
Abstract
The triptans, selective serotonin 5-HT1B/1D agonists, are very effective acute migraine drugs. Soon, seven different triptans will be clinically available at 13 different oral doses, making evidence-based selection guidelines necessary. Triptan trials have similar designs, facilitating meta-analysis. We wished to provide an evidence-based foundation for using triptans in clinical practice, and to review the methodological issues surrounding triptan trials.
We asked pharmaceutical companies and the principal investigators of company-independent trials for the ‘raw patient data’ of all double-blind, randomized, controlled, clinical trials with oral triptans in migraine. All data were cross-checked with published or presented data. We calculated summary estimates across studies for important efficacy and tolerability parameters, and compared these with those from direct, head-to-head, comparator trials.
Out of 76 eligible clinical trials, 53 (12 not yet published) involving 24 089 patients met the criteria for inclusion. Mean results (and 95% confidence intervals) for sumatriptan 100 mg, the first available and most widely prescribed oral triptan, are 59% (57-60) for 2 h headache response (improvement from moderate or severe to mild or no pain); 29% (27-30) for 2 h pain free (improvement to no pain); 20% (18-21) for sustained pain free (pain free by 2 h and no headache recurrence or use of rescue medication 2-24 h postdose), and 67% (63-70) for consistency (response in at least two out of three treated attacks); placebo-subtracted proportions for patients with at least one adverse event (AE) are 13% (8-18), for at least one central nervous system AE 6% (3-9), and for at least one chest AE 1.9% (1.0-2.7).
Compared with these data: rizatriptan 10 mg shows better efficacy and consistency, and similar tolerability; eletriptan 80 mg shows better efficacy, similar consistency, but lower tolerability; almotriptan 12.5 mg shows similar efficacy at 2 h but better sustained pain-free response, consistency, and tolerability; sumatriptan 25 mg, naratriptan 2.5 mg and eletriptan 20 mg show lower efficacy and better tolerability; zolmitriptan 2.5 mg and 5 mg, eletriptan 40 mg, and rizatriptan 5 mg show very similar results.
The results of the 22 trials that directly compared triptans show the same overall pattern. We received no data on frovatriptan, but publicly available data suggest substantially lower efficacy. The major methodological issues involve the choice of the primary endpoint, consistency over multiple attacks, how to evaluate headache recurrence, use of placebo-subtracted proportions to control for across-study differences, and the difference between tolerability and safety. In addition, there are a number of methodological issues specific for direct comparator trials, including encapsulation and patient selection.
At marketed doses, all oral triptans are effective and well tolerated. Differences among them are in general relatively small, but clinically relevant for individual patients. Rizatriptan 10 mg, eletriptan 80 mg and almotriptan 12.5 mg provide the highest likelihood of consistent success. Sumatriptan features the longest clinical experience and the widest range of formulations. All triptans are contra-indicated in the presence of cardiovascular disease.
Introduction
Migraine is a common, multifactorial neurovascular disorder, typically presenting as recurrent disabling attacks of moderate to severe headache, nausea, vomiting, photophobia and phonophobia, and, in up to one-third of patients, neurological aura symptoms (1). For over 80 years, ergotamine and dihydroergotamine have been the most widely prescribed specific acute treatments for migraine attacks. However, their use in clinical practice is complicated by the high affinity for a wide range of different receptors, unpredictable absorption and metabolism, very limited evidence for efficacy, poorly justified dose recommendations, and potent and sustained generalized and coronary vasoconstrictor effects; the use of ergot derivatives is precluded in the presence of cardiovascular disease (2).
Improved understanding of the neurobiology of migraine and 5-HT (serotonin; 5-hydroxytryptamine) receptors has been facilitated by the development of the novel class of selective 5-HT1B/1D agonists, known as the triptans (3). These compounds have three putative main mechanisms of anti-migraine action, which are probably primarily mediated via 5-HT1B/1D receptor agonist activity: cranial vasoconstriction (4), peripheral neuronal inhibition (5), and inhibition of transmission through second order neurones of the trigeminocervical complex (6). The relative importance of each of these mechanisms remains uncertain (7). In comparison with ergots, triptans have several distinct advantages. These include selective pharmacology, simple and consistent pharmacokinetics, evidence-based prescribing instructions, high efficacy, modest side-effects, and a well-established safety record (8). They are, however, like ergot derivatives, also contraindicated in the presence of cardiovascular disease. Despite the higher price, triptans are preferred over ergots in the majority of patients (2, 9). They are now the leading class of prescription migraine medications in many Western Countries.
Given that seven different triptans, five different formulations, and at least 13 oral doses will soon be clinically available, physicians need evidence-based guidelines to select the triptans and doses with the highest likelihood of success. Direct active comparator trials are available for only a few triptans and it is unlikely that they will ever all be compared. Moreover, although head-to-head comparator trials are considered the gold standard for comparing drugs, there are also some important pitfalls, which may complicate the interpretation and generalization of such studies (see below). The clinical trials with triptans are very similar in study design, entry criteria, patient populations, and outcome measurements (10, 11). Meta-analysis of triptan trials is thus feasible and may provide a useful summary of the efficacy and tolerability of the different triptans across studies. Previous triptan meta-analyses were based on summary data from published trials only, and only analysed a few outcome parameters and summary measures, no adverse events, and a limited number of agents and doses (12, 13).
During migraine attacks, the oral absorption of many drugs is delayed (14), favouring non-oral routes of administration. Most patients, however, prefer oral formulations (15) and these formulations account for >80% of all triptan prescriptions (H. Mansbach, GlaxoSmithKline Ltd; personal communication). We shall therefore concentrate on the oral formulations. Sumatriptan is also available in parenteral formulations; these will be discussed.
Here we review the detailed results of a large meta-analysis based on the complete data sets (‘raw data’ of 24 089 patients) of all 53 eligible, randomized, controlled clinical trials (including 12 not yet published studies) involving six of the seven oral triptans that will soon be available. For the seventh triptan, frovatriptan, only summary data presented in congress abstracts could be used. Important efficacy and tolerability (adverse events) parameters were evaluated for all recommended doses in a meta-analysis of all placebo-controlled trials and then contrasted with a separate meta-analysis of 22 direct active comparator trials. Both approaches have complementary strengths and limitations, which will be considered in the discussion. Some main results from the efficacy analysis have been published elsewhere (16), and are recounted here for completeness. Given the importance of triptan use in clinical practice, we have devoted considerable attention here to the methodological details of the meta-analysis and clinical trials included; these have not been covered in depth in our previous account. We further included several additional results on efficacy and tolerability. First, we review the main pharmacokinetic differences between the seven triptans and discuss some important issues and pitfalls in the clinical evaluation of acute migraine trials. We then cover the clinical results from the meta-analysis.
Pharmacokinetic differences of the triptans
The major pharmacokinetic characteristics of the oral triptans are summarized in Table 2. Drug plasma half-lives (T1/2) range from 2 to 25 h; frovatriptan, naratriptan, and eletriptan have the longest T1/2. A long T1/2 was once thought to predict a longer duration of action and lower recurrence rate, although the available data suggest that this may not be correct (17, 18). Indeed, frovatriptan with the longest T1/2 has no demonstrable recurrence benefit over sumatriptan (see below).
The triptans (5-HT1B/1D receptor agonists)

†Previously GlaxoWellcome.
‡F11356 (63).
Comparison of some pharmacokinetic characteristics of triptans∗

∗Doses selected based on Table 1. Smaller font implies a less important route of elimination.
†VML251 or SB209509.
Short time to peak plasma levels (Tmax) is likely to be associated with rapid onset of action. Tmax reflects both the absorption and first-pass metabolism, but interpretation is not always straightforward. First, some triptans, such as sumatriptan and zolmitriptan, show double peaks, with a second peak only slightly higher but significantly later than the first. As a result, the Tmax (to the second peak) may not reflect the rise within the first hour (i.e. primarily time to the first peak). Second, because absorption may be delayed during attacks, only Tmax values during attacks are clinically informative. Claims based on studies conducted outside attacks or in healthy volunteers are potentially misleading. During attacks, rizatriptan shows the shortest Tmax (1 h) of all triptans; for the other triptans ictal Tmax ranges from 2 h to 4 h. No ictal values are available for naratriptan.
Compared with sumatriptan all new triptans, particularly almotriptan and naratriptan, show significantly higher oral bioavailability. This may predict a more consistent efficacy over multiple attacks.
If central sites of action are important, migraine compounds that achieve high CNS levels should have an advantage in efficacy, possibly coupled to a higher risk of CNS adverse events. High lipophilicity, and therefore the potential to cross the blood–brain barrier, and low affinity for the P-glycoprotein pump in the blood–brain barrier, which actively removes compounds from the brain (19, 20), should optimize brain penetration. However, higher relative lipophilicity combined with being a substrate for the P-glycoprotein pump, such as is the case with eletriptan, makes predictions of efficacy and side-effects complex. Moreover, given clinical data from other indications where sumatriptan has been used (21), the degree of brain access by all triptans is unclear.
