of the discrepancy between studies. Treatment with atypical neuroleptics has been shown to reduce cortisol levels [104] and normalize nonsuppression of the DST [96]. For example, studies have reported a lower rate of DST non-suppression in schizophrenia patients following 4-5 weeks of antipsychotic treatment [96, 105]. However, one study has reported no difference in diurnal plasma cortisol level between people with schizophrenia who were either neurolepticnaïve, or using antipsychotics, and controls [73]. The mechanisms underlying the action of neuroleptic medication on the HPA-axis is not yet fully understood. Results from a recent study found that both typical and atypical antipsychotics inhibit CRH gene promoter activity and this may be one mechanism by which antipsychotics dampen HPA-axis activity [106]. There is also evidence to suggest that some antipsychotics inhibit GRmediated gene transcription [107]. Animal studies have shown that treatment with antidepressants can upregulate GR in the brain, decrease stress-induced glucocorticoid secretion and enhance HPA-axis feedback, and studies in vitro have shown that antidepressants can enhance GR function (for review, see [108]). It should also be noted that animal studies have indicated that dexamethasone can cause cell death - particularly in the striatum and CA1 and CA3 regions of the hippocampus [109], and that long-term antidepressant medication can attenuate this damage [110].
In light of the above discussion, it might be more useful for future studies of HPA-axis functioning in individuals with schizophrenia to take into account variables such as duration of illness and level of depressive symptoms. For example, it does not appear to make a great deal of sense to include individuals with a psychotic disorder who are also experiencing heightened levels of depression in the same group as psychotic individuals without comorbid depressive symptoms. This suggestion is supported by reports of a study that assessed the effect of a glucocorticoid antagonist in the treatment of schizophrenia. The results of many of the studies that are reported above suggest that the HPA-axis is potentially an important therapeutic target in psychotic illnesses. Accordingly, Gallagher et al. assessed the effect of a glucocorticoid antagonist - RU486 (mifepristone) - on neurocognitive functioning and symptoms in schizophrenia [111]. Contrary to their hypotheses no effect of the drug was noted in the schizophrenia cohort. This was surprising to the researchers as an earlier study in bipolar patients had shown an ameliorative effect of RU486 on neurocognitive functioning [112]. Gallagher et al. suggested that either the HPA-axis dysfunction seen in association with psychosis might not be attributable to the same underlying neurobiological abnormalities that seen in association with bipolar disorder or differences in HPA-axis functioning between individuals with schizophrenia muted the positive results that might have been seen in a subgroup of patients. They suggested that future studies in schizophrenia should concentrate on those individuals with a demonstrable HPA-axis dysfunction.
The hippocampus and psychotic disorders
Neurocognitive studies
Studies of neurocognition strongly suggest that the hippocampus is impaired in individuals with psychotic disorders. Impairments in attention and memory are commonly experienced by individuals with psychotic disorders[113, 114, 115, 116, 117, 118]. Furthermore, hippocampaldependent verbal memory is thought to be a strong predictor of functional outcome [119].
Studies of brain structure
Studies of hippocampal brain structure provide stronger evidence for a role of the hippocampus in the onset or maintenance of psychosis. One review paper has reported that that psychotic disorders are characterized by reductions in hippocampal and para-hippocampal gyrus volumes, disarray of hippocampal pyramidal neurones, smaller hippocampal neurone size and lower levels of protein in synapses in the hippocampus [120], although some of these findings require further confirmation. Structural neuroimaging studies have repeatedly found decreased hippocampal volumes in psychotic patients[121, 122, 123]. Those findings are supported by spectroscopy, functional imaging and shape analysis studies which have also provided evidence of hippocampal abnormalities in association with schizophrenia[124, 125, 126, 127]. The most robust results come from highresolution MRI studies of chronic schizophrenia patients, using slices of less than 1.5 mm [128, 129], with only one published study of 1.5 mm slices reporting no difference in hippocampal volume between schizophrenia and control subjects [130]. The difference in hippocampal volumes is already apparent when comparing individuals experiencing a first psychotic episode with controls [4], but a recent cross-sectional comparative study suggested that structural changes to medial temporal regions, including the hippocampus, do not occur until after the onset of acute levels of psychosis and that the pattern of structural change differs according to the type of psychosis that develops [131]. This is supported by the results of the only study to date that has been able to assess changes in brain structure during the transition to acute psychosis. This study indicated that left parahippocampal, fusiform and orbitofrontal and cerebellar cortices and cingulate gyri volumes of young people who were identified as being at ‘ultra’ high risk of psychosis were reduced after the onset of acute psychosis [132].
