
Abstract
Bipolar disorder is a severe, persistent mental illness that affects >3% of the population [1, 2]. Few studies have addressed the management of hypomania, despite it being the presenting symptom of many patients in outpatient settings [3]. Oxcarbazepine is a newer antiepileptic drug with proven efficacy in partial seizures as monotherapy and adjunctive therapy for the treatment of adults and children (US ≥ 4 years old, EU ≥ 6 years old). Although an analogue of carbamazepine, there is no known increased incidence of aplastic anaemia or agranulocytosis with oxcarbazepine, but a slightly higher rate of hyponatraemia is reported (2.5%; Physician's Desk Reference
Studies in the 1980s suggested efficacy of oxcarbazepine in bipolar disorder. Studies conducted in Germany suggest that oxcarbazepine has efficacy and tolerability in treating acute mania, with fewer side-effects and drug–drug interactions than carbamazepine [4, 5]. Four 2 week trials found oxcarbazepine superior to placebo, similar in efficacy to lithium and haloperidol and better tolerated than haloperidol [4, 5]. A series of more recent open-label and retrospective studies also support the efficacy of adjunctive oxcarbazepine for reducing mania and depression [5–9] however, a recent double-blind, randomized, placebo-controlled trial ofoxcarbazepine versus placebo in children and adolescents showed no difference between oxcarbazepine and placebo in the treatment ofacute manic or mixed episodes [10].
Comparative studies of oxcarbazepine and divalproex in the treatment of bipolar disorder are limited to date. Divalproex is effective as monotherapy treatment of manic episodes of bipolar disorder [11, 12], and oxcarbazepine had similar efficacy to valproate in a non-randomized, 10 week study and in a retrospective review [4, 13].
In this 8 week study of outpatients with bipolar disorder, currently hypomanic, oxcarbazepine was compared to divalproex. The present study examines the outcomes of treating hypomania (primary outcome), concomitant depressive symptoms, and tolerability with oxcarbazepine compared with divalproex in a randomized, blinded-rater, open-label trial.
Methods
Study overview
The study was approved by the University of Texas Southwestern Medical Center Institutional Review Board and all participants signed an informed consent form. Inclusion criteria were DSM-IV diagnosis of bipolar I, II, or not otherwise specified (NOS); no medications or a stable medication regimen for at least 1 month prior to study entry; age 18–65 years; sodium serum levels between 134 and 146 mEq L–1; and currently experiencing hypomania (YMRS ≥ 12), confirmed on at least two occasions prior to randomization. Exclusion criteria were substance abuse/dependence within the past month; being pregnant or nursing; known hypersensitivity to oxcarbazepine or carbamazepine; severe liver disease and/or dysfunction, or hyponatraemia; or a suspected chronic infectious disease.
A diagnostic interview (modified Structured Clinical Interview for the DSM-IV [14]), physical exam, and blood draw for laboratory evaluation (basic haematology, organ function, and electrolytes), were completed at baseline. Each patient was randomly assigned to receive either oxcarbazepine or divalproex.
Clinical symptoms were evaluated weekly for 4 weeks, then biweekly for 4 weeks, for a total of 8 weeks. A rater blind to treatment assignments completed all ratings. All discussion of study and subjects was kept separate from staff meetings.
Clinical symptoms of hypomania, depression, and overall functioning were rated using the YMRS [15], the IDS-C [16], and Clinical Global Impressions scale for use in bipolar illness (CGI-BP) [17], respectively. The primary outcome measure was change in YMRS score from baseline to end-point. Baseline labs were repeated at visits 4 and 8. Side-effects and adverse events were assessed at every visit. Only side-effects severe enough to cause functional impairment were considered adverse events.
Oxcarbazepine was initiated at 300 mg day−1 and increased to a maximum of 2400 mg day−1, with a target dose of 1200 mg day−1. Divalproex was initiated at 500 mg day−1 and increased to a minimum blood level of 50 mg mL−1. Plasma levels of divalproex were obtained biweekly. A total of 10 mg lorazepam was allowed for acute agitation. No other changes in ongoing medications were permitted.
