
Abstract
Striatal cholinergic interneurons are relatively resistant to ischemic insults. These neurons express hyperpolarization-activated cation current (Ih) that profoundly regulates neuronal excitability. Changes in neuronal excitability early after ischemia may be crucial for determining neuronal injury. Here we report that Ih in cholinergic interneurons was decreased 3 h after transient forebrain ischemia, which was accompanied by a negative shift of the voltage dependence of activation. The inhibition of Ih might be due to the tonic activation of adenosine A1 receptors, as blockade of A1 receptors significantly increased Ih in postischemic neurons, but had no effect on control neurons. Consistent with the inhibition of Ih, postischemic neurons showed a reduction in both spontaneous firing and hyperpolarization-induced rebound depolarization. These findings indicate that Ih may play excitatory roles in striatal cholinergic interneurons. Postischemic inhibition of Ih might be a novel mechanism by which adenosine confers neuronal resistance to cerebral ischemia.
Introduction
Transient cerebral ischemia induces selective neuronal death in certain brain regions; however, its mechanisms are still unclear. In the neostriatum, forebrain ischemia of 20 to 30mins causes irreversible injury of most medium spiny neurons within 24 h, but cholinergic interneurons remain intact (Chesselet et al, 1990; Francis and Pulsinelli, 1982; Pulsinelli et al, 1982). Previous studies have shown that, in addition to excitatory synaptic inputs (Pang et al, 2002; Zhang et al, 2006b), the intrinsic membrane properties are differentially altered between ischemia-sensitive and resistant striatal neurons in response to ischemic insults (Calabresi et al, 1997; Pisani et al, 1999). For instance, during in vitro hypoxia/hypoglycemia, medium spiny neurons are depolarized presumably due to the activation of voltage-dependent Na+ channels and Ca2+ entry, whereas cholinergic interneurons are hyperpolarized by the activation of ATP- and Ca2+-dependent potassium channels (Centonze et al, 2001). Furthermore, the delayed rectifier potassium currents in cholinergic interneurons are enhanced after transient forebrain ischemia and lead to a shortening of spike width (Deng et al, 2005). These results suggest that the intrinsic neuronal excitability may contribute to the different vulnerabilities to cerebral ischemia among striatal neurons.
Striatal cholinergic interneurons express hyperpolarization-activated cation current (Ih) (Jiang and North, 1991; Kawaguchi, 1993). Unlike medium spiny neurons, most of the cholinergic interneurons are spontaneously active during this period Ih provides subthreshold depolarizing currents to drive firings (Bennett et al, 2000; Bennett and Wilson, 1999). Additionally, they exhibit Ih-dependent rebound depolarizations (RD) upon termination of hyperpolarizations (Bennett et al, 2000). Thus, regulation of Ih may cause profound changes in the membrane excitability of cholinergic interneurons. Several lines of evidence have shown that Ih in neurons is very sensitive to ischemic insults (Erdemli and Crunelli, 1998; Gao et al, 2006; Mironov et al, 2000; Ray et al, 2003; Zhang et al, 2006a). However, the role of Ih in ischemia-induced neuronal damage remains to be elucidated. With a focus on the early phase after transient forebrain ischemia in vivo, this study was therefore designed to detect the postischemic changes of Ih in cholinergic interneurons and its possible roles in regulating neuronal excitability.
Materials and methods
Male Wistar rats (weighing 100 to 200 g, Charles River Laboratories, Wilmington, MA, USA) were used in this study. Experimental protocols were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Transient Forebrain Ischemia Model
Transient forebrain ischemia was induced using the four-vessel occlusion method (Pulsinelli and Brierley, 1979) with modifications (Ren et al, 1997). Briefly, the rats were fasted overnight to provide uniform blood glucose levels and anesthetized with a mixture of 1% to 2% halothane, 33% O2 and 66% N2 through a nasal mask. The common carotid arteries were isolated after which an occluding device was placed loosely around each common carotid artery for subsequent occlusion of these vessels. The animal was then placed on a stereotaxic frame and both vertebral arteries were electrocauterized. A tiny temperature probe was inserted beneath the skull in the extradural space, and the brain temperature was maintained at 37°C with a heating lamp connected to a temperature control system (BAT-10; Physitemp, Clifton, NJ, USA). A skull window was opened at 9.5 mm anterior to the interaural line, 3.0 mm from the midline. Glass microelectrodes (5 to 8 μm in diameter of tip, filled with 2mol/L NaCl) were used to record ischemic depolarization, which is a reliable indication of complete ischemia (Ren et al, 1997). The microelectrode was advanced 3.0 mm below dura into the neostriatum. The recordings were performed with a neuroprobe amplifier (Model 1600; A-M Systems, Carlsborg, WA, USA). Transient forebrain ischemia was produced by occluding both common carotid arteries to induce ischemic depolarization for 20 to 22 mins. The duration of ischemic depolarization was determined by measuring the period from the beginning of the extracellular direct current potential reaching −20 mV to the point where the potential started to repolarize after recirculation. The rats were returned to the cages after recovering from ischemia and allowed free access to water and food.