Studies of neuropathology
The neuropathology underlying hippocampal volume reduction in schizophrenia and other psychiatric disorders has not been fully elucidated; however, the reduced neuropil hypothesis suggests that atrophy of cell soma, neuronal processes and synaptic contacts in the hippocampus and prefrontal cortex results in reduced cortical volume at the macroscopic level, which may underlie the functional disturbances observed in schizophrenia [133]. As discussed earlier, excessive exposure to glucocorticoids leads to a reduction in neuronal volume and atrophy of dendrites in the hippocampus [134] and therefore may be a contributing mechanism to the structural changes observed in psychiatric disorders. In line with this suggestion, a decrease in the number of hippocampal neurones has previously been cited as characteristic of schizophrenia [135, 136], although not all studies have supported this finding[137, 138, 139]. Studies examining the size of neuronal cell bodies have produced more consistent results; three relatively large studies reported a decrease in size of pyramidal neuronal cell bodies in the hippocampus of schizophrenia patients[140, 141, 142]. Synaptic and dendritic abnormalities have also been described in the hippocampus of schizophrenia patients. These include decreased expression of synaptophysin and other presynaptic proteins in the hippocampal formation[143, 144, 145, 146] indicating a reduction in synaptic density. Decreased levels of growth-associated-protein 43 (GAP-43) mRNA have also been detected in the hippocampus [147], indicating a reduction in synaptic plasticity. Extensive investigations in psychotic disorders other than schizophrenia have not been yet conducted.
Stress and psychosis
Defining stress
‘Stress’ is a concept that is widely referred to in medicine and psychology in a wide range of contexts. For example, biological or physiological definitions of stress refer to the non-specific response of the body to factors that threaten homeostasis [148], while current psychological conceptualizations of stress refer to a process whereby stimuli are appraised and evaluated and a coping response is developed [149]. Within the psychosis literature, ‘stress’ is generally used to refer to external events that cause the individual to experience a degree of distress or anxiety. Although the experience of psychosis itself, or the process of developing psychosis, has been shown to result in the experience of posttrauma symptoms [150, 151], relatively little research has addressed this process to date. This paper is limited to reviewing the literature that has used the restricted definition of stress as a stimulus and its relationship with the onset or maintenance of psychotic disorders. For a review of the relationship between psychosis and stress considered more broadly, the reader is referred to Philips et al. (in preparation).
The most common method for evaluating the experience of stress by individuals with psychotic disorders is the ‘life events’ approach, whereby the number of stressful events experienced during a certain time period is counted. This approach assumes that feelings of distress are correlated with the number of events that are experienced.
Retrospective studies of life events and psychosis
The first major study investigating the potential relationship between the experience of stressful life events and the onset of a psychotic episode was conducted more than three decades ago [152]. The study reported that individuals with schizophrenia experienced almost double the number of stressful events over the 13-week period immediately preceding the onset of a psychotic episode (mean = 1.74 events) than a healthy comparison group (mean = 0.96 events). Forty-six per cent of the patient group had experienced at least one independent event in the 3-week period immediately preceding episode onset, but only 12% had an event in any of the other 3-week periods under review. A number of more recent studies have replicated Brown and Birley [152] by finding an increase in the number of life events experienced before the onset of an acute episode[101, 153, 154, 155, 156, 157]. However, Chung et al. [158] reported no change in the frequency of life events before onset, and Gruen and Baron [159] reported that only 15.4% of chronic schizophrenia patients experienced stressors at a severe or extreme level during the 12 months before onset of illness. Differences in results between the studies are largely due to methodological or sampling differences, although Jacobs and Myers [160] reported that the role of life events in the onset of schizophrenia was probably ‘marginal’ and they possibly played a ‘small contributory role such as a precipitating or triggering effect’ (p.86) but not a causative role.
Prospective studies of life events and psychosis
The development of reliable criteria to identify individuals at heightened risk of developing a psychotic disorder has enabled prospective investigation of the relationships between the experience of stressors and onset of psychosis. So far, two studies have been conducted within this group. Mason et al. [161] reported that the number of life events experienced by individuals deemed to be at ultra high risk (UHR) of developing a psychosis did not predict onset of disorder. Unpublished results from the PACE Clinic in Melbourne, Australia have indicated that while individuals who meet UHR criteria experienced significantly fewer life events than individuals in a comparison group, they felt they did not cope as well with stressors as a healthy control group and were more distressed by events. Once again, the number of events experienced did not predict onset of psychosis in the UHR group.