Statistical analyses
Means and standard deviations are provided for age and weight at baseline and independent samples t-tests were used to examine group differences. Frequencies and percentages are provided for categorical variables and χ2 or Fisher's exact test were used to test group differences. Primary outcome was change in YMRS. Random regression models were used to assess YMRS, IDS-C, and CGI1c (severity, overall illness) scores with terms for group, time, and group×time interaction, using the baseline value as the covariate. Medians were used to describe the number of side-effects experienced by the two groups and the Kruskal–Wallis was used to test the differences. All p were ≤0.05. SAS version 9.0 (SAS Institute, Cary, NC, USA) and SPSS version 13.0 (SPSS, Chicago, IL, USA) were used for analyses and all statistical tests were two-tailed.
Results
Demographics
Three patients had baseline visits with no follow-up visits (oxcarbazepine, n = 2; divalproex, n = 1). These patients were excluded a priori from efficacy analyses but were included in baseline demographic analyses. The evaluable number of patients for efficacy analyses was 13 for the oxcarbazepine group and 14 for the divalproex group.
Patients in the oxcarbazepine group were younger than those in the divalproex group (30.1±8.0 vs 36.9±9.9 years, respectively; p = 0.05). Other baseline characteristics were similar between groups (Table 1).
Subject characteristics
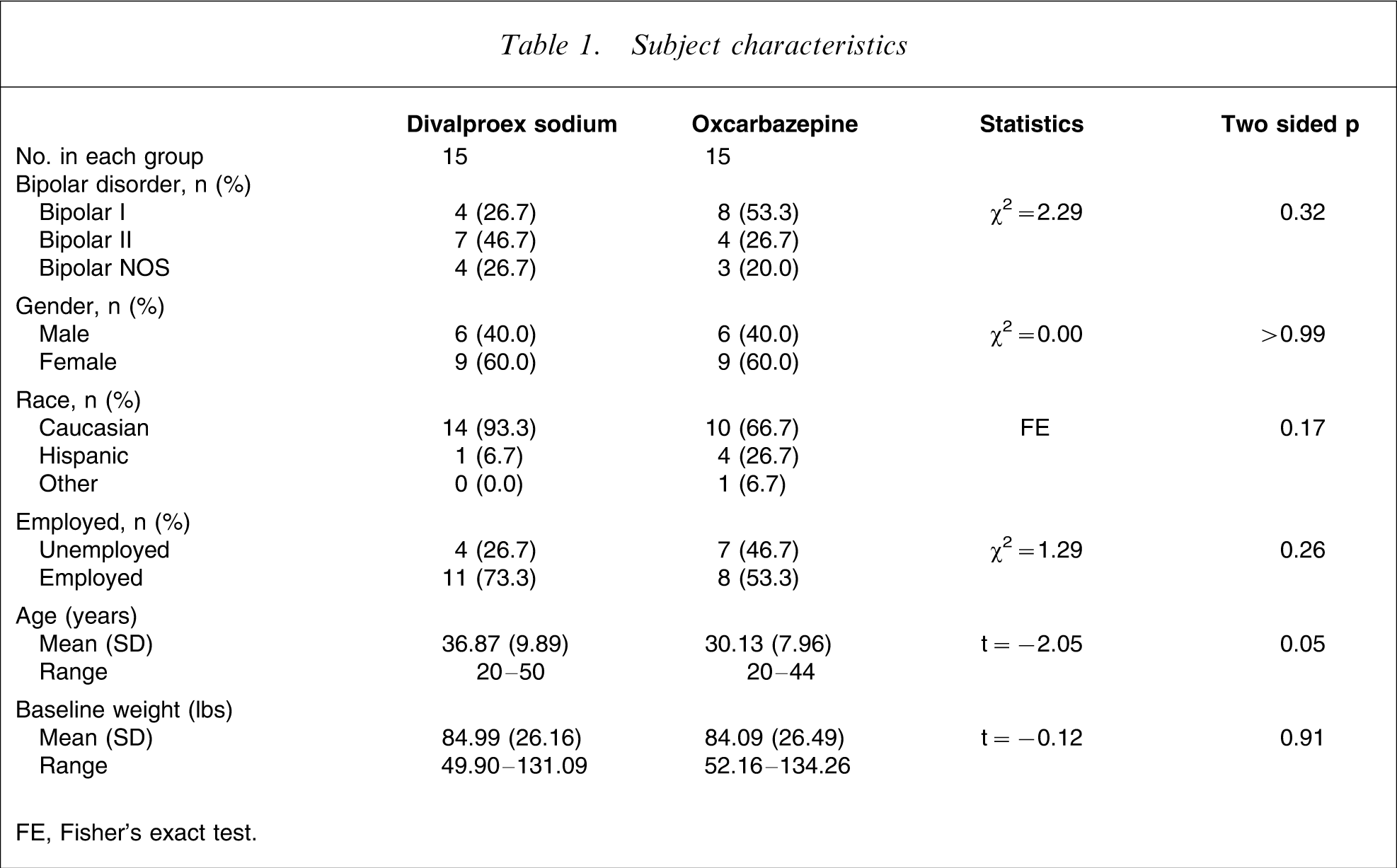
FE, Fisher's exact test.
