
Abstract
Cerebral vascular dysfunction and associated diseases often occur in type-1 diabetes, but the underlying mechanisms are largely unknown. In this study, we sought to determine whether big-conductance, Ca2+-activated K+ (BK) channels were impaired in vascular (cerebral artery) smooth muscle cells (CASMCs) from streptozotocin-induced type-1 diabetic mice using patch clamp, molecular biologic, and genetic approaches. Our data indicate that the frequency and amplitude of spontaneous transient outward currents (STOCs) are significantly decreased, whereas the activity of spontaneous Ca2+ sparks is increased, in diabetic CASMCs. The sensitivity of BK channels to voltage, Ca2+, and the specific inhibitor iberiotoxin are all reduced in diabetic myocytes. Diabetic mice show increased myogenic tone and decreased contraction in response to iberiotoxin in cerebral arteries and elevated blood pressure. The expression of the BK channel β1, but not α-subunit protein, is markedly decreased in diabetic cerebral arteries. Diabetic impairment of BK channel activity is lost in CASMCs from BK channel β1-subunit gene deletion mice. In conclusion, the BK channel β1-subunit is impaired in type-1 diabetic vascular SMCs, resulting in increased vasoconstriction and elevated blood pressure, thereby contributing to vascular diseases in type-1 diabetes.
Keywords
Introduction
Type-1 diabetes mellitus affects more than 1 million people with increasing incidence and mortality. Patients with type-1 diabetes are up to nine times more likely to develop and die from stroke and other cardiovascular diseases (Brands et al, 2004; Ferguson et al, 2005), which may result from abnormal contractile responses of cerebral and other vascular smooth muscle cells (SMCs). An earlier study has shown that myogenic contractions are increased in cerebral and gracilis skeletal muscle arteries from streptozotocin-induced type-1 diabetic rats (Zimmermann et al, 1997; Ungvari et al, 1999). Similarly, vasoconstrictions to high K+ and other agonists are augmented in aortic, carotid, coronary, mesenteric, and tail arteries from type-1 diabetic patients (Ramanadham et al, 1984; Agrawal and McNeill, 1987; Agrawal et al, 1987; White and Carrier, 1988, 1990; Inazu et al, 1991; Taylor et al, 1992; Abebe et al, 1994; Savage et al, 1995; Weber et al, 1996; Chow et al, 2001; Xavier et al, 2003; Tickerhoof et al, 2003). These increased vasoconstrictions are related to the enhanced extracellular Ca2+ influx. Furthermore, pharmacological stimulation of voltage-dependent Ca2+ channels (VDCCs) induces a larger contraction or increase in [Ca2+]i in aortic, gracilis skeletal muscle, mesenteric, and uterine artery SMCs from type-1 diabetic animals and patients (Agrawal and McNeill, 1987; Agrawal et al, 1987; White and Carrier, 1988, 1990; Kamata et al, 1988; Inazu et al, 1991; Ungvari et al, 1999; Fleischhacker et al, 1999; Chow et al, 2001; Burnham et al, 2006). Thus, the enhanced Ca2+ influx through VDCCs may contribute to abnormal increases in [Ca2+]i in type-1 diabetic vascular SMCs.
Big-conductance, Ca2+-activated K+ (BK) channels are exclusively expressed in cerebral arterial and other vascular SMCs. Activation of BK channels causes K+ efflux and produces hyperpolarizing currents, which inhibit the opening of VDCCs and Ca2+ influx across the cell membrane, causing relaxation of cerebral artery smooth muscle cells (CASMCs) (Lu et al, 2006; Ledoux et al, 2006). Presumably, type-1 diabetes may impair BK channels in vascular SMCs, which results in the enhanced Ca2+ influx and contraction, contributing to vascular dysfunction and associated vascular complications. To test this hypothesis, we sought to examine whether the activity of BK channels is impaired, and which BK channel subunits (α-, β1-subunit, or both) are attributed to the impaired activity of BK channels in CASMCs from streptozotocin-induced type-1 diabetic mice using patch clamp, molecular, and genetic approaches. To determine the potential role of impaired BK channels in vascular dysfunctions, we also investigated whether cerebral artery muscle contractions and blood pressure are altered in streptozotocin-induced type-1 diabetic mice using a pressure arteriograph and noninvasive computerized tail—cuff system, respectively.
