
Abstract
GADD34 is expressed in the ischaemic brain and reverses protein synthesis shutdown. Consequently, GADD34 could have neuroprotective potential in stroke. BHK medium, a replication-deficient HSV viral vector (HSV1716) with no insert or containing full-length GADD34, the N terminal or a conserved portion of the gene, was injected into mouse brain before stroke. Infarct size was 1.0±0.26, 1.19±0.36, 1.5±0.36, 1.3±0.36, and 1.1±0.28 mm3, respectively. The increase in infarct size with full-length GADD34 was statistically significant (P<0.05). Immunohistochemistry confirmed viral protein expression. Tissue culture studies revealed GADD34 gene restored virulence in HSV1716, suggesting that HSV virulence, rather than increased GADD34, exacerbated ischaemic damage.
Introduction
One of the major mechanisms influencing cell survival after stroke is inhibition of protein synthesis, which can persist for long periods and may not recover in selectively vulnerable cells (Paschen, 2003). This is particularly pertinent for cells within potentially salvageable penumbral tissue, which, if supported to maintain basic cellular processes including restoration of protein synthesis, could survive, limiting infarct expansion.
GADD34, one of five distinct GADD genes, is a cell-cycle protein inducible by growth arrest and DNA damage (Hollander et al, 1997). GADD34 is upregulated in the rodent and human ischaemic brain (Imai et al, 2002; White et al, 2004) and is known to facilitate reversal of protein synthesis shutdown through the dephosphorylation of eukaryotic initiation factor-2α (eIF2α) (Novoa et al, 2001). Because GADD34 mRNA levels correlate with cell survival during global ischaemia (Doutheil et al, 1999), it is possible that GADD34 expression may be increased to counteract protein synthesis shutdown during cellular stress, thereby promoting cell survival.
The aims of the current study were to generate HSV viral vectors containing full-length, or GADD34 gene fragments, to investigate the consequences of increased GADD34 expression on ischaemic damage in a mouse stroke model. The hypothesis under test was that increased GADD34 expression would limit infarct size.
Materials and methods
Viral Vectors
BHK cells were propagated in modified Eagle's medium with 10% newborn calf serum/10% tryptose phosphate broth and supplemented with glutamine and streptomycin/penicillin. 3T6 cells were maintained in Dulbecco's modified Eagle's medium with 10% newborn calf serum and supplemented with glutamine and streptomycin/penicillin.
The MyD116 sequence was pre-cloned into the pCMV-SPORT 6 plasmid (Invitrogen, Paisley, UK). Full-length, conserved 63 amino acid and N terminal (full-length sequence minus conserved sequence) of MyD116 were PCR-amplified including the CMV IE promoter and contained appropriate start and stop codons. The conserved sequence was amplified by PCR, cloned into the pcDNA 3.1 plasmid to gain a CMV IE promoter, and excised by restriction digest. All three sequences were cloned into the RL1deliresGFP plasmid (provided by Dr P Dunn), which contained an inter-ribosomal entry site and a green fluorescent protein (GFP) reporter gene. The RL1IRESGFP plasmid contains flanking sequences from either side of the herpes simplex viral gene RL1, which codes for ICP34.5.
Plasmid constructs were linearised, incubated with wild-type virus (HSV17+) DNA/calf thymus DNA, and the mixture applied to confluent monolayers of BHK cells for incubation (37°C for 4 h). The plates were then washed, incubated with dimethylsulphoxide (4 mins), overlaid with medium, and maintained at 37°C for 72 h. Plaques formed from recombinant virus identified by GFP signal were harvested and multiple rounds of plaque purification performed until all plaques were fluorescent and pure stocks of recombinant virus were produced. Stocks of each recombinant virus were grown and titred.