With respect to drug metabolism, naratriptan is the least metabolized, and therefore most likely to be free of clinically significant drug interactions on a metabolic basis. Sumatriptan is metabolized by monoamine oxidase (MAO) and thus relatively contraindicated when MAO inhibitors are used. The rare use of MAO inhibitors makes this issue minor and in practice the interaction relatively minor (22). Similarly, there is a theoretical risk of the serotonin syndrome (23) with coexistent use of specific serotonin re-uptake inhibitors (SSRIs), such as fluoxetine (24) and triptans. This syndrome is extraordinarily rare despite widespread co-administration; the risk is increased when triptans are misused. Rizatriptan is metabolized by MAO-A and has a minor active metabolite N 10-monodesmethyl rizatriptan. The metabolism of rizatriptan is affected by propranolol because one of its metabolites competes with rizatriptan for MAO-A. This MAO-A interaction is due to the dimethyl-amino side chain on rizatriptan, a chemical feature shared by sumatriptan and zolmitriptan. As a result, rizatriptan levels are elevated in some patients taking propranolol and a reduction of the primary dose from 10 mg to 5 mg is recommended in these patients. Other β-blockers, such as metoprolol, do not share this route of metabolism and do not affect rizatriptan. Zolmitriptan is metabolized in the liver by cytochrome P450 (CYP1A2) (25), to an active metabolite N-desmethyl-zolmitriptan (183C91) and an inactive N-oxide and indole acetic acid, although some monoamine oxidase metabolism is also involved (26). The latter becomes important in the presence of propranolol at zolmitriptan doses of 15 mg/day, which is, however, not a clinically used dose (27).
Clinical evaluation and pitfalls in acute migraine trials
Endpoints (Table 3)
Definitions of clinical outcome measures used in the present review

In most triptan trials, patients were instructed to treat a migraine headache when pain is moderate or severe on a 4-point pain severity scale (0=no pain; 1=mild; 2=moderate; 3=severe pain) and within 6–8 h of onset of the headache (10). The primary endpoint usually was the proportion of patients with a headache response (i.e. improvement from moderate or severe pain at baseline to mild or no pain 2 h post-dose). More recently, the proportion of patients who become pain-free 2 h post-dose has become the preferred primary endpoint. Pain free is intuitive, robust, less sensitivity to placebo effect, and produces better dose–effect relationships (11, 15). In addition, patients rate rapid onset and complete relief of pain as among the most important attributes of migraine treatment; these measure are more predictive of satisfaction with treatment and improvement in health-related quality of life (15, 28). Some publications present 4 h headache response rates only (29, 30), but this may create a false impression of efficacy due to the self-limiting nature of migraine attacks (31, 32). It also seems questionable to delay rescue medication beyond 2 h. Changes in the associated symptoms and functional disability are correlated with the reduction in pain and therefore are only secondary endpoints. Tolerability and safety are evaluated by reporting of adverse events (AEs) and routine blood, urine tests, and ECG. The important difference between tolerability and safety is discussed below.
Headache recurrence and sustained pain free
In some responders, the headache may return within 24 h of initial relief (= headache recurrence or relapse) requiring re-dosing (17, 32, 33). This is inconvenient to the patient, expensive, and may lead to medication overuse and medication-overuse headaches (34–36). Simple comparison of recurrence rates (as proportion of responders), without accounting for differences in initial relief rates and use of rescue medications, may be misleading. When comparing two drugs, the more effective drug will presumably relieve attacks that would not have responded to the less effective drug. On average, the responders to the more effective drug have headaches that are more difficult to treat than the responders to the less effective drug; they may also be more susceptible to headache recurrence (16, 32, 37). We therefore recommend the use of sustained pain free, a composite measure that is defined as the proportion of patients who are pain free by 2 h post-dose and who do not experience a recurrence of moderate or severe headache and who do not use any rescue headache medication 2–24 h post-dose (16, 32, 37; Table 3). Sustained pain free is easy to explain to patient and physician as ‘the proportion of patients who require only a single dose to abort their attack by 2 h and for at least 24 h’. It captures the central elements of what patients say they want from treatment (15) and captures the factors that predict satisfaction with treatment and health-related quality of life (28). It is sometimes also called ‘complete response’ (38), but this can be a confusing term as many interpret it as complete freedom from all symptoms without any implication of maintenance of relief (39). Although sustained pain free is the ideal efficacy endpoint (15), it is also the hardest to achieve and it may be unrealistic to expect high sustained pain-free rates with the current drugs (32).
Consistency
Most acute migraine trials study only one attack per patient. Because migraine is a chronic disorder with recurrent attacks, patients value highly a predictable, consistent efficacy over many attacks (intra-patient consistency); consistency of response increases the patients' confidence and satisfaction with treatment (15, 28). Demonstration of intra-patient consistency of efficacy over multiple attacks is therefore highly desirable. Ideally, such trials should be conducted in unselected patients, and include at least random insertion of placebo for one attack. Results should be presented as proportion of patients with relief in at least two or three out of three of four actively treated attacks. So far placebo-controlled consistency trials have studied efficacy in at most three attacks. Some publications report the responder rates for each study attack (40–43), which reflects reproducibility of the population response over multiple attacks rather than intra-patient consistency. Long-term open-label extension trials selectively include responders, lack placebo control and usually report population, not intra-patient consistency. Consistency data from such trials must be interpreted cautiously.
Head-to-head active comparator trials
These are considered the gold standard for comparing drugs, but there are some important caveats. These studies must include samples that are representative of the population of interest and must be large enough to detect clinically meaningful differences. When patients are included with previous experience with one of the active comparator drugs, the results should preferably be presented separately for experienced and non-experienced patients. This is to avoid possible bias caused by selection of patients dissatisfied with one of the agents. Such information is, however, rarely available. Another issue might arise when matching placebo tablets are not available. Encapsulation of either or both study drugs and placebo-control may then be used. However, when encapsulation affects drug absorption (44), the results may be influenced. Pharmacokinetic studies showing bioequivalence (45), notably for the pharmacokinetic profile, including the rise of drug plasma concentration in the first hour, and similar response rates as have usually been obtained with the standard formulation, are required to ensure that an encapsulated treatment is identical to the marketed product. Dissolution studies in vitro are insufficient.
Placebo
Medical Ethics Committees are increasingly resistant to the inclusion of placebo arms in acute migraine trials, although their inclusion is scientifically desirable. Relief rates may vary widely depending on the randomization ratio to placebo. In an active comparator trial with a 1 : 16 chance for participants to get placebo, two drugs previously shown to be highly effective failed to achieve superiority due to the very high placebo response (46). Placebo rates may also differ markedly between continents even within the same study design (47, 48). AEs may occur in >30% of migraine patients after placebo. Thus, migraine trials should ideally include adequate placebo control to allow for a historical comparison and extrapolation of the results; to determine the least effective dose; and to evaluate correctly the incidence of AEs. As long as rescue medication is offered no later than 2 h after initial treatment, this approach is considered ethical.
Meta-analysis of oral triptan trials
Procedure
We approached all six pharmaceutical companies that currently market or intend to market a triptan (Table 1). We sent a standard letter explaining the objectives of the study and asked for the ‘raw patient data’ (patient numbers per item) of all randomized controlled trials involving their compound. Five companies (see Acknowledgements) kindly provided virtually all the requested data of both published and, as yet, unpublished trials on a total of six triptans. Vanguard (now Vernalis) declined to disclose any of the raw data on frovatriptan; we therefore used data from publicly available sources (congress presentations and abstracts). Some summary data were published after the current analysis was completed (49). Where possible, we cross-checked all data with published or presented data. In addition, we conducted a systematic review of the (English) literature for triptan trials that were not company-sponsored, and approached the principal investigators with the same request. The companies that provided data were informed about the exact procedures of the meta-analyses. They received the results of the analyses, for their compound only, 2 months prior to the planned submission of the manuscript, with the request that they check the data for accuracy; there were no comments or objections from the companies. We did not provide our interpretations of the data. The database was closed 1 November 2000.
Studies and data included
For inclusion in the meta-analysis, the following standard criteria had to be met: (i) randomized, double-blind, controlled (placebo or active comparator) clinical trial; (ii) treatment of moderate or severe migraine attacks within 8 h of onset, in migraineurs (18–65 years of age) defined according to the International Headache Society (IHS) criteria (50); (iii) treatment with an oral triptan at a recommended clinical dose; and (iv) measurement of the headache on the four-point pain scale (10). If 2-h efficacy results were not available, studies were only included in the AE analysis. These strict criteria were designed to ensure methodological quality and uniformity. We identified and assessed in total 76 clinical trials. Of these, 53 met the eligibility criteria and are summarized in Table 4. Table 5 summarizes the 23 studies that were excluded and the reasons for exclusion; the most common reasons for exclusion were lack of a control group, use of non-recommended drug doses, or selected study populations (e.g. adolescents).
Included trials; numbers of patients based on 2 h headache response rates


P, Parallel; MA, multiple attack; CO, cross over; DC, only included in direct comparison (no placebo included); A, abstract.
∗ASA + Metoclopramide.
†Cafergot two tablets.
‡No data received from pharmaceutical industry, only abstract reports.