Brain Slice Preparation
Brain slices were prepared from animals of control, 3 and 24 h after ischemia using procedures similar to those previously described (Deng et al, 2005). Briefly, the animals were anesthetized with ketamine-HCl (1 mg/kg, intraperitoneally) and perfused transcardially with an ice-cold (4°C) sucrose solution containing (in mmol/L) 230 sucrose, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 10 MgSO4, and 10 glucose, pH 7.4, 290 to 305 mosM/L, equilibrated with 95% O2 and 5% CO2. The brains were quickly removed and transverse striatal slices (300 μm) cut using a vibratome (VT1000S; Leica, Nussloch, Germany) in the sucrose solution. The slices were maintained in an artificial cerebrospinal fluid containing (in mmol/L) 130 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4, 295 to 305mosM/L. The artificial cerebrospinal fluid was continuously equilibrated with 95% O2 and 5% CO2, and slices were incubated for >1h before recording.
Electrophysiological Recording
Recording electrodes were prepared from borosilicate glass (World Precision Instruments, Sarasota, FL, USA) using a horizontal electrode puller (P-97; Sutter Instruments, Novato, CA, USA). Electrodes had resistances of 2 to 4MΩ when filled with an intracellular solution containing (in mM) 120 KMeSO4, 12 KCl, 1 MgCl2, 1 EGTA (ethylene glycol bis (2-aminoethylether)-N,N,N′N′,-tetraacetic acid), 0.2 CaCl2, 10 HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 2 Mg-ATP, and 0.4 Na-GTP, pH 7.4, 295 to 300 mosM/L. Oxygenated artificial cerebrospinal fluid was used as bath solution, and the flow rate was adjusted to 2 to 3mL/min. Neurons in brain slices were visualized with an infrared differential interference contrast microscope (BX50WI; Olympus Optical, Tokyo, Japan) and a charge-coupled device camera. All recordings were performed at 32°C ± 1°C with an Axopatch 200B amplifier (Molecular Devices, Foster City, CA, USA). After tight-seal (> 1 GΩ) formation, the electrode capacitance was compensated. For cell-attached recordings, a voltage-clamp mode was used, and data acquisition was terminated if the seal resistance decreased to < 1 GΩ. During whole-cell recordings, series resistance (8 to 15 MΩ) was monitored periodically, and cells with >15% change were excluded from the analysis. For current-clamp recordings, the fast I-clamp mode was used. Signals were filtered at 2 kHz and digitized at a sampling rate of 5 kHz using a data-acquisition program (Axograph 4.6; Molecular Devices).
Drug Application
All drugs were purchased from Sigma (St Louis, MO, USA) unless otherwise noted and were bath applied. Drugs were dissolved as concentrated stocks in either water or dimethylsulfoxide and stored at −20°C. Working solutions were prepared immediately before use. When dimethylsulfoxide was used to prepare drug solution, equivalent amounts were added to artificial cerebrospinal fluid as controls. To characterize Ih in cholinergic interneurons, 4-ethylphenylamino-1,2-dimethyl-6-methylamino-pyrimidinium chloride (ZD7288, 30 μmol/L; Tocris Bioscience, Ellisville, MO, USA) and CsCl (2 mmol/L) were used as blockers of Ih channels. For recording Ih, CdCl2 (300 μmol/L), 4-aminopyridine (2 mmol/L), and tetraethylammonium (5 mmol/L) were added to bath solution to block voltage-dependent Ca2+ and K+ channels. Tetrodotoxin (TTX, 0.5 to 1 μmol/L) was also used to block Na+ channels. In some experiments, BaCl2 (200 μmol/L) was used to eliminate hyperpolarization-activated inward rectifier K+ currents. To examine the modulation of Ih, membrane-permeable 8-bromo-cyclc AMP (8-Br-cAMP, 100 μmol/L) and Rp-cAMP (50 μmol/L) were used as analog and inhibitor of cAMP signaling pathway, respectively. In addition, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX, 200 nmol/L) was used as a selective antagonist of adenosine A1 receptor. To reduce intracellular Ca2+ concentration, 1,2-bis(2-aminophenoxy) ethane-N,N,N′N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM, 50 μmol/L) was applied in bath solution. Synaptic blockers were not used in this study because the spontaneous activity of cholinergic interneurons is independent of synaptic input (Bennett and Wilson, 1999).
Data Analysis
Recording data were analyzed with Axograph 4.6. To evoke Ih, a series of hyperpolarizing voltage commands (from −60 to −150 in 10 mV steps, 2 sees) were applied from a holding potential of −50 mV. The amplitude of Ih was calculated as the current difference between the instantaneous current (measured just after the decay of capacitive transient) and the steady-state current (mean current of 50 ms before the termination of each voltage step). Voltage-dependent activation of Ih was determined by normalizing the peak amplitudes of tail currents at −50 mV after various hyperpolarizing voltage commands and then by fitting the current-voltage relationship with a Boltzmann function. The time constants of activation were estimated by fitting Ih traces evoked by various hyperpolarizing steps with a single exponential function. To examine the spontaneous activity, the firing rate was obtained from a 2-min sample of a cell-attached recording. The coefficient of variation (CV), a measure of irregularity in interspike intervals, was calculated for cells with a firing rate > 1 Hz. With current-clamp recordings, the amplitude of RD was measured as the voltage difference between the holding potential (approximately −55 mV) and the RD peak. The voltage response to a depolarizing current pulse was measured as the mean voltage of 50 ms (from 100 to 150 ms of current pulse), which was comparable to that of RD reaching the peak amplitude.