A number of longitudinal studies involving individuals with established illness have been conducted to assess the relationship between experience of life events and relapse of illness. An increase in the number of life events experienced in the 4-week period immediately preceding a relapse was reported by Ventura et al. [162] and Pallanti et al. [163]. Hirsch et al. [164] failed to find a significant difference between the number of events experienced in the 4 weeks preceding relapse compared with another 4week period. It should be noted that the validity of the relapse prodrome as a model for the initial prodrome has not been established and the potential influence of factors such as treatment on the relapse prodrome remains unknown.
Minor stressors and psychosis
Norman and Malla [165] suggested that individuals with schizophrenia are more likely to be adversely affected by chronic difficulties and stressors experienced in comparatively normal circumstances than more unusual major life changes and challenges. They demonstrated that the level of distress reported by individuals with schizophrenia was significantly correlated with the number of minor stressors experienced, but not with the number of major events [166, 167]. Beck and Worthen [168] reported that individuals with schizophrenia were likely to attribute symptom exacerbation to stressors of ‘low severity’.
The only other study comparing the influence of major and minor events on the experience of psychotic symptoms was conducted by Hardesty et al. [169]. They reported that major life events experienced by individuals with schizophrenia were often followed by an increase in negative symptomatology, but minor events (often including positive events) were more likely to be associated with a decrease in depressive symptoms. The impact of either major or minor stressors on ‘florid schizophrenic psychopathology’ (positive symptoms) in this study was small.
Experience sampling method and psychosis
Myin-Germeys et al. [170] used the Experience Sampling Method to examine the experience of stress by individuals with schizophrenia compared with their firstdegree relatives and healthy controls. Individuals with schizophrenia reported significantly higher levels of ‘event-related stress’ than the healthy comparison group and significantly higher levels of socially related stress than either their first-degree relatives or the comparison group. They concluded that daily life stress and mood is related in a dose-response fashion with level of genetic or familial risk of psychosis. This suggests that stress reactivity could be viewed as a vulnerability marker for psychosis, but as the study was cross-sectional, this possibility has not been evaluated.
Stress and other psychotic disorders
Few studies have assessed the relationship between the experience of stress and the onset of other psychotic disorders. Bebbington et al. [153] reported an increase in life events of marked or moderate severity over the 6-month period before onset of mania compared with normal controls. They also reported that patients with depressive psychoses experienced more life events in the 3 months before illness onset than were experienced by a control group in 3 months and concluded that all types of psychotic disorder are equally likely to be precipitated by life events. Another study by Clancy et al. [171] reported that individuals suffering from unipolar depression were most likely to experience a significant life event preceding a relapse of symptoms (39%) followed by bipolar patients (27%) and schizophrenia (11%). Chung et al. [158], however, found that in the 26-week period preceding onset of illness, only 14% of manic patients experienced a significant life event compared with 33% of schizophrenia group and 66% of those with schizophreniform psychosis. A ‘kindling’ model of bipolar disorder has been proposed whereby early manic episodes are thought to be associated with the experience of increasing levels of stressors, but later episodes are more autonomous (analogous to the ‘kindling’ model of epilepsy)[172, 173, 174] although some recent studies and reviews have opposed this model [175, 176].
Criticism of stress research
Studies investigating a link between onset of psychotic illnesses and the experience of stress have been criticized for poor study design and inadequate sample sizes. There has also been controversy surrounding the use of the ‘life events’ approach as it may not fully assess an individual's level of stress[177, 178, 179]. However, the research reviewed above suggests that the experience of stressful, or distressing, events and the inability to adequately cope with them may affect the development of psychosis. A meta-analysis of this research has not yet been performed, but might provide some clarity to the issue. Stress management and coping skills training has been incorporated in many psychological treatment approaches for psychotic disorders[180, 181, 182, 183, 184].