Materials and methods
Generation of Type-1 Diabetic Mice
Experiments using animals were conducted in accordance with the approved Animal Care and Use Protocol of Albany Medical College. To create the animal model with type-1 diabetes, male Swiss Webster mice (3-week old, weight = 10 to 15 g) from Taconic Farms (Germantown, NY, USA) received a single intraperitoneal injection of streptozotocin (180 mg/kg). Control mice with the same background, sex, and age received an injection of 0.9% saline solution. Three days after the injection of streptozotocin, blood glucose levels were measured using One Touch Ultra Glucose Meter (Lifescan Inc., Milpitas, CA, USA); mice with blood glucose higher than 280 mg/dl were considered diabetic.
Big-conductance, Ca2+-activated K+ β1-subunit gene deletion (β1−/−) mice were generated and maintained, as reported previously (Brenner et al, 2000). Diabetes in these mice was induced by using the same procedure as described above. Wild-type mice with the same background, sex, and age were used as control.
Preparation of Freshly Isolated Cerebral Artery Smooth Muscle Cells
Freshly isolated CASMCs were obtained using a similar procedure as reported previously (Brenner et al, 2000). In brief, mice were euthanized by sodium pentobarbital (100 mg/kg, intraperitoneally). Cerebral arteries were rapidly dissected in ice-cold (4°C) physiologic saline solution (PSS). The arteries were first digested in low Ca2+ (100 μmol/L) PSS containing (all in mg/mL) 1.0 papain, 0.2 dithioerythritol, and 0.5 bovine serum albumin for 20 min (37°C), and then in PSS containing (mg/mL) 1.0 collagenase H, 1.0 collagenase II, 1.0 dithiothretol, and 0.5 bovine serum albumin for about 18 mins (37°C). After being washed with PSS three times, the arteries were gently triturated to release single myocytes. Cells were stored in ice-cold PSS for daily use. The composition of PSS was (in mmol/L): 130 NaCl, 5.4 KCl, 1 MgSO4, 1.8 CaCl2, 10 HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), and 10 glucose (pH 7.4).
Patch-Clamp Recordings
The nystatin-perforated patch-clamp technique only allows the exchange of monovalent ions between the cytosol and electrode, which may retain the intracellular environment at a nearly physiologic status. Thus, this technique was used to record whole-cell membrane currents, as described previously (Wang et al, 2003; Liu et al, 2007). In these experiments, patch pipettes had a resistance of 3 to 4 ΩM when they were filled with intracellular solution. Nystatin was included in the pipette solution at a final concentration of 200 to 300 μg/mL. Junction potentials between the pipette and bath solutions were compensated just before seal formation. Membrane capacitance and series resistance were compensated. An access resistance of less than 40 ΩM was accepted for current recordings. Voltage-command protocols were generated by the EPC-9 system (Heka Electronics, Lambrecht, Germany). Data were recorded and analyzed using Pulse and Pulsefit software (Heka Electronics). The composition of normal bath solution was the same as the above-mentioned PSS. The pipette (intracellular) solution contained (in mmol/L): 130 KCl, 5 MgCl2, 10 HEPES, 1 CaCl2, and 3 EGTA (ethylene glycol bis(β-aminoethylether)-N,N,N′,N′,-tetraacetic acid) (pH 7.2).
Single BK channel currents were measured in inside-out patches under symmetrical pipette and bath K+ concentrations that contained (in mmol/L): 140 KCl, 1 MgSO4, and 10 HEPES (pH 7.2). Free Ca2+ concentration in bath solution was set at 0.1, 1, or 10 μmol/L using various concentrations of EGTA and CaCl2 with WinMAX program (Chris Patton, Stanford University, http://www.stanford.edu/∼cpatton/maxc.html). Single-channel currents were recorded at digitization rate of 5 kHz and filtered at 1 kHz. Data were analyzed using the Clampfit 9 software (Axon Instruments, Union City, CA, USA).
Measurement of Ca2+ Sparks
Ca2+ sparks were measured using a TCS SP2 laser-scanning confocal microscope (Leica, Mannheim, Germany) with a × 40 oil immersion objective (NA 1.3), as reported previously (Wang et al, 2003; Liu et al, 2007). Cells were loaded with 5 μmol/L Fluo-4 AM (Molecular Probes, Eugene, OR, USA) for 40 mins at room temperature. Fluo-4 was excited with 488 nm light from a krypton/argon laser. The fluorescence emission was measured at 505 nm. Line-scan images were taken to obtain high temporal profiles. Image analysis was performed using Leica Image Examiner and Interactive Data Language software (Research Systems, Boulder, CO, USA).