The data are presented as mean ± s.e.m. Statistical difference was detected using paired or unpaired Student's t-test (StatView 5.0; Abacus Concepts, Berkeley, CA, USA). Changes were considered significant when P <0.05.
Results
In striatal slices of both control and ischemia, only neurons with large somata (> 20 μm in diameter) and thick primary dendrites were selected for recordings (Figure 1A). These neurons exhibited electrophysiological characteristics that are typical of cholinergic interneurons (Jiang and North, 1991; Kawaguchi, 1993). During current-clamp recordings, although injection of depolarizing current evoked repetitive spiking followed by large-amplitude and long-duration after hyperpolarization, negative current induced an initial hyperpolarization followed by a depolarizing sag in the membrane potential (Figure 1B). These morphologic and electrophysiological features show that the recorded cells are cholinergic interneurons.
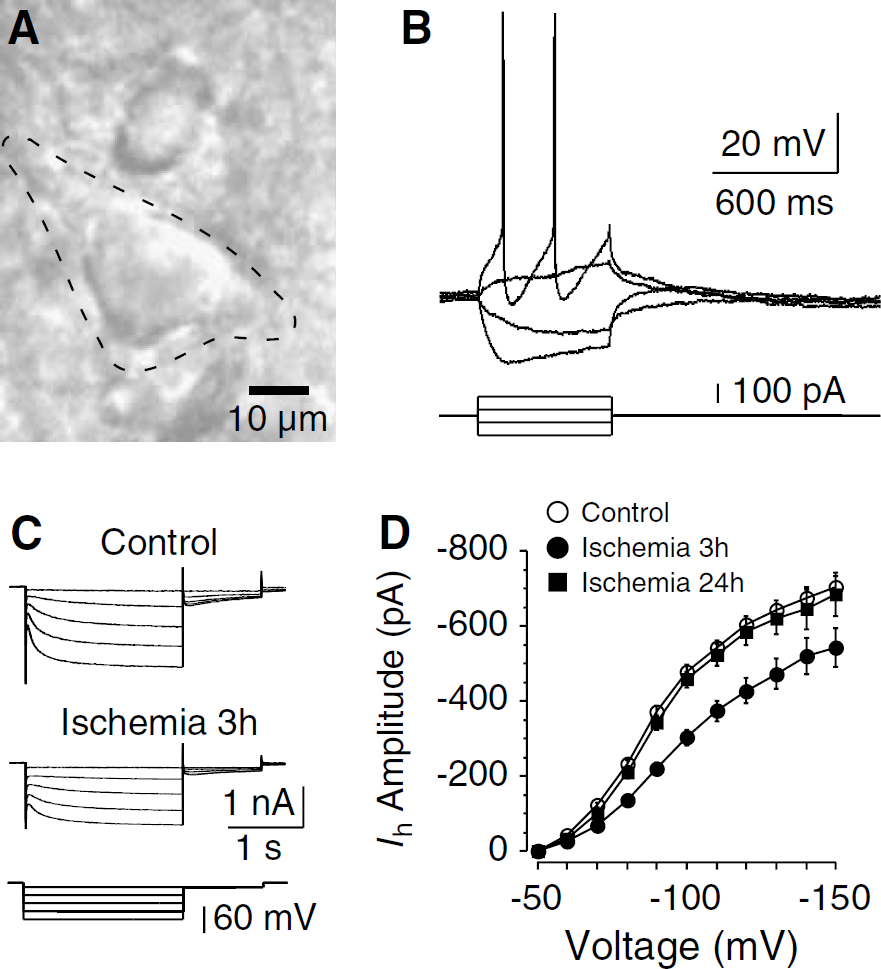
Postischemic decrease of Ih in cholinergic interneurons. (
Postischemic Decrease of Ih in Cholinergic Interneurons
In both control and postischemic neurons, hyperpolarizing voltage steps more negative than −50 mV evoked voltage-dependent, slowly activating inward currents (Figure 1C). Identification of these currents as ih was supported by their high sensitivity to Ib channel blockers such as ZD7288 and Cs+, but not to Ba2+. For example, application of ZD7288 at a concentration of 30 μmol/L blocked >90% of the currents (n = 5).
To test whether Ih was altered after ischemia, we first examined the amplitude of Ih in cholinergic interneurons. Notably, the Ih amplitude was decreased in neurons 3 h after ischemia and then recovered to control levels by 24 h (Figures 1C and 1D). When evoked with a command voltage of −90mV, the amplitudes were 372.6 ± 15.2 pA in control neurons (n = 45), 220.7 ± 14.8 pA in neurons 3 h after ischemia (n = 38; P <0.01 compared to control), and 343.5 ± 19.2 pA in neurons 24 h after ischemia (n = 13; P > 0.05 compared to control). It is possible that changes in rapidly activating inward rectifier K+ currents might affect the measure of Ih amplitude. We therefore performed another set of experiments, in which Ba2+ (200 μmol/L) was applied to reduce the contamination of instantaneous currents. Consistent with the above data, a significant reduction of Ih amplitude was detected in neurons 3 h after ischemia (by 37.7% when evoked at −90mV, n = 11 in each group; P <0.01). These results show an inhibition of Ih early after ischemia. Thus, the following experiments were focused on neurons of control and 3 h after ischemia.