Summary
The research that has been summarized above suggests that the experience of stress, HPA-axis dysfunction and hippocampal damage are all associated with at least some cases of psychotic disorders. Most of this research has focused on schizophrenia, but it is possible that similar processes underlie other psychotic disorders. To date, however, the published research has addressed the association between these factors and psychosis in isolation - surprisingly a fully integrated model has not yet been tested. We propose three models of the development of psychosis that attempt to synthesize these strands of research. The first draws extensively on the glucocorticoid cascade hypothesis developed by Sapolsky et al. [185, 186], which proposes that excessive cortisol levels result in damage to neurological regions, such as the hippocampus. The second model suggests that insufficient GR activity can be damaging to the brain [187]. The final model proposes that prefrontal cortex pathology might impact on HPA-axis functioning and the hippocampus to influence the development of psychotic symptoms. Although described separately, these models might not act exclusively.
Model 1: overactive glucocorticoids
This model builds on the suggestion that there is more than one period of heightened vulnerability for the development of psychotic illness across the life span [1, 188, 189]. The first ‘window’ of vulnerability occurs during the perinatal period when an early traumatic event or genetic liability impacts on neurological development (Fig. 2). (A full discussion of the concept of vulnerability to psychotic disorders is outside of the immediate focus of this review. Papers that have addressed this issue include [8, 190, 191]). Similarly research regarding evidence for a genetic basis for psychosis is also available elsewhere (e.g. [192, 193, 194]). In the case of psychosis, the neuronal damage or abnormal development is thought to particularly affect the hippocampus. In addition, early life factors that compromise the development of the HPA-axis may lead to a long-lasting, if not permanent, neurobiological vulnerability to stress later in life. The set point of HPA-axis activity in adulthood is determined, firstly, by genotype and subsequently, by the environment, particularly during the early stages of life [16], and it has been hypothesized that an imbalance in MR and GR or a change in the set point of the HPA-axis may increase vulnerability for stress-related psychiatric disorders [195]. This model is supported by studies with rats that have demonstrated variations in maternal care (such as frequency of licking and grooming) influence the development of behavioural and endocrine responses to stress in the offspring [196]. This variation in stress reactivity has been linked to permanent alterations of GR expression, via epigenetic mechanisms, resulting in changes in the MR/GR balance (or set point [197, 198]). It is speculated that a similar process might occur in humans.