Confluent monolayers of BHK and 3T6 cells were infected with HSV17+, HSV1716 (replication-deficient vector lacking the RL1 gene encoding the virulence protein ICP34.5 (Brown et al, 1994)) or one of the recombinant viruses at 0.1 PFU per cell and absorbed at 37°C for 1 h. The cells were overlaid with medium for incubation periods of 6, 24, 48, or 72 h at 37°C and then scraped from the plate and stored at −70°C. The cells were thawed and sonicated for 5 mins to disrupt cells and release progeny virus. Confluent monolayers of BHK and 3T6 cells were infected with serial dilutions of each virus for 45 mins and overlaid with methyl cellulose for 72 h at 37°C. Finally, cells were Giemsa stained and the plaques counted on analogous plates.
Animal Procedures
Procedures were performed under licence from the UK Home Office. Male C57BL/6 mice (22 to 30 g) were anaesthetised with halothane (2% to 3%) in 70% N2O/30% O2 via a face mask and placed in a stereotaxic frame. Body temperature was maintained at 37±0.5°C and subcutaneous fluids given post-operatively to prevent dehydration.
Stereotaxic co-ordinates (2 mm lateral, 0.5 mm anterior to bregma, 2 mm ventral to dura) were used to deliver viral vector (1 × 106 PFU in 0.17 to 0.3 μL) and (24 h later) endothelin-1 (ET-1, 400 pmol, 0.5 μL, Alexis Biochemicals, Nottingham, UK) into the striatum at 0.05 μL per min.
Mice were randomly assigned to five groups (n=8): Group 1: BHK medium (vehicle for viral vectors); Group 2: HSV1716 (replication-deficient vector); Group 3: HSVGADD34 (HSV1716 containing full-length GADD34 gene); Group 4: HSVGADD34 conserved (HSV1716 containing GADD34 conserved sequence); and Group 5: HSVGADD34 N terminal (HSV1716 containing GADD34 N-terminal sequence). Groups 1 and 2 served as controls. The experiment was terminated by perfusion fixation (4% paraformaldehyde in phosphate-buffered saline) 72 h later.
Approximately eight sections collected throughout the lesion were stained with haematoxylin and eosin and examined by an investigator, blind to treatment allocation, to quantify lesion volume. Infarcted tissue was identified microscopically, transcribed onto line diagrams and measured by image analysis to generate infarct volume.