§No. 2-h response data available, included for direct comparison and adverse event analysis.
Excluded trials for efficacy analysis

Data from placebo-controlled trials, those both with and without an active comparator, were combined in the meta-analysis (per patient only the first study attack). Data from direct active comparator trials were also analysed separately. For rizatriptan, two oral formulations are available: traditional tablets and soluble wafers. As the study designs were identical and formulation did not influence results, we combined them.
Statistical analysis
Differences in all endpoints between triptans and placebo were assessed with random effect models as proposed by DerSimonian and Laird (51). Random effect models incorporate potential heterogeneity of the endpoints among different studies by assuming that each study estimates a unique endpoint (52). Homogeneity of observed endpoints was assessed using χ2 tests of independence (53). Because of the relatively low power of tests to detect heterogeneity, we used a very conservative α of 0.1 instead of 0.05, thus increasing the likelihood of detecting heterogeneity by reducing the threshold for statistical significance. The results for these tests are summarized for placebo-subtracted data in Table 6.
Homogeneity of placebo-subtracted data

Note that there was a conservative cut-off point for heterogeneity (P<0.1).
Homogeneity was very good for pain-free, clinically the most relevant and most robust outcome measure. Here, heterogeneity was only found for some of the ‘secondary doses’.
Figure 1 presents a funnel plot of the individual study estimates (for the ‘main doses’ only) for pain-free, visualizing the excellent homogeneity of the individual study estimates. For eletriptan 80 mg there was heterogeneity, but this actually caused a conservative bias (the summary estimate from the random effect model was lower than when calculated with a fixed effects model, thus decreasing the difference from the reference dose). For the other two endpoints there were heterogeneities for some of the doses, which were usually caused by one outlying study. Re-analysis of the data after exclusion of such an outlier, however, never resulted in a significant change of the differences between the summary estimates for the various drugs and doses: previously statistically significant differences remained, as did previously non-significant differences.

Funnel plot of the individual study estimates (and 95% confidence intervals) for the placebo-subtracted pain-free data as well as the summary estimates for all trials per drug as calculated by using a random effect model (for the ‘main doses’ per drug only). Note that only eletriptan 80 mg showed heterogeneity, but that this is actually causing a conservative bias.
When between-studies variance is zero, the study is homogeneous for that triptan dose and endpoint, a random effect model is identical to a fixed effect model. Therefore, random effect models were used for all endpoints. Differences between treatment regimes are presented as means with 95% confidence intervals (CI).
The study designs and eligibility criteria are remarkably similar across the triptan trials. However, even small differences in these factors may influence comparisons of treatment effects across studies. Although such differences will not affect the internal validity of controlled trials (because active drug is compared with a control), they may influence comparisons of treatment effects across studies in a meta-analysis. Three strategies are generally recommended for controlling those influences: (i) the risk or rate ratio (i.e. divide for each individual study the response to the active drug by the placebo response); (ii) the placebo-subtracted proportion or therapeutic gain (i.e. subtract for each individual study the placebo response from the response to the active drug); and (iii) the number needed to treat (NNT), which is the reciprocal of the therapeutic gain. Using the rate ratio approach assumes that the relationship between active drug and placebo is multiplicative; the placebo-subtraction and NNT approaches assume an additive relationship. The additive model is more intuitive for most clinicians. The multiplicative model has at least two disadvantages. First, the commonly used statistical models overestimate the prevalence ratios when the ‘rare disease assumption’ is violated as it is in migraine. In addition, as placebo rates increase (approaching 50%) the maximum ratio is limited (only 2).
We analysed the data by using all three strategies as well as by comparing the absolute values. Results were similar using all four methods. The homogeneities for the outcome measures were virtually identical for both the multiplicative and additive models. Because most clinicians in headache (12) and pain (54) management are familiar with placebo-subtracted rates (or NNT), we elected to present the additive model for comparing the efficacy measures. Likewise, subtracting the placebo AE rate from the active drug AE rate can help to correct the differences in the methods of collection and definitions of AEs among studies (therapeutic harm).
A similar pattern for the differences in treatment effects between the active agents when using absolute proportions and when using placebo-subtracted proportions increases the confidence in the validity of the results. We therefore present data both ways. There are tables for both the efficacy data and the adverse event data in the Appendix.
Results
Reference dose for oral sumatriptan
World-wide, there are two primary oral doses for sumatriptan: 100 mg in most European countries and 50 mg in North America and some other countries. In a head-to-head study, the two doses did not differ (55). For simplicity, we selected the 100 mg dose as the single reference dose, based on the following advantages over the 50 mg dose: (i) 100 mg was the initially developed dose, thus far more clinical trials and patient data are available, resulting in tighter CIs; (ii) there is a better consistency over time and across trials for the usual primary efficacy endpoint, response at 2 h (16); (iii) consistency rates for response are higher (see Fig. 4); (iv) many patients in Europe starting with 50 mg end up with 100 mg; and (v) in a patient preference trial, patients preferred the 100 mg dose over the 50 mg dose (56).

Consistency results. Headache response and pain free at 2 h in at least one out of three attacks, at least two out of three, and three out of three attacks for each of the triptans. Data are presented as group result and 95% confidence intervals. For each agent the white bar indicates the consistency rate for placebo. For rizatriptan this could not be calculated due to the different staggered placebo design (see text). (N.A. indicates not available.)
Headache response at 2 h
Headache response at 2 h is the primary per protocol endpoint in nearly all triptan trials. The mean absolute and placebo-subtracted rates (95% CIs) are depicted in Fig. 2a. Compared with sumatriptan 100 mg (mean=59%; 95% CI 57–60), rizatriptan 10 mg and eletriptan 80 mg show higher, and naratriptan 2.5 mg, eletriptan 20 mg, and frovatriptan 2.5 mg (data from abstracts only) inferior response rates. Zolmitriptan 2.5 mg is just better, while sumatriptan 50 mg, zolmitriptan 5 mg, and rizatriptan 5 mg just miss significance. There are no differences for the other doses and drugs. Placebo-subtracted values show wider CIs and overlap between most triptans (mean for sumatriptan 100 mg = 29%; 95% CI 26–34). A significant positive difference persists for eletriptan 80 mg (mean=42%; 95% CI 36–48) and a negative difference for frovatriptan (mean=17%; 95% CI 13–20).
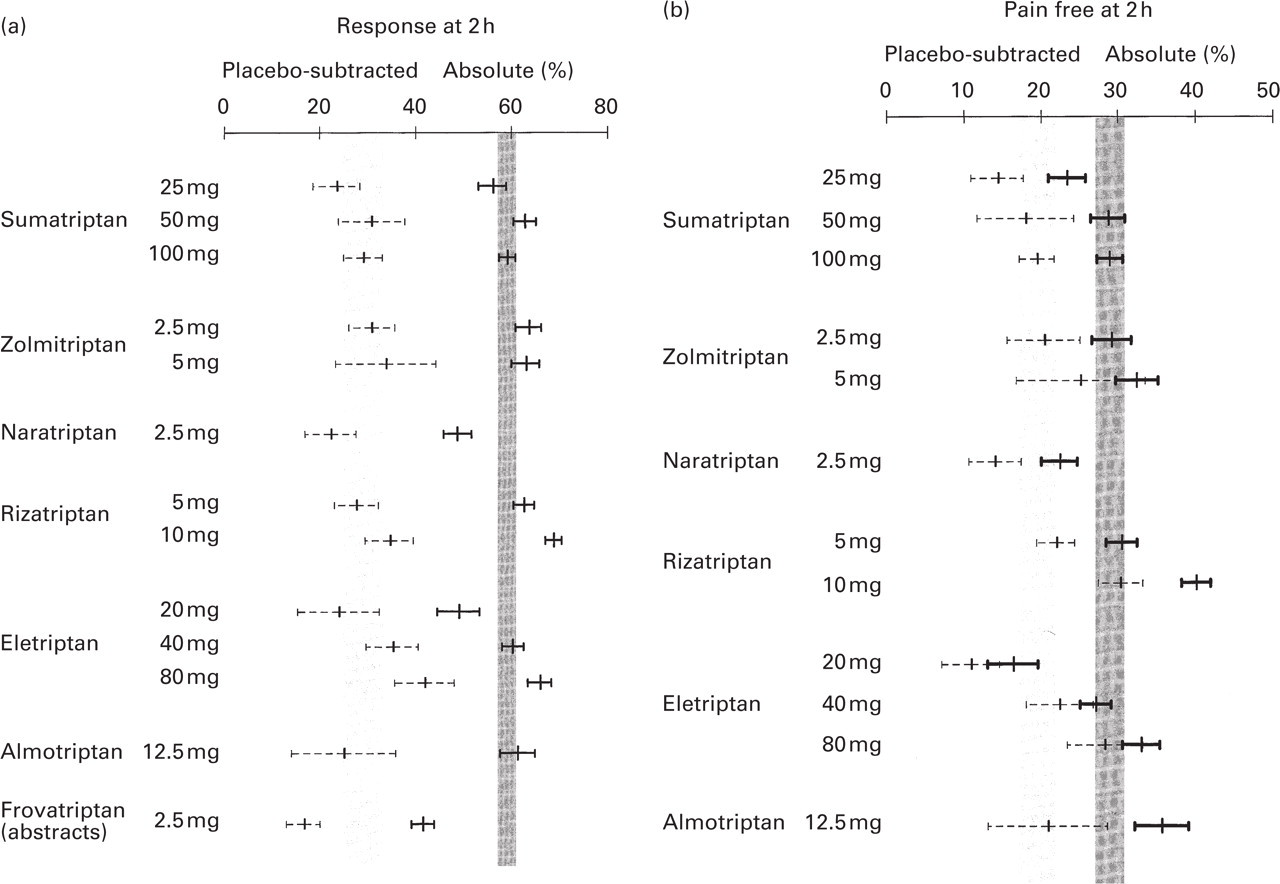
Data (mean and 95% confidence intervals) for headache response at 2 h (a) and pain free at 2 h (b) are shown for each triptan. Absolute and placebo subtracted outcomes are presented with the hatched region being the 95% confidence interval envelope for sumatriptan 100 mg.