Of the 30 patients enrolled, 17 completed the study. Six were lost to follow up (oxcarbazepine, n = 3; divalproex, n = 3), four had worsening mood symptoms (oxcarbazepine, n = 2; divalproex, n = 2), one discontinued for lack of improvement, one discontinued due to side-effects, and one patient withdrew for personal reasons.
Medications
The mean peak dose of oxcarbazepine was 1350 mg (450–2400 mg). The mean peak dose of divalproex was 1167 mg (750–2000 mg), and the mean peak serum level was 91.7 mg (52–147 mg). Sixteen patients used study drugs as monotherapy (oxcarbazepine, n = 7; divalproex, n = 9), and 14 as add-on treatment (oxcarbazepine, n = 8; divalproex, n = 6). Medication use at baseline for both groups was similar (Table 2).
Medication use at baseline (n = 30)
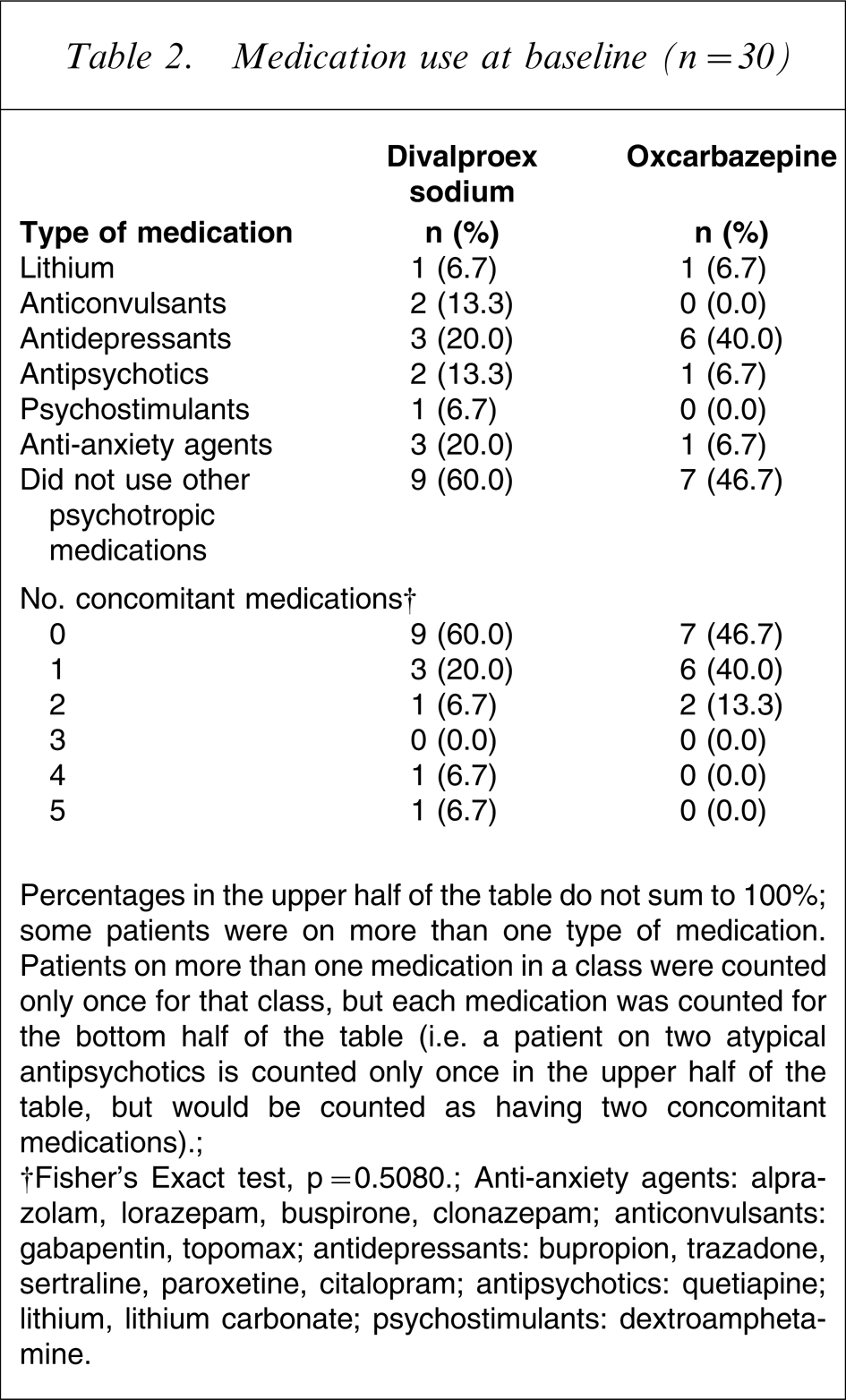
Percentages in the upper half of the table do not sum to 100%; some patients were on more than one type of medication. Patients on more than one medication in a class were counted only once for that class, but each medication was counted for the bottom half of the table (i.e. a patient on two atypical antipsychotics is counted only once in the upper half of the table, but would be counted as having two concomitant medications).
†Fisher's Exact test, p = 0.5080.
Anti-anxiety agents: alprazolam, lorazepam, buspirone, clonazepam; anticonvulsants, gabapentin: topomax; antidepressants: bupropion: trazadone, sertraline, paroxetine, citalopram; antipsychotics: quetiapine; lithium, lithium carbonate; psychostimulants: dextroamphetamine.