We then examined the voltage dependence and kinetics of Ih activation. Analysis of the tail currents revealed a negative shift of the half-activation voltage (control: −87.3 ± 2.1 mV, n = 17; ischemia: −94.0 ± 1.7 mV, n = 18; P <0.01; Figure 2A). In both control and postischemic neurons, the activation kinetics of Ih was voltage dependent, as the time constant of activation decreased sharply with more hyperpolarization. However, when evoked with the same voltage, the activation of Ih in postischemic neurons was much slower than that of control. At a voltage step of −90 mV, the activating time constants were 391.2 ± 21.3 ms in control (n = 27) and 645.6 ± 20.8 ms in postischemic neurons (n = 23; P <0.01; Figures 2B and 2C). The above data show coincident changes in the amplitude and the voltage-dependent activation of Ih after ischemia, and suggest that the negative shift of voltage dependence might contribute to the postischemic decrease of Ih amplitude.
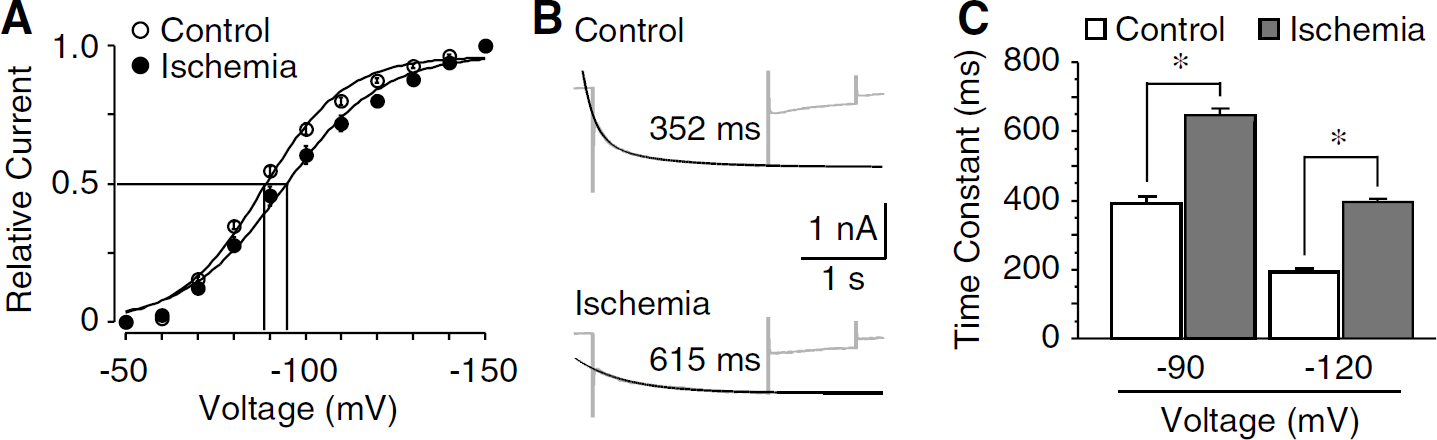
Ischemia-induced changes in voltage dependence and kinetics of Ih activation. (
Differential Modulation of Ih by Intracellular cAMP and Ca2+
Intracellular cAMP modulates the voltage-dependent activation of Ih (Frere et al, 2004). It is reasonable to speculate that cAMP might be involved in the postischemic decrease of Ih in cholinergic interneurons. To test this hypothesis, we examined the modulation of cAMP on Ih amplitude in control and postischemic neurons. Application of 8-Br-cAMP (100 μmol/L), an analog of cAMP, led to a dramatic increase in the Ih amplitude in both control (by 44.0% ± 5.6%, n = 6; P <0.01) and postischemic neurons (by 21.1% ± 3.4%, n = 5; P <0.01; Figures 3A and 3D), with less efficacy after ischemia (P<0.05). However, application of Rp-cAMP (50 μmol/L), an inhibitor of cAMP pathway, caused a decrease in Ih amplitude in control neurons (by 31.7% ± 4.4%, n = 5; P <0.01), but not in postischemic neurons (by 1.0% ± 5.2%, n = 5; P > 0.05; Figures 3B and 3D). These results indicate that the cAMP modulation of Ih was tonically suppressed in cholinergic interneurons after ischemia, which might underlie the postischemic decrease of Ih.