Pain free at 2 h
Although the pain-free response is currently the primary endpoint recommended by the IHS Clinical Trial Committee, it was a secondary endpoint in most trials. Data are depicted in Fig. 2b. Compared with sumatriptan 100 mg (mean=29%; 95% CI 27–30), sumatriptan 25 mg, naratriptan 2.5 mg and eletriptan 20 mg show lower absolute pain-free rates, whereas eletriptan 80 mg, almotriptan 12.5 mg, and rizatriptan 10 mg show higher values. The other triptans and doses do not differ from sumatriptan 100 mg. Placebo-subtracted values (mean for sumatriptan 100 mg = 19%; 95% CI 17–22) are significantly higher for rizatriptan 10 mg and eletriptan 80 mg.
Recurrence and sustained pain free
These are depicted in Fig. 3a, b. Compared with sumatriptan 100 mg (mean=30%; 95% CI 27–33), recurrence rates are lower for eletriptan 40 and 80 mg, and higher for rizatriptan 5 and 10 mg. Naratriptan 2.5 mg appears to show a lower recurrence rate, but this is based on 4 h rather than on 2-h response rates and therefore not directly comparable. Other recurrence rates overlap. Note that isolated comparison of recurrence rates may be misleading and that comparison of sustained pain-free rates is preferred. These were calculated, post hoc, for those trials where the following raw data were available: pain free at 2 h, headache recurrence and use of analgesics 2–24 h post-dose. Use of analgesics 2–24 h post-dose was not assessed in some trials and recurrence was sometimes based on 4-h rather than on 2-h response data requiring reanalysis of the data (29, 30, 55). Compared with sumatriptan 100 mg (mean=20%; 95% CI 18–21), sustained pain-free rates are higher for rizatriptan 10 mg, eletriptan 80 mg, and almotriptan 12.5 mg, and lower for eletriptan 20 mg. Sumatriptan 25 mg and naratriptan 2.5 mg tend to show lower values, while no differences were found for the other triptans. The interpretation of recurrence after a response following placebo is unclear, so no placebo-subtracted sustained pain-free rates have been calculated. These rates would have been predictably very small and associated with wide confidence intervals resulting in a low power to detect differences. Patients with sustained pain free could still have had a recurrence of mild headache, but as this did not prompt the use of rescue medication (by definition), the recurrence was unlikely to be clinically significant.
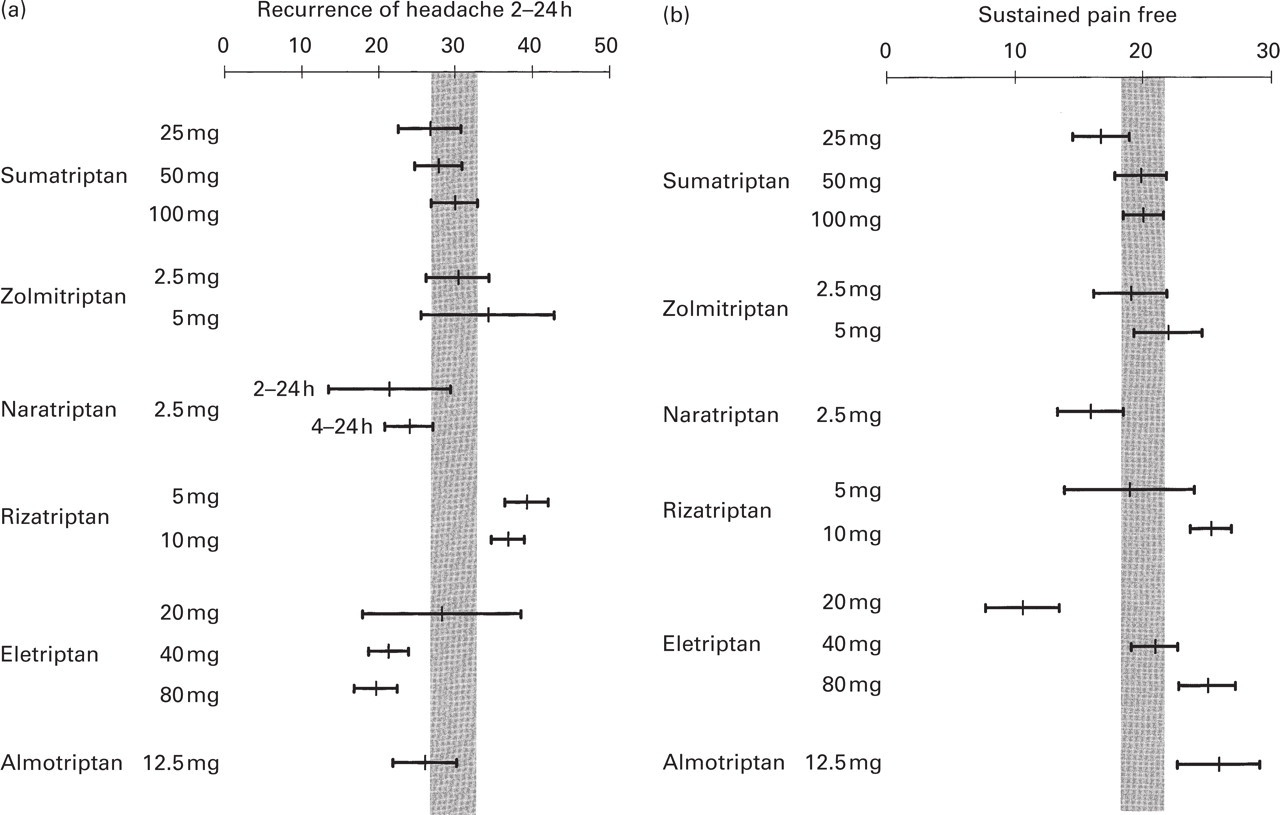
Data (mean and 95% confidence intervals) for headache recurrence from 2 h to 24 h (a) and sustained pain free (b) are presented with the hatched region being the 95% confidence interval envelope for sumatriptan 100 mg. For naratriptan the recurrence rate is given for the time period 4–24 h post-dose (as presented in the original publications) and for 2–24 h post-dose (after recalculating the data).
Intra-patient consistency
Placebo-controlled intra-patient consistency of efficacy over multiple attacks was investigated in only a few studies. No such studies are available for sumatriptan 25 and 50 mg, zolmitriptan 2.5 and 5 mg, and rizatriptan 5 mg, although data have been presented for zolmitriptan 2.5 mg in the context of a comparison with zolmitriptan nasal spray (57). Results are depicted in Fig. 4. All drugs (except rizatriptan 10 mg; see below) were tested in a parallel-group design, treating three consecutive attacks with either active or placebo. These studies show that consistent lack of response is rare: response in at least one of three treated attacks occurs in 79–89% of patients (placebo approximately 50%) and pain free in 51–59% (placebo 18%). Response in at least two of three treated attacks occurs in 47–72% of patients (placebo 17–33%) and pain free in 14–42% (placebo 3–13%); highest consistency rates are for sumatriptan 100 mg and almotriptan 12.5 mg (but here placebo rates are also highest); lowest rates are for naratriptan 2.5 mg and sumatriptan 25 mg. Response in all three attacks occurs in 16–47% of patients (placebo up to 9%) and pain free in 1–17% (placebo<2%); highest consistency rates are for sumatriptan 100 mg and almotriptan 12.5 mg (with highest placebo rates).
The consistency of rizatriptan 10 mg was evaluated in a novel double-blind, randomized, cross-over design over four attacks, with placebo in one attack interspersed at random in four of five patient groups; the fifth group received rizatriptan 10 mg for four attacks (42). The different design complicates the comparison with the other consistency rates, although it seems unlikely that it would have increased consistency. Consistency rates over three attacks are the highest of all triptans: response (and pain-free) rates are 96% (77%) in at least one of three, 86% (48%) in at least two of three and 60% (20%) in all three actively treated attacks (58). In the subgroup of 125 patients who treated three consecutive attacks with rizatriptan, without prior exposure to placebo, the results were very similar: response (and pain-free) rates were 87% (42%) in at least two of three and 50% (16%) in all three attacks.