Overactive glucocorticoid model of the onset of psychosis. DA, dopamine; HPA, Hypothalamic-Pituitary-Adrenal.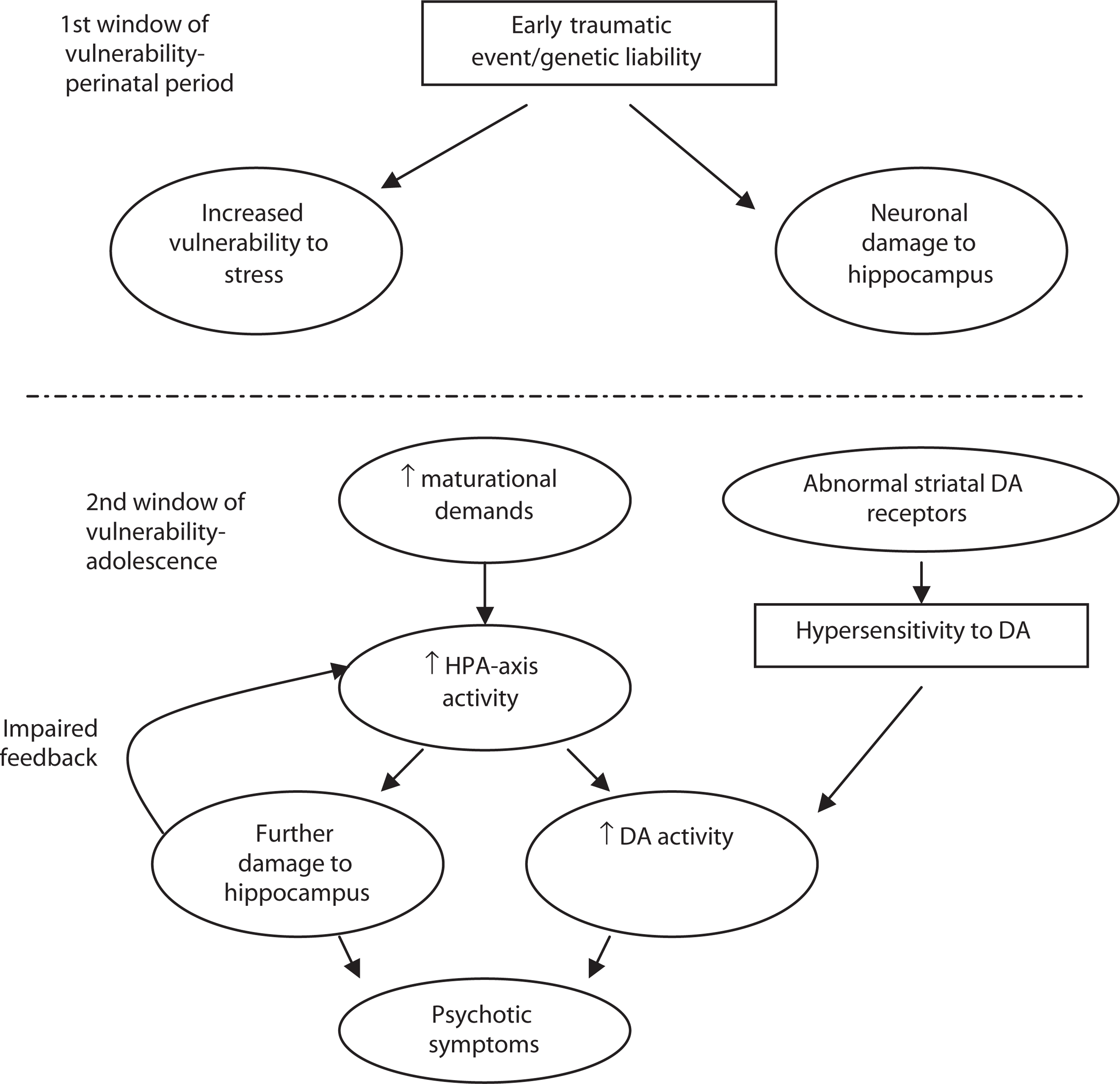
The second window of vulnerability is thought to occur during adolescence. At this time, the brain is undergoing immense maturational changes such as a decrease in grey matter and an increase in white matter [199], increased activity and metabolic demand in the frontal cortex [200, 201] and synaptic pruning and reorganization [202]. The latter are implicated in a widely supported model of the development of psychotic symptoms [202, 203, 204]. Adolescence is also a particularly vulnerable time of life when people are typically negotiating the transition to adulthood [205], including identity formation, education and career development and the formation of an independent peer group [206]. Increased HPA-axis activity has been documented during adolescence [207], possibly associated with normal maturation processes. The model suggests that the prolonged overproduction of glucocorticoids at this time, possibly exacerbated by an altered set point of HPA-axis activity, can have a negative effect on an already damaged hippocampus. In turn, this is thought to have a negative impact on the regulation of HPA-axis activity and a feed-forward circuit has been proposed in which glucocorticoid production is driven into overdrive under these circumstances. These hormonal and neuronal modifications are thought to unmask the underlying vulnerability to psychosis, paving the way for the development of psychotic symptoms. For example, as described earlier, heightened HPA-axis activity has been associated with enhanced DA activity in subcortical brain regions [3], and excessive levels of circulating cortisol lead to damage of neuronal processes and reduced neurogenesis, particularly in the hippocampus [208]. This could potentially lead to the development of psychotic symptoms. Subsequent experiences of stress might exacerbate this process, increasing the intensity and frequency of psychotic symptoms that develop.
A parallel process that would contribute to this chain of events might occur if an individual had a hypersensitivity to DA because of abnormalities in striatal DA receptors [3]. During adolescence when HPA activity is augmented, such individuals could experience enhanced HPA activity and hippocampal damage (Fig. 2). This might interact with the process described above to influence the development of psychotic symptoms.
Model 2: insufficient glucocorticoid signalling
A second model suggests that reduced GR signalling might play a role in the development of psychosis. Reduced GR signalling could arise from a primary (genetic) alteration or a secondary alteration, for example, if there are low levels of circulating glucocorticoids (hypocortisolism), low GR numbers, or the binding affinity or functional capacity of GRs is reduced because of prolonged exposure to elevated cortisol levels [187].