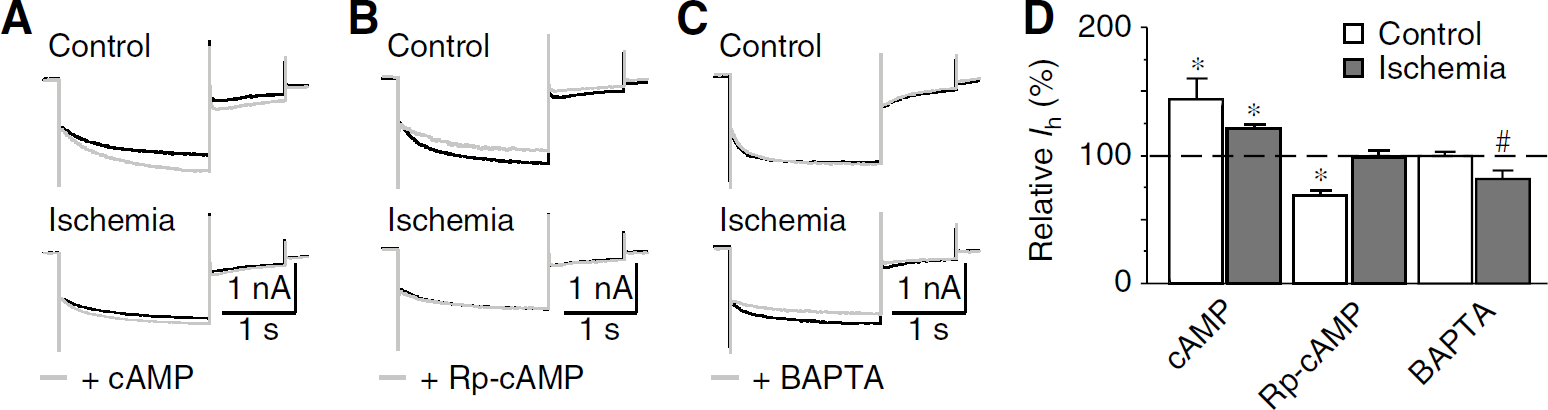
Regulation of Ih by intracellular cAMP and Ca2+. (
We also tested whether Ca2+ was involved in the postischemic changes of Ih, considering that intracellular Ca2+ regulates Ih in other neurons (Luthi and McCormick, 1998). As shown in Figures 3C and 3D, membrane-permeable Ca2+ chelator BAPTA-AM (50 μmol/L) significantly decreased Ih in postischemic neurons (by 17.9% ± 6.3%, n = 5; P <0.05), but not in control neurons (by 0.2% ± 3.3%, n = 5; P > 0.05). These data suggest that, rather than reducing, intracellular Ca2+ upregulates Ih in cholinergic interneurons after ischemia.
Adenosine Modulation of Ih in Cholinergic Interneurons
Striatal cholinergic interneurons express adenosine A1 receptors (Song et al, 2000). Activation of A1 receptors has been shown to reduce Ih in mesopontine cholinergic neurons and geniculocortical neurons, presumably by inhibiting adenylyl cyclase activity and thus decreasing the intracellular cAMP levels (Pape, 1992; Rainnie et al, 1994). It is possible that the same pathway might be responsible for the ischemia-induced decrease in Ih. As expected, the application of selective A1 receptor antagonist DPCPX (200nmol/L) led to a significant increase in Ih in postischemic neurons (by 23.3% ± 5.9%, n = 7; P <0.01), but not in control neurons (by 3.0% ± 6.1%, n = 7; P > 0.05; Figure 4). In addition, application of DPCPX caused a positive shift of the half-activation voltage of Ih in postischemic neurons (5.1 ± 1.9mV, n = 5; P <0.05). Furthermore, when DPCPX (200nmol/L) was applied in the presence of 8-Br-cAMP (100mmol/L), no obvious increase in Ih was detected in postischemic neurons (by 1.2% ± 2.2%, n = 5; P > 0.05; Figure 4B). This is in agreement with the fact that the actions of A1 receptors were mediated by cAMP pathway. Thus, our findings strongly support the conclusion that through a cAMP pathway, tonic activation of adenosine A1 receptors contributes, at least in part, to the postischemic decrease of Ih in cholinergic interneurons.
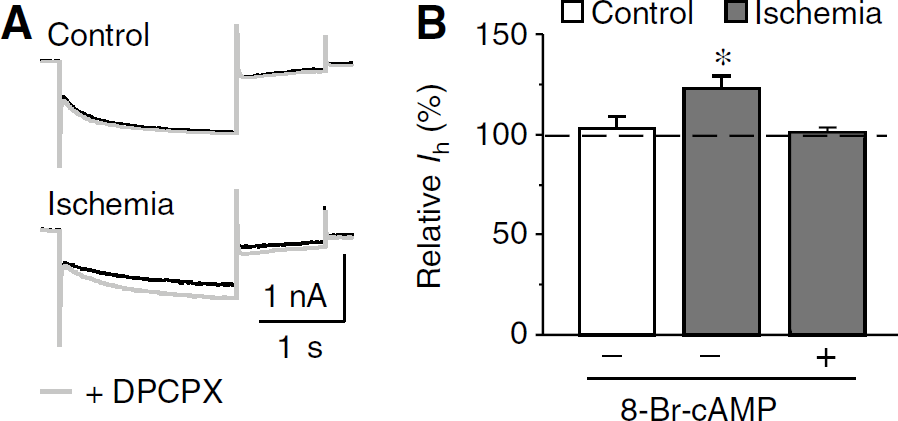
Involvement of adenosine A1 receptor in Ih inhibition after ischemia. (
Postischemic Changes of Neuronal Excitability in Cholinergic Interneurons
Activity of Ih is critical to the intrinsic membrane properties (Robinson and Siegelbaum, 2003). Previous studies have shown that the spontaneous firing in cholinergic interneurons is largely controlled by Ih. Specifically, a partial blockade of Ih causes a decrease in both firing rate and regularity (Bennett et al, 2000; Deng et al, 2007). Consistent with the inhibition of Ih, we found a significantly decreased firing rate in postischemic neurons (control: 3.08 ± 0.33 Hz, n = 19; ischemia: 1.20 ± 0.38 Hz, n = 5; P <0.01), accompanied by an increase in the CV of interspike interval (control: 0.29 ± 0.02, n = 19; ischemia: 0.53 ± 0.14, n = 5; P <0.01; Figure 5). Changes of TTX-sensitive Na+ currents may also modulate spontaneous firing. However, these currents have been shown to be unchanged in cholinergic interneurons in response to ischemic insults (Pisani et al, 1999). Therefore, inhibition of Ih might be the major mechanism underlying the postischemic suppression of spontaneous activities, although other membrane conductances (e.g., K+ currents) cannot be completely excluded.