Consistency of the efficacy with sumatriptan 25 and 50 mg was also tested in two parallel-group trials vs. an active comparator, but without placebo; consistency with zolmitriptan 2.5 and 5 mg was only tested in active comparator trials without placebo. In these active comparator trials with sumatriptan and zolmitriptan, the consistency rates for response (and pain free) were 88–90% (47–59%) in at least one out of three, 65–71% (18–32%) in at least two out of three, and 29–43% (4–15%) in three out of three attacks. The consistency rates of both zolmitriptan doses are slightly higher than those of sumatriptan 25 mg, and similar to those of sumatriptan 50 mg. Compared with the consistency rates in the placebo-controlled trials, the response rates for sumatriptan 25 and 50 mg were 13–16% higher. No such difference was observed for pain free.
Tolerability
AEs with triptans are relatively frequent, but usually mild and short-lived. The most frequent ‘typical triptan AEs’ include tingling, paraesthesias, and warm sensations in the head, neck, chest, and limbs; less frequent are dizziness, flushing, and neck pain or stiffness. Of more relevance are the much rarer ‘central nervous system (CNS) AEs’ (asthenia, abnormal dreams, agitation, aphasia, ataxia, confusion, dizziness, somnolence, speech disorder, thinking abnormal, tremor, vertigo, and other focal neurological symptoms) and notably the ‘chest-related AEs’ (chest pressure, chest pain, radiating pain in arm, other chest feelings, heavy arms, shortness of breath, palpitations, and anxiety).
Differences among studies in the methods of collecting AEs and in their definitions complicate comparisons. In the early sumatriptan studies AEs were collected retrospectively without diary cards. In the zolmitriptan trials AEs were collected for 24 h post-dose and not thereafter. Almirall-Prodesfarma and Pfizer did not provide us with any methodological information except that in the eletriptan studies CNS AEs do not include asthenia and fatigue. The latter is also the case in the rizatriptan studies.
Figure 5a–c depicts the placebo-subtracted proportions of patients with: (i) at least one AE (any AE), irrespective of their nature and intensity, and irrespective of whether or not the study physician judged the AE to be drug-related; (ii) at least one ‘chest AE’; and (iii) at least one ‘CNS AE’. Values greater than zero indicate that AEs occur in more patients for active drug than for placebo; values with narrow 95% CIs which do not cross the zero line indicate placebo-like incidences.
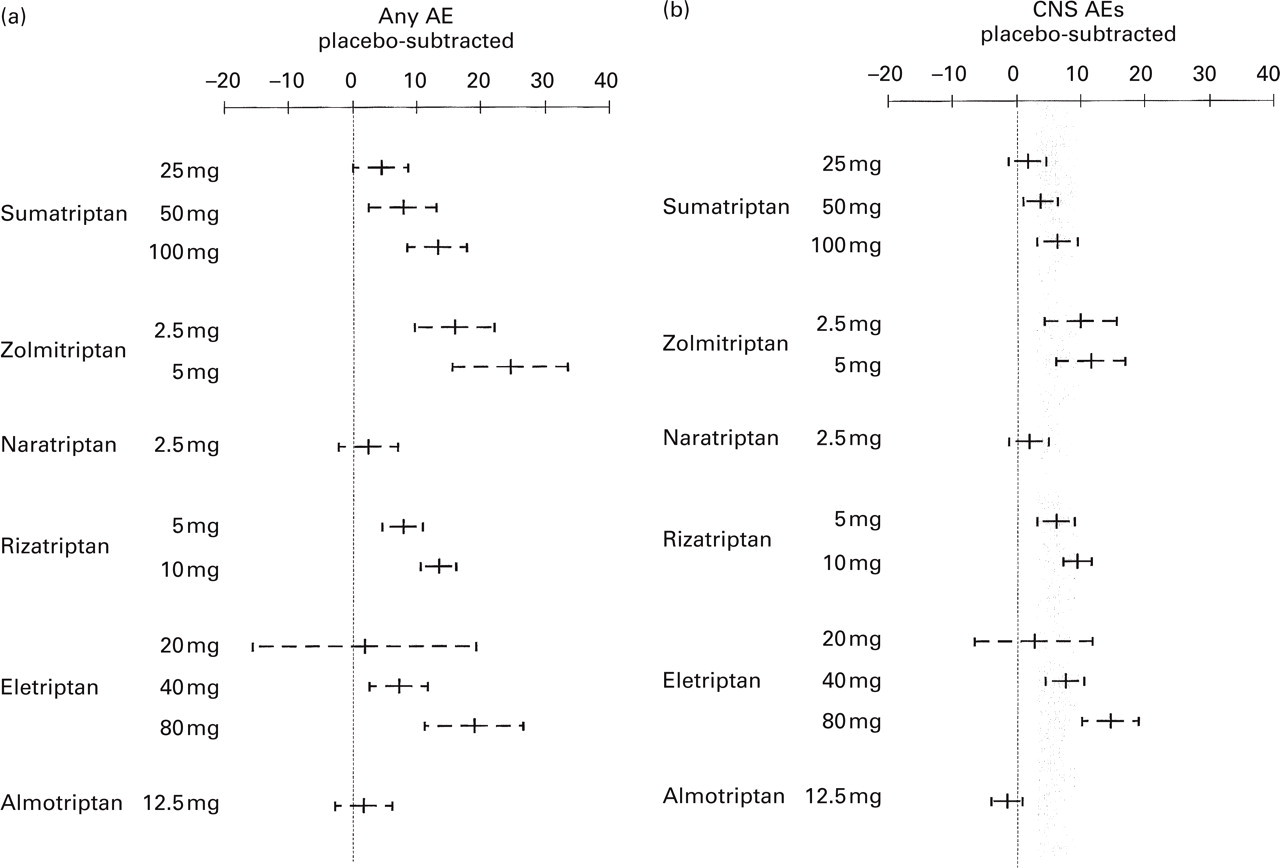
Placebo subtracted adverse event (AE) data (mean and 95% confidence interval) for each triptan for any AE (a), central nervous system (CNS) AE (b), and chest AE (c). The hatched region is the 95% confidence interval envelope for sumatriptan 100 mg.
Sumatriptan 100 mg had a mean placebo-subtracted rate of any AEs of 13% (95% CI 8–18). Rates for other triptans overlap, except for lower values for naratriptan 2.5 mg and almotriptan 12.5 mg; these rates also do not differ from placebo (95% CI is narrow and crosses the zero difference line). Some other AE rates also cross the zero line but the associated 95% CIs are wide. Within compounds, there are clear trends for increasing AE incidences with increasing doses. A similar pattern emerges when only AEs are included which were (blindly) considered by the trial investigator as drug-related; both almotriptan 12.5 mg and naratriptan 2.5 mg, however, still demonstrate the lowest AE rates (data not shown).
CNS AE rates largely overlap with those of sumatriptan 100 mg (mean=6%; 95% CI 3–9), except for higher values for eletriptan 80 mg and lower values for almotriptan (also not different from zero). Some other AE rates also are not different from zero but with wide 95% CIs and overlap with sumatriptan.
For Chest AEs, compared with sumatriptan 100 mg (mean=1.9%; 95% CI 1.0–2.7) almotriptan 12.5 mg has the lowest incidence of ‘chest symptoms’ which is also not different from zero. All other incidences overlap. The incidences for sumatriptan 25 mg, naratriptan 2.5 mg, rizatriptan 5 mg (just), and eletriptan 20 mg and 40 mg do not differ from zero but with wide CIs.
Results for placebo and sumatriptan by company
Consistent differences in designs, populations, and definitions and methods of collecting for AEs among the studies conducted by the different companies might have influenced the meta-analytic comparisons and even head-to-head comparisons of drugs. To identify such differences, we compared the results for placebo and sumatriptan by company as internal standards (Fig. 6). These results may differ from those in Figs 2, 3 and 4, as these reflect the overall results.

Placebo and sumatriptan 100 mg outcomes (mean and 95% CI) in GlaxoWellcome-conducted sumatriptan studies (suma), AstraZeneca-conducted zolmitriptan studies (zolmi), GlaxoWellcome-conducted naratriptan studies (nara), MSD-conducted rizatriptan studies (riza), Pfizer-conducted eletriptan studies (ele), Almirall-conducted almotriptan studies (almo) and overall (all). Outcomes for headache response and pain free at 2 h, sustained pain free, and adverse events (AEs) are plotted separately.
The average placebo rates (and 95% CI) are 29% (28–31) for headache response, 8% (7–9) for pain free, 6% (5.0–6.4) for sustained pain free, and 27% (25–28) for any AE. Almirall-Prodesfarma-conducted almotriptan studies show the highest placebo rates for efficacy and the lowest for any AE; Pfizer-conducted eletriptan studies show the lowest placebo rates for efficacy and the highest for any AE. The other placebo rates are remarkably consistent across companies.