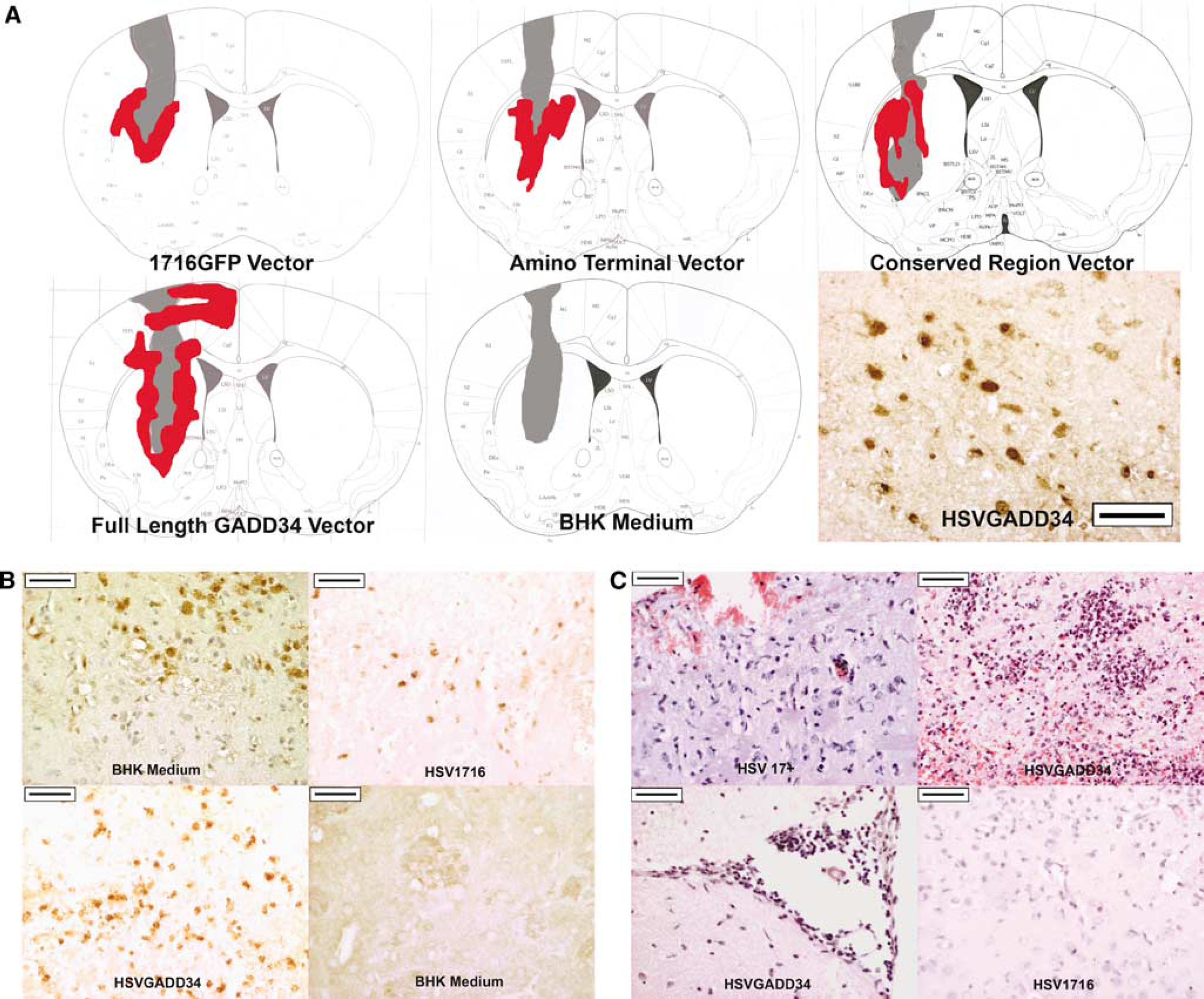
Immunohistochemistry: striatal sections were dewaxed, dehydrated, and microwaved (10 mins, 10 mmol/L citric acid, pH 6.0). HSV rabbit polyclonal antibody (DAKO, Cambridge, UK; 1:2,000) or GADD34 (sc-8327, Santa Cruz, Heidelberg, Germany; 1:100) incubated overnight at 4°C followed by a biotinylated goat anti-rabbit secondary antibody (1:100; Vector Laboratories, Peterborough, UK) used with avidin/biotinylated horseradish peroxidase complex (ABC kit, Vector Laboratories) and 3,3′-diaminobenzidine (Vector Laboratories). Green fluorescent protein monoclonal antibody (Clontech, Saint German En-Laye, France; 1:100) was used with a biotinylated goat anti-mouse (1:100; Vector Laboratories) secondary antibody. HSV staining mapped onto line diagrams (Figure 1A).
(
Statistical Analysis
Infarct volume in viral vector groups was compared with the vehicle group using repeated measures analysis of variance with a Dunnett's post hoc test to determine statistical significance. All data are expressed as mean±s.d.
Results
Viral Protein Expression In Vivo
In vivo HSV antigen was detected in cell bodies within the infarct core and peri-infarct region in all but the vehicle group, confirming spread throughout the target zone (red shading in Figure 1A).
Increased GADD34 immunoreactivity was present in the peri-infarct region of all groups in both the cortex and striatum. However, ischaemia-induced endogenous GADD34 (vehicle group, left-hand panel, Figure 1B) could not be differentiated from viral vector-derived GADD34. Therefore, GFP was used to confirm expression of new viral proteins. Green fluorescent protein was detected in cell bodies within infarct and peri-infarct regions in all recombinant virus groups, but not in the vehicle group, confirming cells containing the viral vector had expressed gene products (Figure 1B).
Effect GADD34 on Infarct Size
ET-1 causes local vasoconstriction resulting in a small, reproducible striatal infarct, extending along the needle tract into external capsule and cortex (Figure 1A).