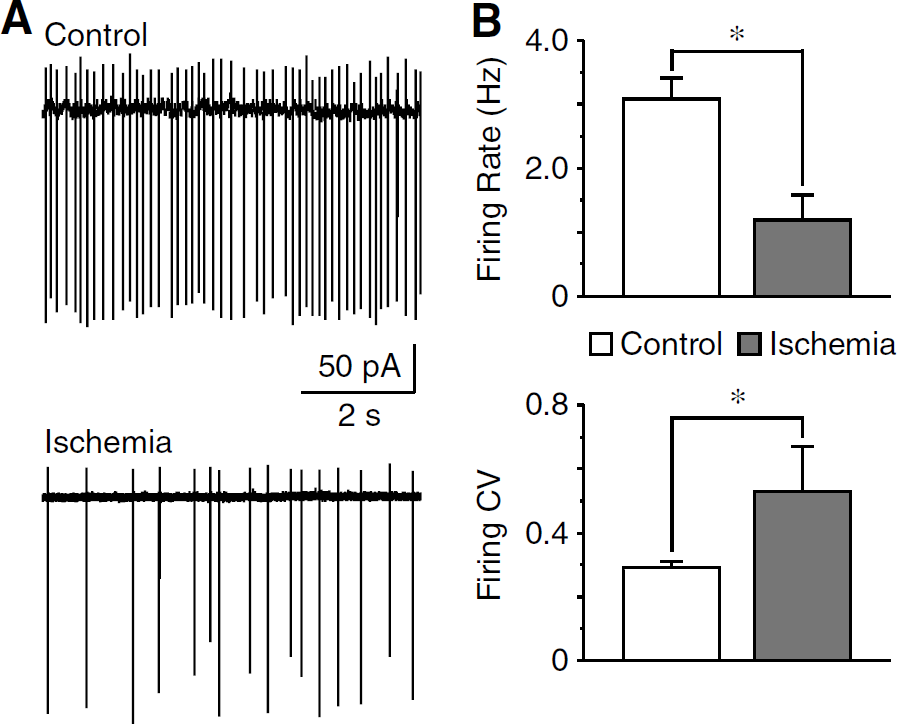
Suppression of spontaneous activity after ischemia. (
In cholinergic interneurons, hyperpolarizing currents can induce rebound excitation (i.e., RD) with increasing firings (Deng et al, 2007; Reynolds et al, 2004). The RD is primarily produced by Ih because these neurons do not express T-type Ca2+ currents (Bennett et al, 2000). To test whether the rebound excitation was altered after ischemia, the RD amplitude was measured in the presence of TTX (0.5 μmol/L) to eliminate the contribution of Na+ currents and to suppress spontaneous spikes. With the continuous application of TTX, the membrane potentials were −61.4 ± 2.4 mV (n = 11) and −63.7 ± 3.8mV (n = 7) in control and postischemic neurons (P > 0.05), respectively. The neurons were then held at −55 mV to ensure that most Ih channels were deactivated before the onset of hyperpolarization. Under these conditions, a prominent RD could be evoked by the offset of a hyperpolarizing current pulse in both control and postischemic neurons (Figure 6A). Moreover, the RD could be completely abolished with the Ih channel blocker ZD7288 (30 μmol/L; n = 6 in each group). Because membrane resistance at depolarizing voltages (more than −55 mV) may affect the amplitude of RD, the voltage responses to depolarizing current pulses (30 to 120 pA) were compared between control and postischemic neurons, and no obvious change was detected after ischemia. For example, when elicited with a current of 90 pA, the voltages were 11.6 ± 1.8 mV (n = 5) in control and 11.4 ± 1.5 mV in postischemic neurons (n = 6; P>0.05; Figures 6A and 6B). We then examined the amplitude of RD evoked by hyperpolarizing pulses (from −30 to −300 pA) with different durations (120, 600, and 1,000ms). In both control and postischemic neurons, the RD amplitude was positively related to the amplitude and duration of hyperpolarizing pulses, increased with increasing amplitude or duration of current pulse (Figures 6C and 6D). Most importantly, the RD amplitude was dramatically decreased after ischemia (by 45 to 60%, n = 6 in each group; P < 0.01; Figures 6C and 6D). Together with previous studies (Bennett et al, 2000; Reynolds et al, 2004), our data indicate that postischemic inhibition of Ib is capable of reducing rebound excitation in cholinergic interneurons.