As a result, the inhibitory feedback loops of the HPAaxis would not be activated to restrain the stress response, resulting in further release of circulating glucocorticoids, which would result in increased DA levels (as described by Imperato et al. [25]) and the development of psychotic symptoms (Fig. 3). This model is similar to the corticosteroid receptor hypothesis of depression, which suggests that impaired corticosteroid receptor signalling results in the increased production of CRH in various regions of the brain that are thought to be associated with the aetiology of depression [22]. In major depression, for example, it is unknown whether a primary abnormality in GR function leads to CRH hyper-secretion (because of impaired negative feedback), or alternatively, whether CRH hyper-secretion (perhaps caused by chronic stress) leads to impaired GR function. Using transgenic mice, one study showed that impaired GR function present from early embryonic life does not result in CRH hypersecretion in adulthood, thus lending support to the latter notion [209]. The GR signalling model has not yet been fully explored in schizophrenia and other psychotic disorders. It should be noted that glucocorticoids exert their adaptive effects (such as changes in energy metabolism, gene transcription and so forth) through MR and GR and thus, if GR signalling is reduced, this will influence, not only negative feedback inhibition, but also the effect of glucorticoids on cellular processes and will result in increased signalling at MR.
Insufficient glucocorticoid signalling model. DA, dopamine; GR, glucocorticoid receptors; HPA, Hypothalamic-Pituitary-Adrenal.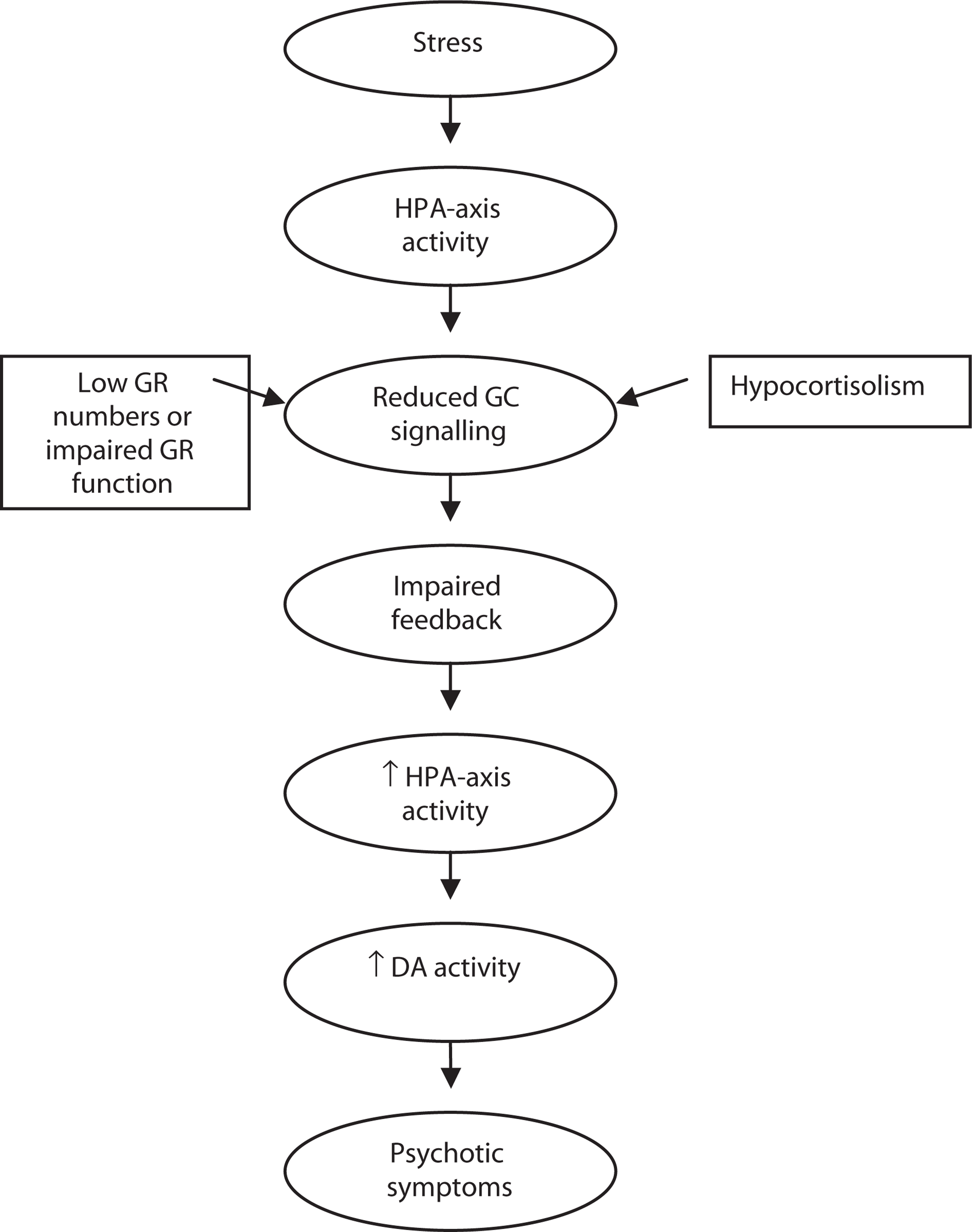
Model 3: involvement of prefrontal cortex
Another process that might explain the hippocampal volume loss and associated dysfunction that is apparent in schizophrenia, and the onset of psychosis during adolescence, has been described by Grace [210]. He has suggested that pathology of the prefrontal cortex might, in turn, impact on the hippocampus ultimately resulting in the expression of psychotic symptoms. A level of pathology in the prefrontal cortex of individuals with psychotic disorders is suggested by evidence of reduced performance on neuropsychological tasks that reflect prefrontal cortical functioning such as tests of executive functioning and working memory [211, 212]. This level of dysfunction might be accounted for by a reduction of up to 33% in DA innervation of the prefrontal cortex in the brains of individuals