For sumatriptan 100 mg, the average rates (and 95% CI) are 59% (57–61) for headache response, 29% (27–31) for pain free, 20% (18–21) for sustained pain free, and 39% (37–41) for any AEs. The efficacy rates are remarkably consistent across companies except for substantially lower pain-free and sustained pain-free rates in the Pfizer-conducted eletriptan-sumatriptan comparator studies. In these studies sumatriptan 100 mg performed less well than in studies conducted by other companies. As sumatriptan was encapsulated in these trials for blinding purposes, comparison of the pharmacokinetic profiles of the encapsulated and non-encapsulated normal tablets of sumatriptan could shed some light on this under-performance (44), although perhaps not provide the entire explanation. The AE rates vary markedly among the company-sponsored studies: AE rates are highest in the MSD-conducted rizatriptan and AstraZeneca-conducted zolmitriptan programmes, while AE rates are lowest in the Almirall-Prodesfarma-conducted almotriptan and GlaxoWellcome-conducted sumatriptan studies.
Direct comparator trials
Table 7 summarizes all 22 eligible trials which compared one triptan with another, or with ergotamine, by listing the main efficacy and AE differences (and 95% CI) between the two indicated compounds; the primary study endpoints and appropriate statistics are indicated with grey boxes. As most comparisons were with sumatriptan, most trials are listed under this drug: a positive difference indicates superiority of sumatriptan, a negative difference indicates inferiority.
Efficacy and tolerability in direct comparator trials.



For each trial, the differences (and 95% CI) between the two indicated compounds are listed for: response at 2 h; pain free at 2 h; sustained pain free; and proportion of patients with at least one AE in the categories any AE, CNS AE, and chest AE. The primary efficacy endpoint is indicated with the appropriate statistic, either as one of the standard parameters in bold, or as a text in the last column if the primary endpoint is different from the standard parameters. The different agents, doses, and trials are listed under the triptan against which the comparison was done (=comparator triptan, mostly sumatriptan 100 or 50 mg): a positive difference indicates superiority of the comparator triptan (e.g. sumatriptan 100 mg), a negative difference indicates inferiority. If more than one trial was conducted comparing the same compounds and doses, the studies are also combined (total).
Study endpoints:
Response=proportion of patients with headache response at 2 h.
Pain free=proportion of patients with pain free at 2 h.
Sustained pain free=proportion of patients with sustained pain free.
ANY-AE=proportion of patients with at least one adverse events (AE).
CNS-AE=proportion of patients with at least one central nervous system AE.
Chest-AE=proportion of patients with at least one chest AE.
Agents: Suma, sumatriptan; Z and zolmi, zolmitriptan; Nara and N, naratriptan; Riza and R, rizatriptan; Ele and E, eletriptan; Almo and A, almotriptan.
Primary endpoints (indicated with
Response all 2 attacks=headache response at 2 h over (all) 2 attacks.
Response all 6 attacks=headache response at 2 h over (all) 6 attacks.
Complete response=headache response at 2 h without moderate or severe recurrence within 24 h of dosing (note difference with sustained pain free: a patient who responded but took rescue medication, but did not indicate recurrence would be counted as a complete response).
Consistency of response=percentage of patients with response in 3 out of 3 attacks.
Time to relief/response/pain free=time-to-relief analysis (129). This is a variant of survival analysis to estimate the likelihood that a patient will respond sooner to drug A than to drug B over the first 2 h following treatment. The results are expressed as a hazard ratio. A ratio, e.g. 1.30, indicates that patients on drug A are 30% more likely to respond at any give instant in time over the first 2 h than those taking drug B. Patients who relapse within the first 2 h post-dose should be excluded from the analysis as the model assumes no early relapse. As long as the incidence of relapse is balanced between the two study populations this should not significantly influence the results.
Statistics:
†Based on 4 h data; no 2 h data available.
For totals, the differences are assessed with random effect models:
∗ P<0.05;
∗∗0.05<P<0.005;
∗∗∗0.005<P<0.0005.
References for unpublished data:
aGlaxoWellcome data on file (trial number 2004).
bGlaxoWellcome data on file (trial number 3002).
cGlaxoWellcome data on file (trial number 4003).
dAstraZeneca data on file (trial number 070).
eAstraZeneca data on file (trial number 071).
fAstraZeneca data on file (trial number 073).
gMerck Sharp and Dohme data on file (trial number 052).
hPfizer data on file (trial number 104).
Differences are generally small, as to be expected when comparing active compounds, but the overall pattern is similar to that in the meta-analysis. Compared with sumatriptan 100 mg: Cafergot 2 mg shows lower efficacy but fewer CNS AEs; zolmitriptan 5 mg shows no differences; naratriptan 2.5 mg shows lower efficacy at 4 h (only when the results of two studies are combined) and fewer AEs; rizatriptan 10 mg is superior in one of two studies for both the primary study endpoint (time to response) and pain free, and for pain free in the combined results; eletriptan 40 mg is superior in one of two studies as well as in the combined results; eletriptan 80 mg is superior in both of two studies for all parameters, but also causes more AEs (note that sumatriptan 100 mg under-performs for pain free in these studies compared with other trials); finally, almotriptan 12.5 mg is no different on the efficacy endpoints, but causes fewer AEs.
Compared with sumatriptan 50 mg: zolmitriptan 2.5 mg is just superior on the primary study endpoint (response over six attacks) in one of two studies, but on none of the other standard parameters; zolmitriptan 5 mg is no different on all parameters; rizatriptan 5 mg is no different in three studies, except for slightly more AEs; rizatriptan 10 mg is superior on the primary study endpoint (time to response) in one of two studies, but on none of the other parameters; eletriptan 40 mg is superior in one of two studies and on the combined results, but also causes more AEs; eletriptan 80 mg is superior in two studies on all parameters, but also causes more AEs.
Compared with sumatriptan 25 mg: zolmitriptan 2.5 mg and 5 mg (on all parameters), rizatriptan 5 mg and 10 mg (on most parameters in two studies), and eletriptan 80 mg (all parameters) are superior; zolmitriptan 5 mg and eletriptan 80 mg also cause more AEs; eletriptan 40 mg is no different for efficacy but causes more CNS AEs.
There are also a few trials in which triptans are compared with specific anti-migraine treatments other than sumatriptan. Zolmitriptan 2.5 mg has similar efficacy to naratriptan 2.5 mg at 4 h (no 2-h data available), but causes more AEs. Rizatriptan 10 mg is superior to zolmitriptan 2.5 mg on the primary endpoint (time to pain free), but only after exclusion of patients who relapsed within 2 h (balanced in both treatment groups), and causes fewer AEs. Rizatriptan 10 mg is superior to naratriptan 2.5 mg on all parameters but also causes more AEs. Eletriptan 40 and 80 mg are superior to Cafergot 2 mg on all parameters.
Discussion
We used two complementary approaches for comparing the efficacy and tolerability of the oral triptans: a large meta-analysis of all the eligible, high-quality, randomized, placebo-controlled clinical trials, and a separate analysis of all direct comparative studies. Both approaches give very similar results. The meta-analysis uses studies of a fundamentally similar design so that summary estimates of the efficacy and tolerability of the full range of compounds can be derived. By using placebo-subtracted measures one can partially adjust for the methodological differences among studies, which may influence the results. The great strength of randomized head-to-head comparator trials is their internal validity. However, factors such as patient selection, study size, and encapsulation of a drug may limit the generalizibility of the results into clinical practice. Furthermore, it is unlikely that triptans will ever all be compared. The remarkable similarity of the results from the meta-analysis of the placebo-controlled trials (both for the absolute and placebo-subtracted rates, and active/placebo ratios, i.e. using the multiplicity model) and from the direct head-to-head comparator studies, reinforces our confidence in the overall interpretation of the relative merits of the oral triptans.
Tolerability refers to medically unimportant but clinically irritating and often relatively frequent AEs, whilst safety refers to medically significant, and usually rare, side-effects. Safety can thus usually only be reliably assessed after large-scale and long-term clinical exposure. Although less so than with the ergots (2, 9), the main concern with all triptans has been their potential of coronary vasoconstriction (59). Given the now very substantial well-documented total human exposure to triptans over the past decade, there have been very few reports of clinically significant myocardial ischaemia, and invariably in patients with cardiovascular disease or risk factors (8). Thus, in appropriately selected patients, the triptans are very safe. As there are no clinically significant differences in coronary vasoconstriction effects, no triptan is demonstrably safer than the others. Concerns about cardiovascular safety have been exacerbated by the occurrence of non-cardiac chest symptoms that sometimes resemble pectoral angina (60). The usual underlying mechanism is not myocardial ischaemia; other mechanisms such as oesophageal spasm are much more likely (32, 61). When patients are warned about these events, they rarely cause problems (60).
Tolerability was assessed by calculating the proportions of patients with at least one AE, irrespective of their number, nature or intensity. Thus, as trivial and significant AEs were pooled, differences in total AE rates must be interpreted cautiously. In addition, slight differences among the company-sponsored studies in the study populations and methods of collecting AEs and their definitions may complicate the comparison of tolerabilities even further. This is reflected in the differences in any AE rates for placebo and sumatriptan by company (Fig. 6). The most remarkable outlier here is the very low AE rate in the Almirall-Prodesfarma-conducted almotriptan studies. This might indicate that their study population had a higher threshold for reporting AEs.