HSVGADD34 caused a significant increase in infarct volume compared with vehicle (1.5±0.36 versus 1.0±0.25 mm3, P<0.05). In contrast, HSV1716, HSVGADD34 conserved and HSVGADD34 N terminal (1.19±0.36, 1.3±0.35, and 1.1±0.28 mm3, respectively) had no significant influence on infarct volume.
The HSVGADD34 group displayed a greater spread of HSV immunostaining compared with other groups (Figure 1A), similar to a wild-type HSV17+ phenotype.
Additional animals, which did not undergo ischaemia, were subsequently injected with HSV17+ or the recombinant vectors to check for viral virulence and evidence of associated pathology. HSV 1716, HSVGADD34 conserved, and HSVGADD34 N-terminal constructs induced a mild inflammatory response around the needle tract (Figure 1C, bottom right-hand panel) and mice showed no signs of ill health. As expected, 72 h after HSV17+ injection, histology showed evidence of encephalitis characterised by perivascular inflammation, leptomeningeal inflammation, neuronophagia, and necrotising encephalitis (Figure 1, top left panel). A similar pattern of pathology was observed with HSVGADD34 (Figure 1C top right and bottom left panels). HSV17+ and HSVGADD34 injected mice displayed behavioural signs of ill health such as hunching, shaking, inactivity, and hypersensitivity, indicative of a wild-type HSV infection.
Virus Replication
Multicycle growth curves provided a replication profile of the three recombinant viruses, wild-type HSV17+ and HSV1716. BHK cells were permissive for HSV17+ and HSV1716 and this was also true for the recombinant viruses (data not shown). 3T6 cells were permissive to HSV17+ but were non-permissive for HSV1716, HSVGADD34 conserved, and HSVGADD34 N-terminal. However, HSVGADD34 showed efficient growth in 3T6 cells where titres reached similar levels to HSV17+ (Figure 2).
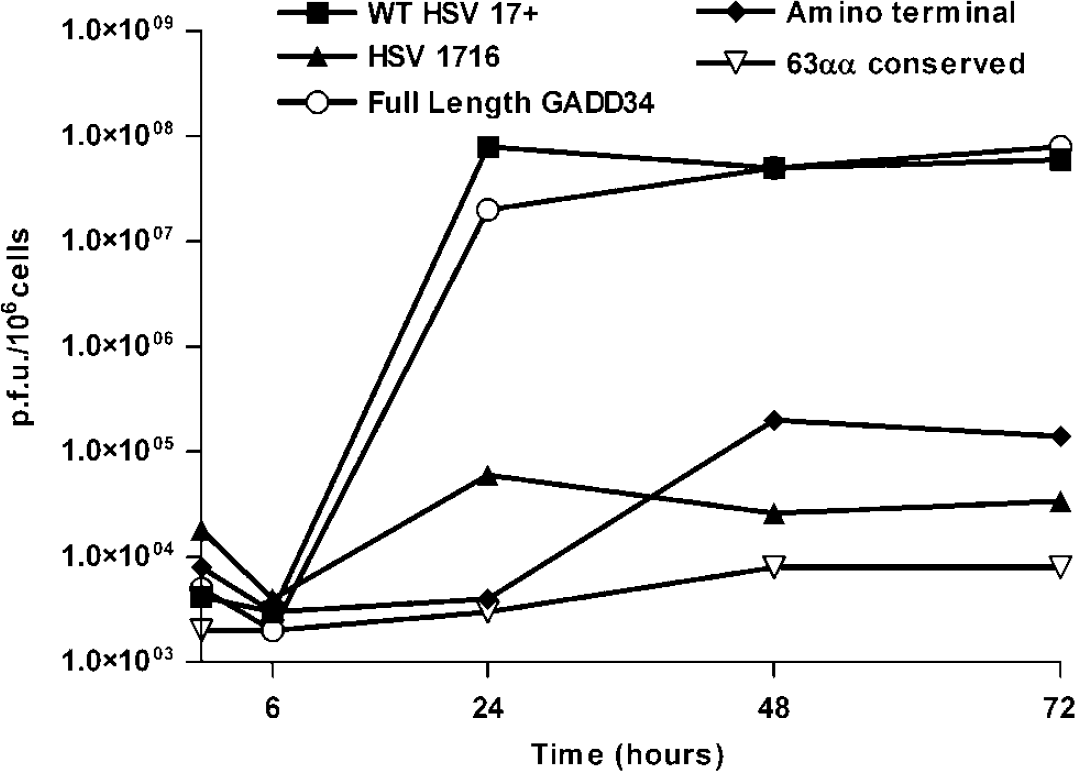
Growth curves for full-length GADD34, N terminal and 63 amino acid conserved sequence in RL1iresGFP viruses in 3T6 cells. 17+ refers to wild-type HSV virus while 1716 refers to the replication-deficient HSV viral vector containing no gene insert.