The number of types of concomitant medications across groups was found to be non-significant (p = 0.56) with a median of 1 in the oxcarbazepine group and 0 in the divalproex group. The majority of patients (n = 9) receiving combination treatment were receiving only one other medication (oxcarbazepine, n = 6; divalproex, n = 3). Nine patients were receiving antidepressants for depression or insomnia (oxcarbazepine, n = 6; divalproex, n = 3); all nine patients maintained their antidepressant therapy over the course of the study.
Patients receiving antidepressants continued based on clinical need and if it could be clearly determined from medical history and duration of treatment that these agents were not causal of hypomanic symptoms. Of the nine patients receiving antidepressants, seven received selective serotonin re-uptake inhibitors (citalopram, n = 2; sertraline, n = 4; paroxetine, n = 1) and two received bupropion SR. The two patients receiving trazadone took this as a sleep aid adjunctive to other antidepressant medication. Patients receiving antidepressants had been stable on medications for a minimum of 6 months. The mean duration of treatment was 21 months (SD = 24), with a range of 6–84 months, and a median duration of 14 months.
Symptom ratings
Figure 1 (a) shows the mean±standard errors by group for the YMRS total scores. Mean YMRS scores at baseline were 22.07±5.86 and 20.53±6.02 for the oxcarbazepine and the divalproex groups, respectively. Mean scores at end-point were 8.8±7.2 (n = 13) and 8.1±9.2 (n = 14) for the oxcarbazepine and divalproex groups, respectively. Using the baseline value as a covariate, a random regression analysis showed that the overall difference between baseline and final visit YMRS scores was significant (F(1, 22) = 59.08, p < 0.001), but there were non-significant between group differences for the YMRS total score (p = 0.95) and IDS-C total score (p = 0.82). There was no group×score interaction for YMRS and IDS-C.