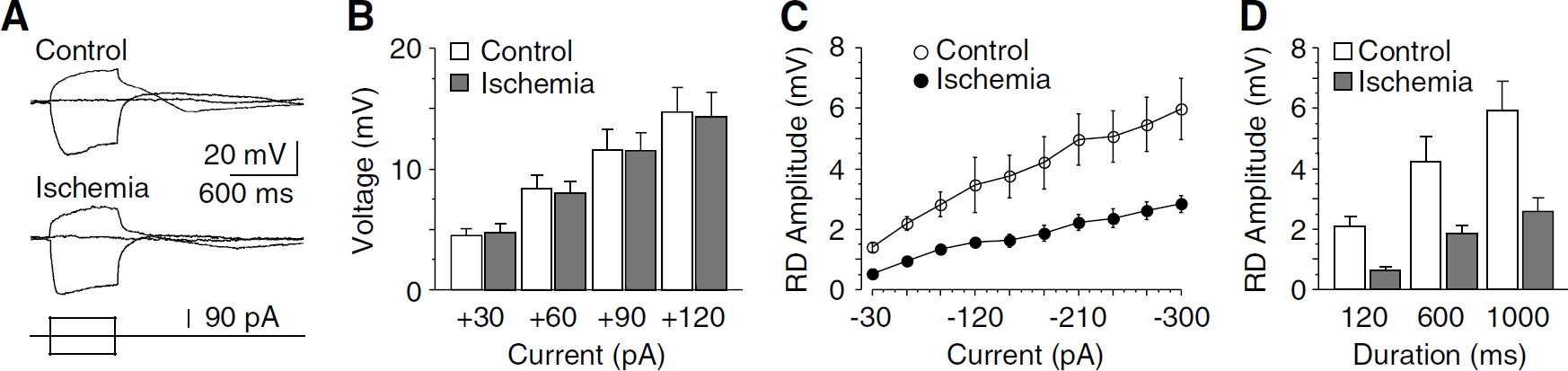
Postischemic reduction of RD in cholinergic interneurons. (
Discussion
The major findings of this study are that Ih in striatal cholinergic interneurons is decreased early after transient forebrain ischemia, which results, at least in part, from the tonic activation of adenosine A1 receptors via a cAMP-dependent pathway. Postischemic inhibition of Ih is well correlated with the decreased neuronal excitability, suggesting that adenosine-mediated Ih inhibition may be one of the mechanisms underlying neuronal resistance to cerebral ischemia.
Mechanisms of Postischemic Decrease of Ih in Cholinergic Interneurons
It has long been recognized that striatal cholinergic interneurons express Ih, which is essential for neuronal excitability (Bennett et al, 2000; Jiang and North, 1991; Kawaguchi, 1993). Several studies have shown that the activity of Ih was altered in other neurons in response to hypoxia. For instance, Ih in thalamocortical neurons was increased during hypoxia in vitro, which might be due to a positive shift of the voltage dependence and an acceleration of activation (Erdemli and Crunelli, 1998). On the contrary, acute hypoxia caused a disappearance of Ih in inspiratory brainstem neurons and dorsal root ganglion neurons (Gao et al, 2006; Mironov et al, 2000). In addition, a partial inhibition ofIh, without significant shift of voltage-dependent activation, was observed in hippocampal CA1 pyramidal neurons after neonatal hypoxia in vivo (Zhang et al, 2006a). Different from the above observations, this study revealed that Ih in striatal cholinergic interneurons was significantly decreased early (3h) after transient forebrain ischemia and suggested that the postischemic inhibition of Ih might be caused by a negative shift of the voltage dependence of Ih activation. It is therefore likely that the responses of Ih to ischemia/hypoxia vary dramatically among particular neurons and may depend on specific insults.
Although little is known about the mechanisms underlying the changes of Ih in response to ischemic insults, altered intracellular cAMP levels might be involved in the postischemic inhibition of Ih in striatal cholinergic interneurons. It has been well established that intracellular cAMP regulates Ih by shifting its voltage-dependent activation (Frere et al, 2004; Robinson and Siegelbaum, 2003). In particular, decreases in the intracellular cAMP levels cause negative shifts of the voltage-dependent activation and thus reduce Ih at a given voltage. Morphologic studies have shown that striatal cholinergic interneurons express cAMP-sensitive Ih channel (HCN) subunits, including HCN2–4 (Notomi and Shigemoto, 2004; Santoro et al, 2000). Indeed, Ih in these neurons is enhanced in the presence of membrane-permeable cAMP analog (8-Br-cAMP), whereas application of the cAMP pathway inhibitor (Rp-cAMP) decreases the current amplitude (Deng et al, 2007). Furthermore, as detected in this study, the modulation of Ih through cAMP pathway was remarkably suppressed in postischemic neurons. These observations strongly support the idea that a decrease of intracellular cAMP levels is involved in the ischemia-induced Ih inhibition in cholinergic interneurons. On the basis of our previous results, it is further suggested that the postischemic inhibition of Ih might be independent of protein kinase A, as its selective inhibitors failed to affect the cAMP-mediated modulation of Ih (Deng et al, 2007). If a reduction of intracellular cAMP levels was the only mechanism mediating the postischemic inhibition of Ih, the application of 8-Br-cAMP would produce a greater effect on Ih in postischemic neurons. However, our data revealed that 8-Br-cAMP was less effective in postischemic neurons, suggesting that other pathways (cAMP-independent) might also contribute to the postischemic inhibition of Ih. One of the possible candidates is phosphatidylinositol-4,5-bisphosphate, because this phosphoinositide modulates Ih channels independent of cAMP, and a decrease of phosphoinositides may inhibit Ih in neurons (Zolles et al, 2006). It is also possible that ischemic insults may cause a loss of functional Ih channels, leading to a decrease in Ih amplitude.