with schizophrenia [213]. Maturation of the prefrontal cortex is not thought to be completed until well into adolescence [214], which may explain the severity of executive function deficits in schizophrenia [189, 215]. It is thought that some higher order memory functions that are mediated by the prefrontal cortex when it is fully matured are actually mediated by the hippocampus until that time [216]. However, if the prefrontal cortex does not fully mature or is dysfunctional, it might not be able to take over these tasks. In support of this notion, Grace cites data indicating that some working memory tasks that are usually mediated by the prefrontal cortex in adults are mediated by the hippocampus in individuals with schizophrenia [210]. He hypothesizes that at times of increased stress, the hippocampus of such individuals is under even higher levels of functional demand. Reports of increased ‘premorbid’ hippocampal volume in a UHR cohort who later developed an acute psychotic episode compared with other individuals in the UHR cohort who did not develop psychosis are consistent with this hypothesis [131, 217].
Grace goes on to suggest that under these circumstances, glucocorticoid release could be augmented through a chain of events whereby the prefrontal dysfunction impacts on functioning of the amygdala which in turn impacts on the hippocampus [210]. Stress responses, including glucocorticoid release, would be increased because of the hyperactivated subcortical DA system. Therefore, hippocampal damage could result from the combination of increased release of glucocorticoids resulting from the impact of dysfunctional prefrontal cortex and increased functional demand. Hippocampal mediation of the stress response would then be compromised leading to the ‘glucocorticoid cascade’ hypothesis referred to earlier. This in itself would lead to further hippocampal damage and so forth (Fig. 4). While appealing, this model is yet to be fully investigated.
Model of psychosis onset incorporating prefrontal cortex pathology. DA, dopamine; PFC, prefrontal cortex.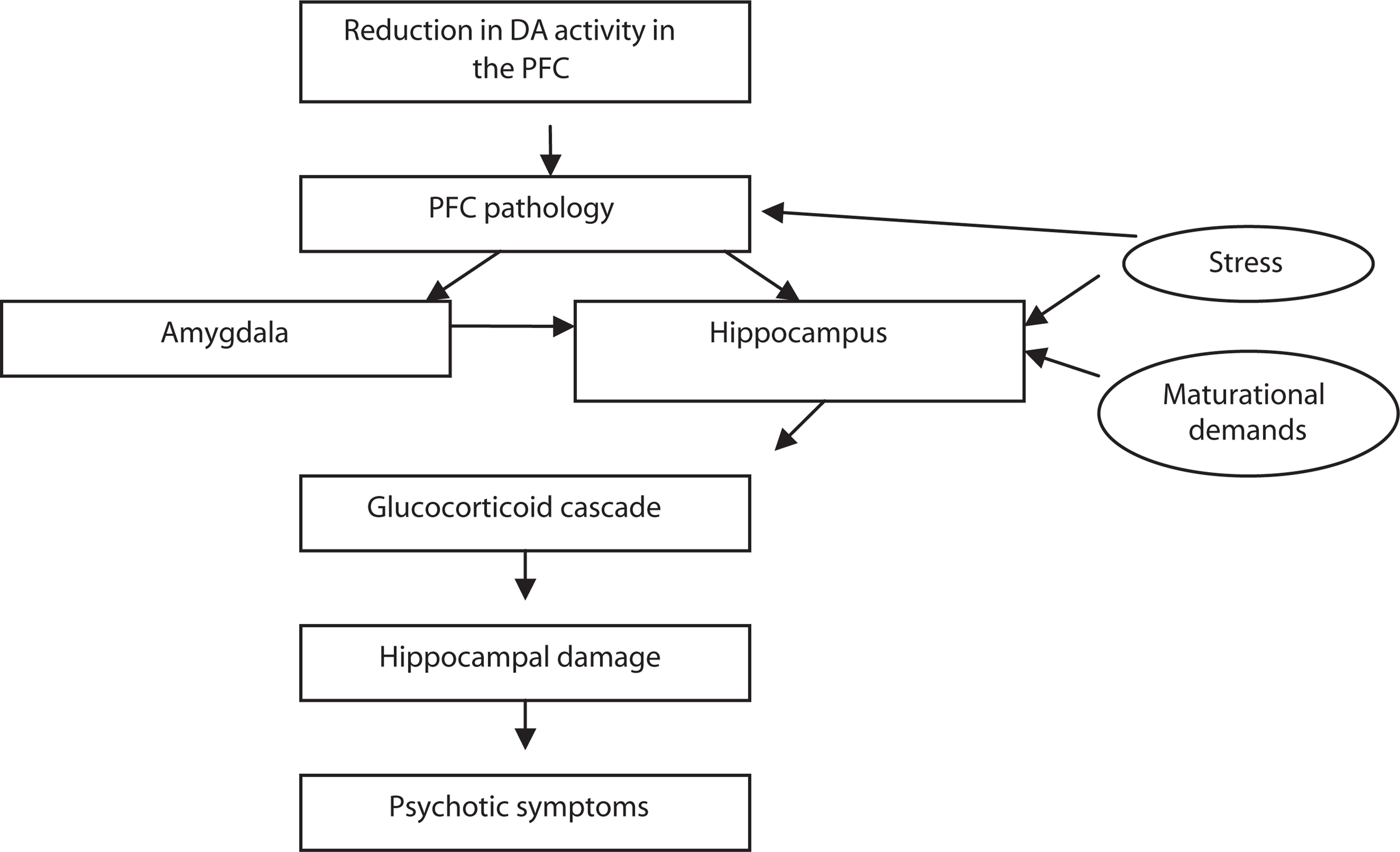
Conclusion and future research opportunities