(a) Mean Yound Mania Rating Scale (YMRS) Total Score across visits by group; (b) mean Inventory of Depressive Symptoms–Clinician Version (IDS-C) Total Score across visits by group. Bar = ±1 standard error.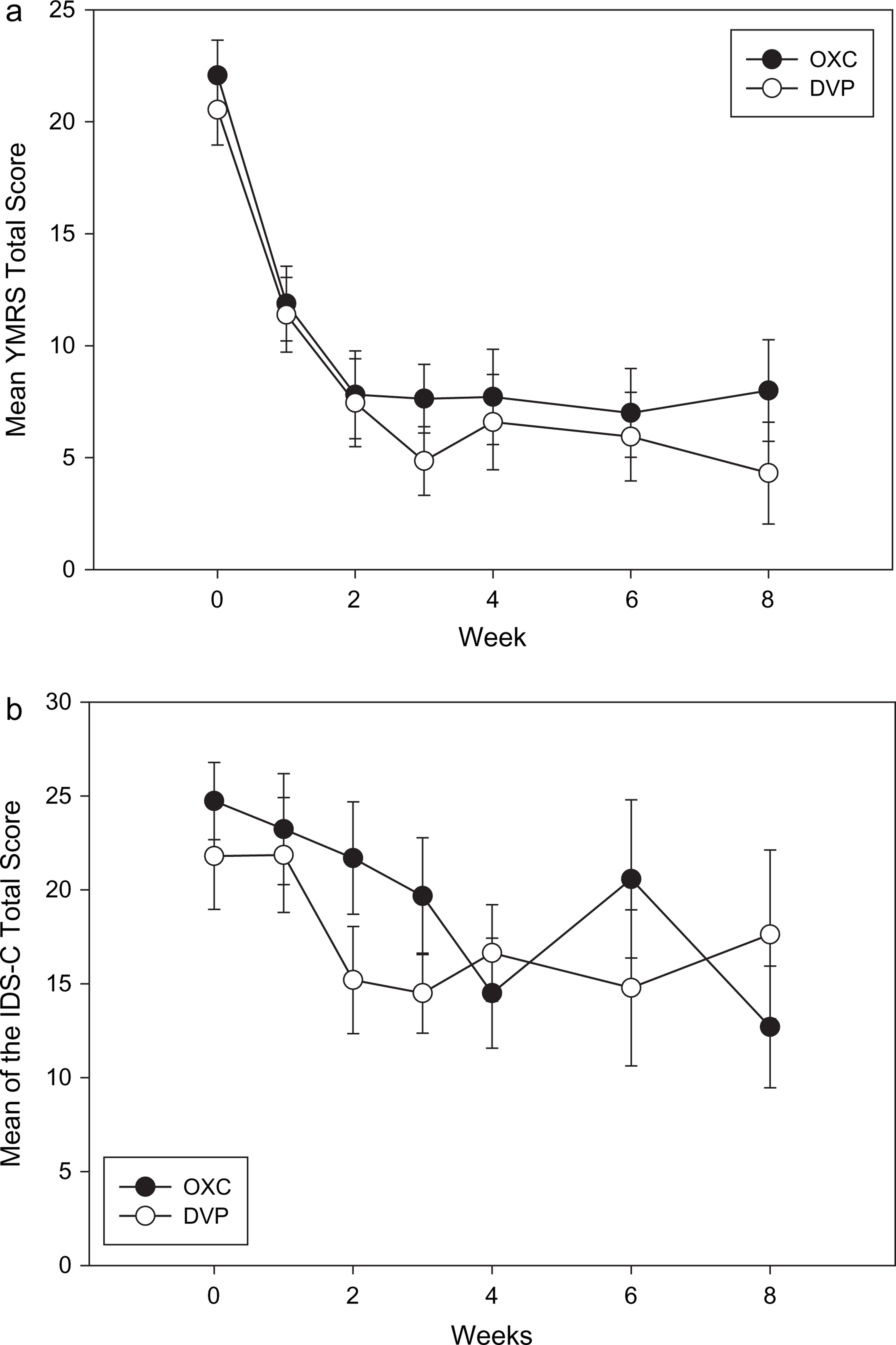
For the YMRS, the mean percent reduction from baseline to week 8 was smaller for the oxcarbazepine group compared to the divalproex group (63.8% vs 79.0%, respectively), and both groups achieved a mean reduction >50% at week 2.
For the IDS-C, the mean percent reduction from baseline to week 8 for the oxcarbazepine group was greater than for the divalproex group (48.7% vs 19.7%, respectively) with neither group achieving a >50% mean reduction during the 8 weeks (Figure 1b).
Mean baseline scores on the CGI1c (severity of illness) for the oxcarbazepine and divalproex groups were 4.08±0.86 and 4.00±0.68, respectively, and mean scores at termination were 2.00±1.63 and 2.75±1.75, respectively. Using the baseline value as a covariate, a random regression analysis showed non-significant group differences for the CGI1c score (p = 0.37). There was no group×score interaction for CGI1c.