In their marketed doses, all the oral triptans are substantially superior to placebo. Consistent lack of response is rare, as the great majority of patients will have a response in at least one of three treated attacks. Differences among the triptans may seem relatively small but are clinically relevant for the individual patient. Depending on the effect parameter, success rates were 10–38% higher with some of the newer triptans compared with those with the reference dose (see below: Which triptan to select). Sumatriptan was the first clinically available triptan; it has the longest clinical experience and is the most widely prescribed oral triptan. Table 8 compares the main clinical characteristics of the new oral triptans with those of oral sumatriptan 100 mg, based on the present meta-analysis and the direct comparator studies (Table 7). Three compounds show favourable results: rizatriptan 10 mg (better efficacy and consistency; similar tolerability), eletriptan 80 mg (better efficacy, but slightly more AEs), and almotriptan 12.5 mg (better sustained pain free, consistency, and notably tolerability). There are, however, three potential caveats that deserve further discussion.
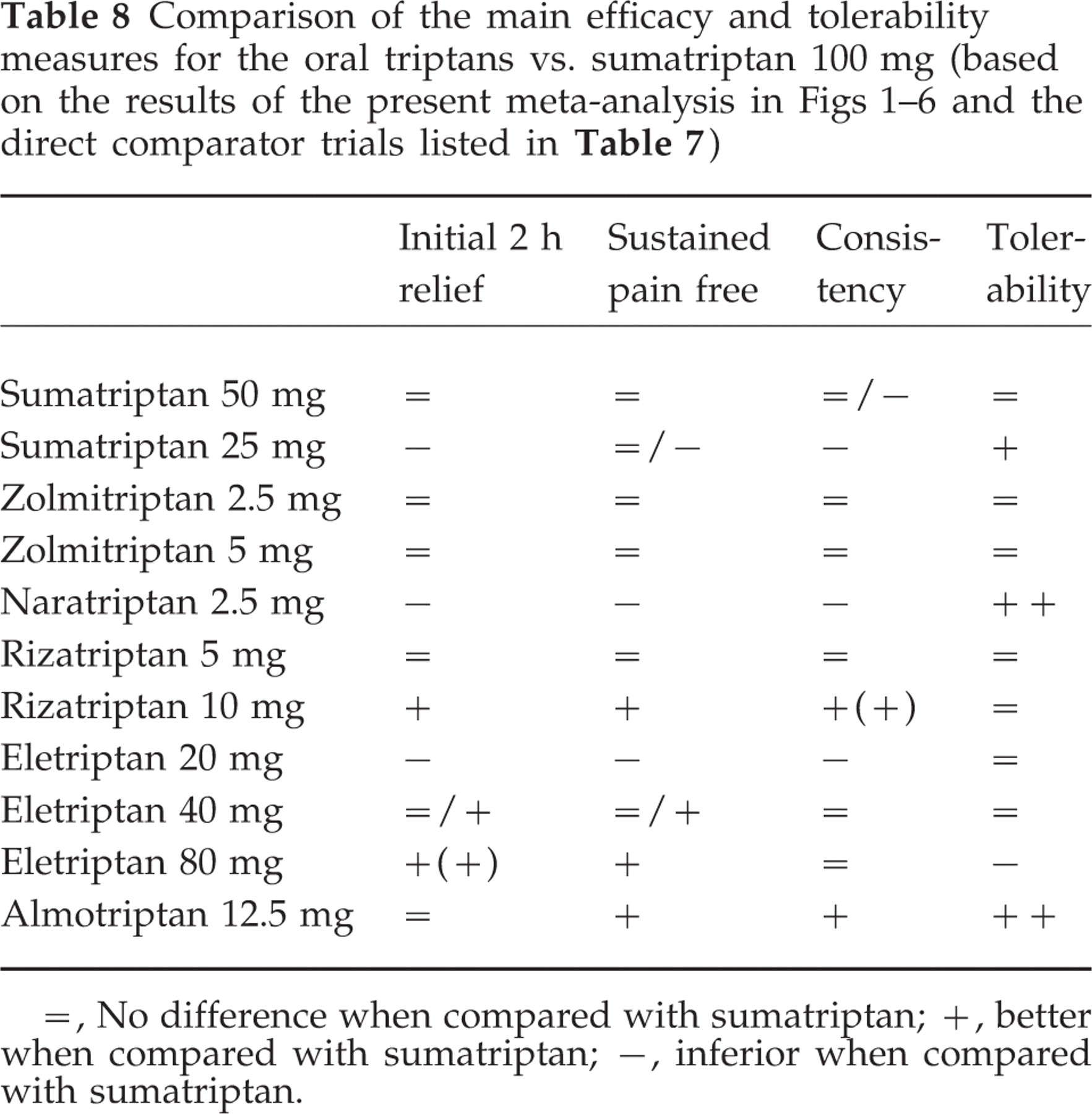
=, No difference when compared with sumatriptan; +, better when compared with sumatriptan; −, inferior when compared with sumatriptan.
The higher rates for consistency with rizatriptan should be interpreted with some caution because of the different study designs (cross-over with placebo interspersed at random in the rizatriptan study compared with a parallel design for the other triptans). The main difference here was the risk for the patients of getting placebo: 80% chance to get placebo in one of four treated attacks in the rizatriptan study compared with 50% chance of getting placebo in all three treated attacks in the consistency studies with the other triptans. This difference might have influenced the outcome, although the direction is difficult to predict. We feel, however, that it is unlikely that the difference in study designs has had a major influence on the results. This is supported by the finding that in the subgroup of 125 patients who treated three consecutive attacks with rizatriptan, without prior exposure to placebo (i.e. in the same way as in a parallel design study), the results were virtually the same as those obtained in the overall study population.
In the almotriptan trials placebo efficacy is high and placebo and sumatriptan AE rates are very low (Fig. 6). This suggests that the patients in these studies were more therapy-responsive and had a higher threshold to report AEs; however, almotriptan retained its tolerability advantage in a head-to-head study with sumatriptan 100 mg (Table 7).
In the direct comparator trials vs. eletriptan, sumatriptan 100 mg was encapsulated (for blinding purposes) and under-performed for pain free compared with other trials. In a direct comparison of sumatriptan and encapsulated sumatriptan, the early absorption of the encapsulated form was delayed but the open label 2-h response was equivalent to the normal sumatriptan (44).
For the other compounds, differences are minor and sometimes favour sumatriptan 100 mg. Sumatriptan 50 mg, zolmitriptan 2.5 and 5 mg, rizatriptan 5 mg, and eletriptan 40 mg have efficacy and tolerability profiles very similar to sumatriptan 100 mg. Sumatriptan 25 mg, naratriptan 2.5 mg and eletriptan 20 mg have inferior efficacy, but better tolerability.
Frovatriptan is not discussed because the sponsor declined to provide the relevant data and published clinical trials are not available. Based on congress presentations, the estimated results are well below those of the other triptans: 41% (therapeutic gain 20%) for headache response and 12% (therapeutic gain 9%) for pain free. Recurrence rates are comparatively low, but the clinical significance is limited by the low initial relief rates, the modest patient numbers, and the wide CIs; in a direct comparative study, recurrence rates for frovatriptan (25%) and sumatriptan 100 mg (32%) were not significantly different. Sustained pain-free data could not be calculated. AE rates seem very similar to those of other triptans. A claim for a higher degree of cardiovascular safety has been based on the 5-HT7-mediated coronary vasodilator effects of frovatriptan and on a safety study in 75 migraine patients with cardiovascular disease, or risk factors, or both (International Headache Research Seminar, Copenhagen, 3–5 November 2000). This study failed to show a difference in cardiovascular event rates between frovatriptan and placebo. It should be noted, however, that this safety study was severely underpowered: the post hoc power was only 8% to detect any significant difference and at least 1000 patients would have been required for an 80% power. Thus, in the absence of published evidence and extensive clinical experience, and taking into account that the 5-HT7-mediated coronary vasodilator effects only occur at doses well above those recommended for clinical use, a claim that frovatriptan has a higher degree of cardiovascular safety is unsustainable and potentially hazardous.
Which triptan to select?