Western Blot Analysis
Western blotting was performed as described previously (Zheng et al, 2004). Isolated mouse cerebral arteries were homogenized in ice-cold radioimmunoprecipitation assay buffer. The homogenate was centrifuged to obtain total membrane fraction. The protein concentration was determined using Bio-Rad Protein Kit (Bio-Rad, Hercules, CA, USA). The proteins were transferred to a polyvinylidene fluoride membrane using Semi-Dry Electrophoresis Transfer Cell (Bio-Rad). The nonspecific binding sites on the membrane were blocked by Tris-buffered saline Tween-20 buffer containing 5% nonfat dry milk. The membrane was probed with specific antibodies against α- and β1-subunit mouse BK channels (Alomone Labs, Jerusalem, Israel and Affinity BioReagents, Golden, CO, USA, respectively) at 1:500 dilution, followed by incubation with horseradish peroxidase-conjugated secondary antibody, and then developed using enhanced chemiluminescence reagents. Then, the membrane was re-stripped and probed with anti-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 1:1,000 dilution as loading controls. The BK channel α- and β1-subunit protein expression levels were normalized to β-actin expression levels.
Measurement of Vasoconstriction in Cerebral Arteries
Vasoconstrictions in posterior cerebral arteries were assessed using a pressure arteriograph (Living Systems Instrumentation, Burlington, VT, USA), as described previously (Nelson et al, 1995). Arteries with a diameter of ∼100 μm were dissected in ice-cold PSS containing (in mmol/L): 119 NaCl, 4.7 KCl, 24 NaHCO3, 1.2 KH2PO4, 1.6 CaCl2, 1.2 MgSO4, 0.023 ethylenediaminetetraacetic acid, and 11 glucose (pH 7.4). Vessels were cannulated with glass micropipettes, and perfused with PSS (gassed with 95% O2 and 5% CO2, at 35°C). After an equilibration period of 60 mins, vessels were subjected to a series of pressure steps between 20 and 100 mm Hg to induce myogenic vasoconstrictions. Then, vessels were perfused with nominally Ca2+-free and 3 mmol/L EGTA bath solution for 45 mins, and re-subjected to various pressure steps to obtain passive responses. Myogenic vasoconstrictions were calculated as the percentage difference in diameters observed for Ca2+-containing versus Ca2+-free bath solution at each pressure step.
To examine iberiotoxin- or tetraethylammonium-induced vasoconstrictions, vessels were pressured at 60 mm Hg and then exposed to either of these BK channel blockers.