Side-effects
The results from the two-way ANOVA with one between-subjects factor (group) and one within-subjects factor (visits) showed non-significant effects for group, visit, and the group×visit interaction for all patients with the last observation carried forward (p = 0.81, p = 0.25, and p = 0.31, respectively). The median number of side-effects was 2.0 for oxcarbazepine and 3.0 for divalproex. This difference was not statistically significant (Mann–Whitney U-test, p = 0.29). The most common side-effects for patients in both groups (n ≥ 5) were drowsiness or sedation; other common side-effects (n ≥ 5) were dizziness or lightheadedness, blurred vision, increased thirst, and headaches in the oxcarbazepine group and tiredness, increased appetite, and weight gain in the divalproex group. No patient developed hyponatraemia, and one patient discontinued medication (divalproex) due to side-effects.
Discussion
Oxcarbazepine and divalproex sodium show similar antimanic efficacy in this single-blind, randomized study. Despite the low YMRS threshold of 12 for hypomania, the mean YMRS was ≥20, consistent with moderate to severe hypomania or mild mania. Thus, this group of patients presented with clinically relevant symptomatic illness.
The results from the present study are consistent with recent retrospective, non-randomized, and add-on studies of oxcarbazepine [4–9, 13]. These studies report varying degrees of efficacy in patients with bipolar disorder, in most cases with concomitant medications. Our research is consistent with European data suggesting that oxcarbazepine may be primarily effective as an antimanic [5–7, 13].
This pilot study found similar efficacy and tolerability to an established antimanic, divalproex, and no significant between group differences on change from baseline in hypomanic or depressive symptoms. Good tolerability with no significant differences was seen in somatic complaints between the two groups. Longer duration studies are needed to fully assess side-effect issues.
A power analysis was done in an attempt to estimate what sample size would be needed to obtain a significant difference in the overall group YMRS means. The observed F-ratio was small so that several thousand subjects (n > 3000) would be needed. This is unsurprising, because both groups improved significantly, and relatively equally, over time. A second power analysis was done to determine what sample size would be necessary to show a significant group×visit interaction on the YMRS. Although the interaction was similarly non-significant, it was larger in absolute value, allowing for a more reasonable estimate. In particular, a total of approximately 130 subjects would be needed to achieve significance at the 0.05 level, and approximately 200 subjects would be need at the 0.01 level.
Limitations of the present study were the open-label treatment design, the add-on study design, the small sample size, and the relatively brief period of observation. Although randomization and blinded raters are strengths, double-blinded studies are needed.
The results from the present pilot study support a comparable degree of efficacy in treating hypomania in patients with bipolar disorder with reasonable tolerability for oxcarbazepine as compared to divalproex. Further controlled studies of oxcarbazepine in bipolar disorder are recommended.
Disclosure
Dr. Suppes receives clinical grants or medications for grants from Abbott Laboratories, AstraZeneca, GlaxoSmithKline Pharmaceuticals, JDS Pharmaceuticals, Janssen Pharmaceutica, National Institute of Mental Health, Novartis Pharmaceuticals, Pfizer Inc, The Stanley Medical Research Institute, and Wyeth; she serves on on the advisory board or as a consultant for Abbott Laboratories, AstraZeneca, Eli Lilly Research Laboratoroies, GlaxoSmithKline Pharmaceuticals, Novartis Pharmaceuticals, and Pfizer Inc; she serves on the speaker's bureau for AstraZeneca and GlaxoSmithKline Pharmaceuticals; and receives royalties from Compact Clinicals Publishers. Dr. Sureddi is a speaker for Cyberonics, Wyeth, Avanir Pharmacuticals, Bristol Myers Squibb, Pfizer, GlaxoSmithKline, Janssen, and Sanofi-Aventis; and is a stock shareholder of Johnson and Johnson, GlaxoSmithKline, Pfizer, AstraZeneca, and Bristol Myers Squibb. Dr. Snow is a speaker for Bristol Myers Squibb and Eli Lilly. Drs. Hynan and Foster, Ms. Kelly, and Mr. Curley have no financial conflicts to disclose.
Footnotes
Acknowledgements
Thanks to Novartis, who provided funding for this research and editorial assistance on the final manuscript.