Adenosine is a potential modulator of striatal cholinergic interneurons (Song et al, 2000). Extracellular adenosine concentration is dramatically increased during and after ischemia in vitro (Pearson et al, 2006). In addition, it appears that adenosine A1 receptors in the neostriatum are tonically active early (4 to 6h) after transient forebrain ischemia (Pang et al, 2002). One of the signaling pathways of A1 receptors is inhibition of adenylyl cyclase, leading to a decrease in cAMP (Ralevic and Burnstock, 1998). This pathway has been believed to be a mechanism by which adenosine inhibits Ih (Pape, 1992). In this study, we found that the blockade of A1 receptors with a selective antagonist (DPCPX) produced an increase of Ih in postischemic neurons but had no effect in control neurons. Moreover, the effects of DPCPX could be completely occluded by a cAMP analog 8-Br-cAMP. These findings indicate that the tonic activation of A1 receptors might contribute to the postischemic inhibition of Ih by decreasing intracellular cAMP. Interestingly, our findings also suggest that Ih in postischemic neurons was substantially upregulated by intracellular Ca2+. Given the decreased neuronal excitability of striatal cholinergic interneurons after ischemia, the Ca2+-mediated modulation of Ih might be related to an intracellular Ca2+ mobilization rather than an extracellular Ca2+ influx. If this is true, A1 receptors might also play a role in this process because their activation is able to stimulate the release of Ca2+ from intracellular stores, such as endoplasmic reticulum, by activating phospholipase C (Ralevic and Burnstock, 1998). Overall, it seems that activation of A1 receptors may exhibit opposite effects on Ih through distinct signaling pathways, with an overwhelming inhibition in striatal cholinergic interneurons after ischemia.
However, it remains uncertain as to whether the postischemic inhibition of Ih is solely mediated by adenosine. Neurotransmitters and neuromodulators that regulate cAMP levels may produce a reduction of Ih in postischemic neurons. Activation of dopamine D2 receptors has been shown to inhibit Ih in cholinergic interneurons (Deng et al, 2007); however, it is unlikely that dopamine may contribute to the postischemic inhibition of Ih, as the elevated dopamine release is returned to control levels within 30 mins after ischemia (Globus et al, 1988). Also unlikely is serotonin, considering its lack of effect on Ih in these neurons (Blomeley and Bracci, 2005). Nevertheless, our findings support the fact that adenosine, at least partially, mediates the postischemic inhibition of Ih in striatal cholinergic interneurons.
Functional Implications of Ih in Ischemia-Induced Neuronal Damage
Accumulating evidence indicates that Ih is involved in neurologic disorders such as seizure and stroke. In addition to regulating intrinsic membrane properties, including the resting membrane potential and the input resistance, Ih modulates neuronal responses to synaptic inputs (Robinson and Siegelbaum, 2003). The functional significance of Ih in neurologic disorders may depend on the subcellular compartmentalization and property of Ih channels, the nature and location of synaptic inputs, and the electrophysiological activity of particular neurons (Santoro and Baram, 2003). For instance, Ih may exhibit opposite roles in epileptogenesis. An enhanced Ih in CA1 pyramidal neurons (most likely in soma) can convert the potentiated synaptic inhibition to hyperexcitability (i.e., an increase of rebound firing) in a frequency-dependent manner, which promotes epileptogenic process (Chen et al, 2001). In contrast, an increase of dendritic (but not somatic) Ih attenuates the summation of excitatory synaptic inputs from distal dendrites, leading to a decrease of neuronal excitability and hence to the prevention of epileptogenesis (Poolos et al, 2002).
Studies on the involvement of Ih in cerebral ischemia are less extensive. This study has shown that the spontaneous activity and hyperpolarization-induced RD in striatal cholinergic interneurons were dramatically suppressed after ischemia. As both spontaneous firing and RD are largely controlled by Ih in these neurons, our findings indicate that postischemic inhibition of Ih is sufficient to decrease neuronal excitability and that Ih inhibition may reduce hyperexcitability induced by inhibitory synaptic inputs in postischemic neurons. In cholinergic interneurons, Ih may have little impact on excitatory synaptic inputs because Ih channels are mainly located in the soma, as shown by the immunostaining against HCN3 (Notomi and Shigemoto, 2004). Additionally, both membrane potentials and voltage responses to depolarizing currents were not changed significantly after ischemia. The simplest explanation is that cholinergic interneurons have less negative membrane potentials (more than −64 mV in the presence of TTX), and thus only a small proportion of Ih is constitutively active. The roles of Ih in cholinergic interneurons may differ from those in other neurons. Both ischemia-sensitive and -resistant neurons express Ih. The functional significance of Ih in neuronal damage after cerebral ischemia may be related to the nature of its changes (i.e., excitatory or inhibitory). It is reasonable to speculate that changes of Ih with inhibitory effects on neuronal excitability might be neuroprotective.
One of the most intriguing findings in this study is that a tonic activation of A1 receptors produced an inhibition of Ih after ischemia, leading to a suppression of neuronal excitability. Endogenous adenosine is believed to be neuroprotective against cerebral ischemia (Fredholm et al, 2005). Activation of A1 receptors has been shown to cause the presynaptic inhibition of excitatory neurotransmission (Pang et al, 2002; Pearson et al, 2006). However, intrinsic membrane excitability is also critical to neuronal injury after ischemic insults. Therefore, the postischemic inhibition of Ih may represent a novel mechanism by which adenosine protects neurons from cerebral ischemia.
Disclosure/Conflict of Interest
The authors declare no competing financial interests.