Measurement of Blood Pressure
Blood pressures were measured using a noninvasive computerized tail—cuff system (Kent Scientific Corporation, Torrington, CT, USA). Mice were placed in a plastic holder at 30°C. The occlusion and sensor cuff were both positioned on the base of the tail. After mice were allowed to accustom to the system for 7 days, blood pressure was measured for at least three consecutive times in each mouse.
Chemicals
Unless otherwise stated, all chemicals used in this study were obtained from Sigma Chemical Corp. (St Louis, MO, USA).
Statistical Analysis
Data are expressed as mean ± s.e. Student's t-tests were used for comparison of paired or unpaired data. P < 0.05 was considered statistically significant.
Results
The Activity of Spontaneous Transient Outward Currents is Decreased in Type-1 Diabetic Mouse Cerebral Artery Smooth Muscle Cells
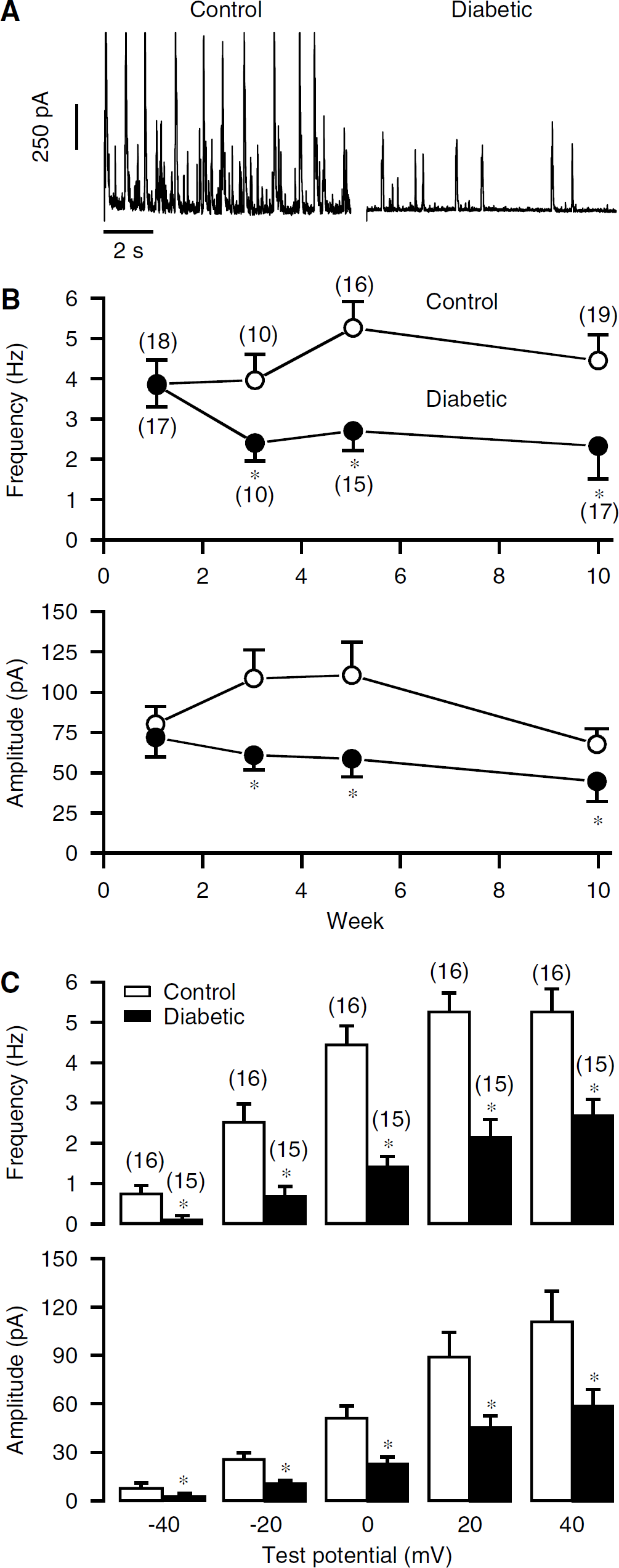
As spontaneous transient outward currents (STOCs) reflect the activity of BK channels, we first examined and compared their activity in CASMCs from control and streptozotocin-induced type-1 diabetic mice. Our data indicate that the frequency and amplitude of STOCs were unaltered in cells from mice with type-1 diabetes for 1 week. However, a significant decrease in the frequency and amplitude was found in cells from mice with type-1 diabetes for 3 weeks. The reduction of STOC frequency and amplitude were even greater in cells from mice with diabetes for 5 weeks. Figure 1A shows representative recordings of STOCs in a control and diabetic cell. The diabetic effects were sustained up to 10 weeks. The changes in the frequency and amplitude of STOCs at a holding potential of +40 mV in CASMCs from mice with type-1 diabetes for different time periods are summarized in Figure 1B. The mean frequency and amplitude decreased from 5.3 ±0.5 Hz and 110.6 +18.3 pA in control cells (n = 16 from seven mice) to 2.7 ± 0.4Hz and 57.5 ± 8.2pA in 5-week diabetic cells (n = 15 from six mice), respectively (P < 0.05). Moreover, the diabetic decrease in the frequency and amplitude of STOCs was observed at holding potentials ranging from −40 to + 20 mV (Figure 1C). Because mice with diabetes for 5 weeks showed the greatest reduction in the activity of STOCs, this time period was used for the rest of the experiments in this study.