Patients' characteristics and preferences vary, and individual responses to a triptan cannot be predicted. As a consequence, optimizing therapy involves trial-and-error; if the first triptan is not successful one may successfully switch to another. This approach has, however, not yet been tested in controlled trials. Physicians thus need more than one triptan in their repertoire to treat migraine patients optimally. Differences among the oral triptans at optimal doses may seem relatively small, but are clinically relevant for individual patients, i.e. provide clinically relevantly higher likelihood of success. For example, compared with sumatriptan 100 mg, rizatriptan 10 mg provides clinically significantly higher rates for response (60% for sumatriptan vs. 70% vs. rizatriptan =+17% relative improvement), pain free (29% vs. 40% =+38%) and sustained pain free (20% vs. 25% =+25%). Similarly, eletriptan 80 mg provides clinically significantly higher rates for response (60% vs. 66% =+10%) and sustained pain free (20% vs. 25% =+25%). Finally, almotriptan 12.5 mg provides clinically significantly higher rates for pain free (29% vs. 36% =+24%) and sustained pain free (20% vs. 26% =+30%) and a clinically significantly lower risk of AEs (33% for sumatriptan vs. 14% for almotriptan =−57%), although this latter advantage may have been exaggerated by the inclusion of different study populations (see above). The present analysis thus offers an indication as to which of the oral triptans are associated with the highest likelihood of success. Rizatriptan 10 mg (especially when consistent and rapid freedom from pain is desired), eletriptan 80 mg (especially when high efficacy and low recurrence are favoured over tolerability) and almotriptan 12.5 mg (especially when high tolerability and good efficacy are favoured) offer the highest likelihood of success. The lower doses of these agents (rizatriptan 5 mg; eletriptan 40 mg) may be good starting doses in many patients. Sumatriptan 100 mg and 50 mg provide good efficacy and tolerability and have by far the longest clinical experience. Sumatriptan also, and uniquely, offers rectal, nasal, and subcutaneous formulations, allowing tailor-made treatments for individual patients. Subcutaneous sumatriptan (6 mg) clearly is the most effective acute treatment for migraine attacks (response=76% and pain free=48% at 1 h post-dose), but is also associated with more intense AEs and the need of self-injection (62). Naratriptan 2.5 mg offers very good tolerability coupled to a slower onset of improvement; this can be useful in patients with mild or moderate migraine. Zolmitriptan 2.5 mg and 5 mg are good alternatives in many patients; they offer no specific advantages or flaws. Frovatriptan cannot be fully judged in view of the lack of data but does not seem to offer any particular advantage.
Conflict of interest
Drs Ferrari, Goadsby and Lipton are consultants to, and have received travel and grant supports from all the pharmaceutical companies involved in the manufacturing and marketing of the drugs discussed in the present paper. They, like Dr Roon, do not hold any equity, stock, or shares in the discussed pharmaceutical industries, nor in any competing company. The salaries of the authors are all fully covered by their employers.
Footnotes
Acknowledgements
The authors thank the following pharmaceutical companies and persons for providing data and for patiently answering all related questions: GlaxoWellcome now GlaxoSmithKline (sumatriptan and naratriptan): Diane J. Boswell, Scott B. McNeal, Gayla P. Putman, Jane Saiers, Reijo Salonen; AstraZeneca (zolmitriptan): David Lee, James Sawyer, Andrew A.M. Stone; Merck Sharp & Dohme (rizatriptan): Christopher Lines, Eric Sandquist; Pfizer (eletriptan): Scott Haugie, Neville Jackson, Phil Poole, John A. Schalhoub; Almirall (almotriptan): Xavier Carabarrocas, Pau Ferrer, Jose Palacios. We are also grateful to Ton de Craen (Department of Clinical Epidemiology, LUMC) and Douglas C McCrory (Center for Clinical Health Policy Research, Duke University Medical Center, Durham, NC, USA) for statistical and methodological advice. The work of P.J.G. is supported by the Migraine Trust and the Wellcome Trust. P.J.G. is a Wellcome Senior Research Fellow.
Appendix
Adverse events—placebo subtracted
| Any | CNS | Chest | ||||
| % | 95% CI | % | 95% CI | % | 95% CI | |
| Suma 25 mg | 4.4 | (0.1; 8.8) | 1.7 | (−1.2; 4.7) | 0.8 | (−1.0; 2.6) |
| Suma 50 mg | 7.8 | (2.6; 13.1) | 3.7 | (1.0; 6.4) | 1.9 | (0.4; 3.3) |
| Suma 100 mg | 13.2 | (8.6; 17.8) | 6.3 | (3.2; 9.5) | 1.7 | (0.8; 2.5) |
| Zolmi 2.5 mg | 15.9 | (9.6; 22.1) | 9.9 | (4.3; 15.5) | 2.0 | (0.7; 3.3) |
| Zolmi 5 mg | 24.5 | (15.5; 33.5) | 11.5 | (6.1; 16.8) | 2.9 | (1.2; 4.6) |
| Nara 2.5 mg | 2.4 | (−2.2; 7.0) | 1.9 | (−1.2; 5.0) | 0.4 | (−0.8; 1.6) |
| Riza 5 mg | 7.9 | (4.7; 11.1) | 6.1 | (3.2; 9.0) | 0.9 | (−0.04; 1.8) |
| Riza 10 mg | 13.5 | (10.6; 16.3) | 9.4 | (7.2; 11.6) | 1.5 | (0.8; 2.3) |
| Ele 20 mg | 1.9 | (−15.5; 19.3) | 2.6 | (−6.6; 11.7) | −0.3 | (−3.1; 2.6) |
| Ele 40 mg | 7.3 | (2.7; 11.8) | 7.5 | (4.5; 10.6) | 0.9 | (−0.2; 2.0) |
| Ele 80 mg | 18.9 | (11.2; 26.6) | 14.6 | (10.2; 19.0) | 2.6 | (0.6; 4.5) |
| Almo 12.5 mg | 1.8 | (−2.7; 6.2) | −1.5 | (−3.9; 1.0) | −0.4 | (−1.6; 0.8) |

| Response 2 h | Pain free 2 h | Recurrence | Sustained
pain free |
|||||||||
| Absolute | TG | Absolute | TG | Absolute | Absolute | |||||||
| % | 95% CI | % | 95% CI | % | 95% CI | % | 95% CI | % | 95% CI | % | 95% CI | |
| Suma 25 mg | 56.0 | (53.1; 58.9) | 23.6 | (18.7; 28.5) | 23.4 | (21.0; 25.9) | 14.4 | (11.0; 17.8) | 26.7 | (22.6; 30.7) | 16.7 | (14.5; 18.9) |
| Suma 50 mg | 62.7 | (60.4; 65.1) | 30.9 | (23.9; 37.9) | 28.7 | (26.5; 30.9) | 18.0 | (11.7; 24.3) | 27.8 | (24.7; 30.9) | 19.8 | (17.8; 21.8) |
| Suma 100 mg | 59.0 | (57.3; 60.8) | 29.1 | (25.7; 33.7) | 28.9 | (27.2; 30.5) | 19.5 | (17.3; 21.8) | 29.9 | (26.9; 32.9) | 20.0 | (18.2; 21.3) |
| Zolmi 2.5 mg | 63.5 | (60.8; 66.2) | 30.9 | (26.1; 35.8) | 29.1 | (26.6; 31.7) | 20.4 | (15.6; 25.1) | 30.3 | (26.2; 34.4) | 19.0 | (16.1; 21.8) |
| Zolmi 5 mg | 62.8 | (60.0; 65.6) | 33.8 | (23.4; 44.2) | 32.4 | (29.7; 35.1) | 25.2 | (16.9; 33.5) | 34.2 | (25.6; 42.8) | 21.9 | (19.3; 24.6) |
| Nara 2.5 mg | 48.6 | (45.7; 51.4) | 22.2 | (16.9; 27.5) | 22.4 | (20.0; 24.7) | 14.1 | (10.7; 17.5) | 21.4
24.0 |
(13.4; 29.3)
2–24 h (20.9; 27.2) 4–24 h |
15.9 | (13.4; 18.5) |
| Riza 5 mg | 62.4 | (60.2; 64.5) | 27.6 | (23.0; 32.2) | 30.5 | (28.4; 32.5) | 22.0 | (19.5; 24.5) | 39.3 | (36.5; 42.2) | 18.9 | (17.0; 27.3) |
| Riza 10 mg | 68.6 | (66.9; 70.4) | 34.6 | (29.6; 39.6) | 40.1 | (38.3; 42.0) | 30.4 | (27.5; 33.2) | 36.9 | (34.8; 39.1) | 25.3 | (23.7; 26.9) |
| Ele 20 mg | 48.9 | (44.5; 53.3) | 23.9 | (15.3; 32.5) | 16.4 | (13.2; 19.7) | 11.0 | (7.2; 14.7) | 28.4 | (18.1; 38.7) | 10.6 | (7.7; 13.5) |
| Ele 40 mg | 60.2 | (58.0; 62.4) | 35.2 | (29.8; 40.7) | 27.2 | (25.2; 29.2) | 22.5 | (18.1; 26.8) | 21.4 | (18.8; 24.0) | 20.9 | (19.1; 22.7) |
| Ele 80 mg | 65.8 | (63.3; 68.3) | 42.0 | (35.8; 48.2) | 33.0 | (30.5; 35.4) | 28.4 | (23.5; 33.3) | 19.8 | (17.0; 22.7) | 25.0 | (22.8; 27.2) |
| Almo 12.5 mg | 61.2 | (57.6; 64.8) | 25.0 | (14.1; 35.9) | 61.2 | 21.0 | (13.3; 28.7) | 26.2 | (22.1; 30.3) | 25.9 | (22.7; 29.1) | |
| Frova 2.5 mg∗ | 41.5 | (39.3; 43.8) | 16.6 | (13.1; 20.1) | ||||||||
∗Data from published abstracts; when the number of patients responding to frovatriptan was not given, the number of patients was calculated from the percentage and total number treated given.