Discussion
One of the most critical events induced by cerebral ischaemia is endoplasmic reticulum stress resulting in protein synthesis shutdown. The mechanism involves upregulation of PKR-like endoplasmic reticulum kinase resulting in phosphorylation of eIF2α, inhibition of eIF2B, and cessation of protein synthesis translation (Paschen, 2003). Although, generalised protein synthesis is shut down, certain proteins are upregulated through open reading frame bypass scanning (Kojima et al, 2003). ATF4, an endoplasmic reticulum stress-induced protein increased after eIF2α phosphorylation, induces several downstream genes including GADD34 (Blais et al, 2004) which dephosphorylates eIF2α, restoring protein synthesis (Novoa et al, 2001).
We have reported upregulation of GADD34 in neurones and microglia in the peri-infarct region in rodent focal ischaemia (Imai et al, 2002) and in the human brain after cardiac arrest (White et al, 2004). Because upregulation is within peri-infarct tissue and GADD34 is implicated in DNA repair, ischaemic tolerance, and restoration of protein synthesis (Garcia et al, 2004; Novoa et al, 2001), GADD34 (or specific fragments) levels were increased by viral vector gene transfer to determine its influence on ischaemic damage.
HSV1716 successfully delivered and increased protein products of inserted genes, which was confirmed by GFP immunohistochemistry, because viral GADD34 could not be distinguished from ischaemia-induced endogenous GADD34.
Unexpectedly, HSVGADD34 significantly increased infarct volume. The fact that HSV immunoreactivity spread further in the HSVGADD34 group (Figure 1A) alongside behavioural signs of ill health and growth curve data (Figure 2) suggested that the full-length GADD34 gene restored virulence to HSV1716.
Histology from animals that received HSVGADD34 without ischaemia confirmed evidence of encephalitis and comparable neuropathology to HSV17+ animals, both groups exhibiting behavioural signs indicative of HSV infection. In contrast, HSV1716, HSVGADD34 conserved, and HSVGADD34 N-terminal groups displayed only a mild inflammatory response around the injection tract with no signs of ill health.
HSV1716 lacks the virulence protein ICP34.5 (McKay et al, 1993) that prevents protein synthesis shutdown on viral infection, allowing viral replication to occur (Chou and Roizman, 1994). By binding to protein phosphatase-1α, the C terminal of ICP34.5 removes phosphate from eIF2α in response to a variety of eIF2α kinases, including PKR and endoplasmic reticulum kinase (Cheng et al, 2005; He et al, 1997). The full-length GADD34 gene appears to compensate for the lack of ICP34.5 and restore virulence to HSV1716. Therefore, the increased infarct volume seen with HSVGADD34 is most likely due to GADD34 restoring virulence to HSV1716 and not to increased tissue levels of GADD34 per se. To avoid this problem, HSV amplicons or adenoviruses could be used as alternative delivery vehicles for GADD34 or in studies conducted in GADD34 knockout mice (Kojima et al, 2003) used to provide further insights into the role of GADD34 in stroke.
Footnotes
Acknowledgements
The authors thank Professor David Graham and Dr William Stewart for help in the neuropathological assessment of brain tissue. This work was funded by the Neuroscience Foundation, Scottish Enterprise Proof of Concept Scheme, SHERT (to FW), and a Wellcome Trust VIP award (to CM).