As outlined extensively above, little is currently known about the functioning of the HPA-axis before the development of psychosis. In a similar vein, little research has been performed addressing how the experience of stress might influence the development of psychotic symptoms - despite this model being widely adopted as a rationale for treatment. Moreover, there has been no research that has integrated these various strands of research into a comprehensive model of the onset of psychosis like those described above. It is possible that a combination of these models operate. Existing research linking HPA-axis and DA activity as well as the emerging evidence of hippocampal volume change around the time of onset of a first psychotic episode leans towards Models 1 and 3 being important. However, it is noted that very little investigation of glucocorticoid receptor functioning in relation to psychotic disorders has been conducted.
The development of well-validated criteria to identify young people at UHR of developing a psychotic disorder within a relatively short time period[218, 219, 220] provides an opportunity to evaluate the models that have been proposed. To further investigate the role of HPA-axis functioning in the development of acute psychosis in the UHR group, future studies could examine negative feedback mechanisms and GR function, as well as the responsiveness of HPA-axis to challenge such as CRH or an acute stress. This would help to shed light on whether HPA hyperactivity or insufficient glucocorticoid signalling is involved during the prodromal period. Furthermore, these measures of HPA function along with psychological measures of stress could be linked with measures of hippocampal and other brain volumes obtained through neuroimaging techniques to determine whether greater HPA dysfunction/stress is associated with a more severe loss of brain volume during the transition to psychosis.
This review has highlighted other aspects of the relationship between psychosis and HPA-axis activity that warrant further attention. These include differences in level of HPA-axis dysfunction associated with various psychotic disorders other than schizophrenia, the profile of HPA-axis activity associated with different subtypes of schizophrenia, and determining whether the therapeutic effect of antipsychotic medication is mediated, in part, by normalizing HPA activity.
Given the potential link between stress, HPA dysfunction, hippocampal damage and psychotic symptoms, the response to stress and the HPA-axis could be a potentially important therapeutic target in psychosis. Both psychosocial and pharmacological interventions may have a role to play. Although still in its early stages, a greater understanding of the relationship between HPA-axis function and the development of psychosis could provide the opportunity for a range of novel early intervention and preventive treatments.