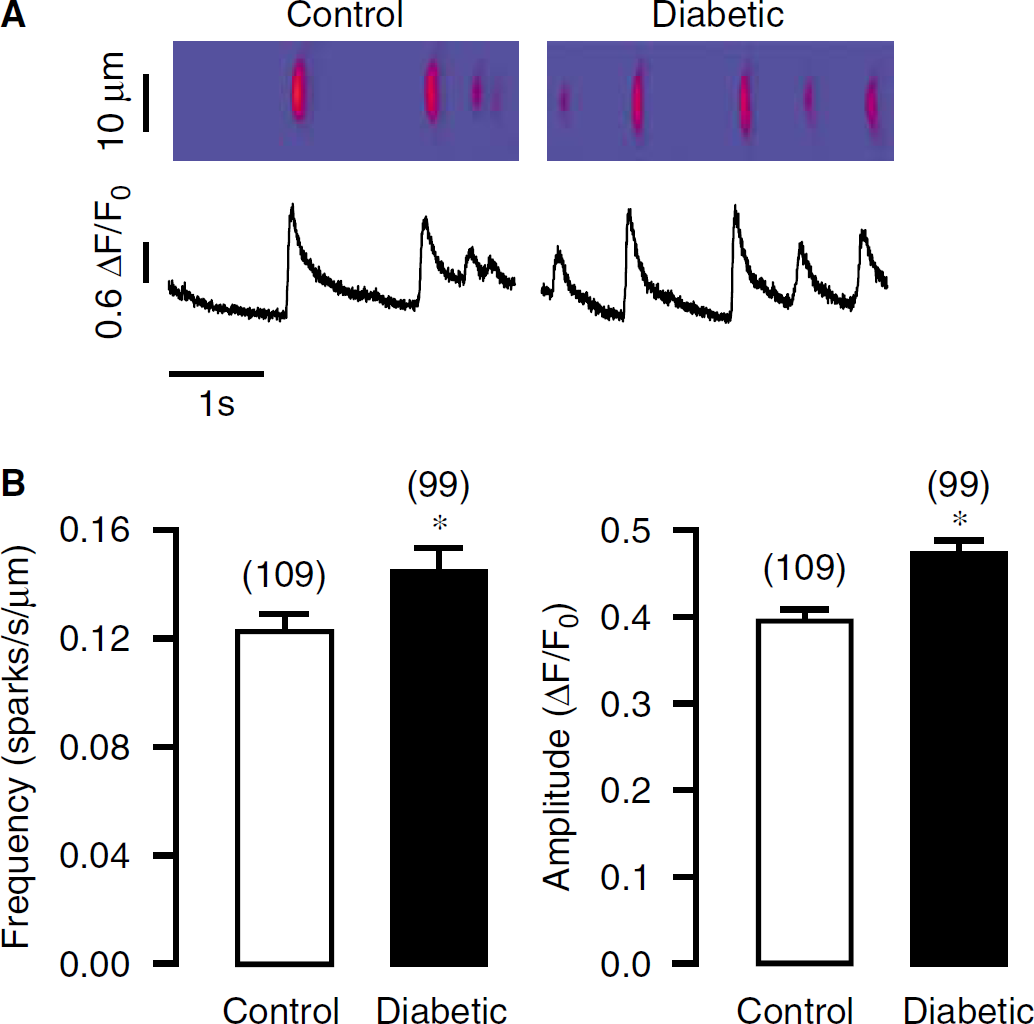
Frequency and amplitude Ca2+ sparks are increased in freshly isolated CASMCs from mice with streptozotocin-induced type-1 diabetes for 5 weeks. (
Ca2+ sparks are increased in type-1 diabetic mouse Cerebral Artery Smooth Muscle Cells
Spontaneous transient outward currents are generally believed to result from Ca2+ sparks in vascular SMCs. Thus, we wondered whether the decreased activity of STOCs was due to a reduction in Ca2+ sparks. Figure 2A shows Ca2+ sparks in a control and diabetic CASMC, indicating that the frequency and amplitude of Ca2+ sparks were both higher in a diabetic than control myocyte. As summarized in Figure 2B, the mean frequency was increased from 0.12 ± 0.01 sparks/sec per μm in control cells (n = 109 from seven mice) to 0.15 ± 0.01 sparks/sec per μm in diabetic cells (n = 99 from seven mice, P < 0.05), whereas the mean amplitude was 0.40 ± 0.01 ΔF/F0 in control cells and 0.47 ± 0.01 ΔF/F0 in diabetic cells (P < 0.05). These data suggest that the inhibited activity of STOCs in diabetic CASMCs is not attributed to the changes in Ca2+ sparks.
Frequency and amplitude of STOCs are decreased in freshly isolated CASMCs from streptozotocin-induced type-1 diabetic mice. (
Whole-Cell big-Conductance, Ca2+-Activated K+ Channel Currents are Inhibited in Type-1 Diabetic Mouse Cerebral Artery Smooth Muscle Cells
Whole-cell BK channels currents were induced by depolarization pulses from −20 to 60 mV with 10-mVincrements for 500 ms. 4-Aminopyridine (5 mmol/L) was used to block delayed rectifier K+ currents (Wang et al, 1997). Compared with control cells, currents in diabetic CASMCs were significantly smaller (Figure 3A). The currents at +60 mV were decreased from 59.5 ± 5.5 pA/pF in control cells (n = 6 from four mice) to 25.2 ± 3.4 pA/pF in diabetic cells (n = 6 from five mice, Figure 3B). In addition, there was no difference in membrane capacitance (an indicator of cell size) in control and diabetic cells (7.9 ± 0.7 versus 6.8 ± 0.7 pF).
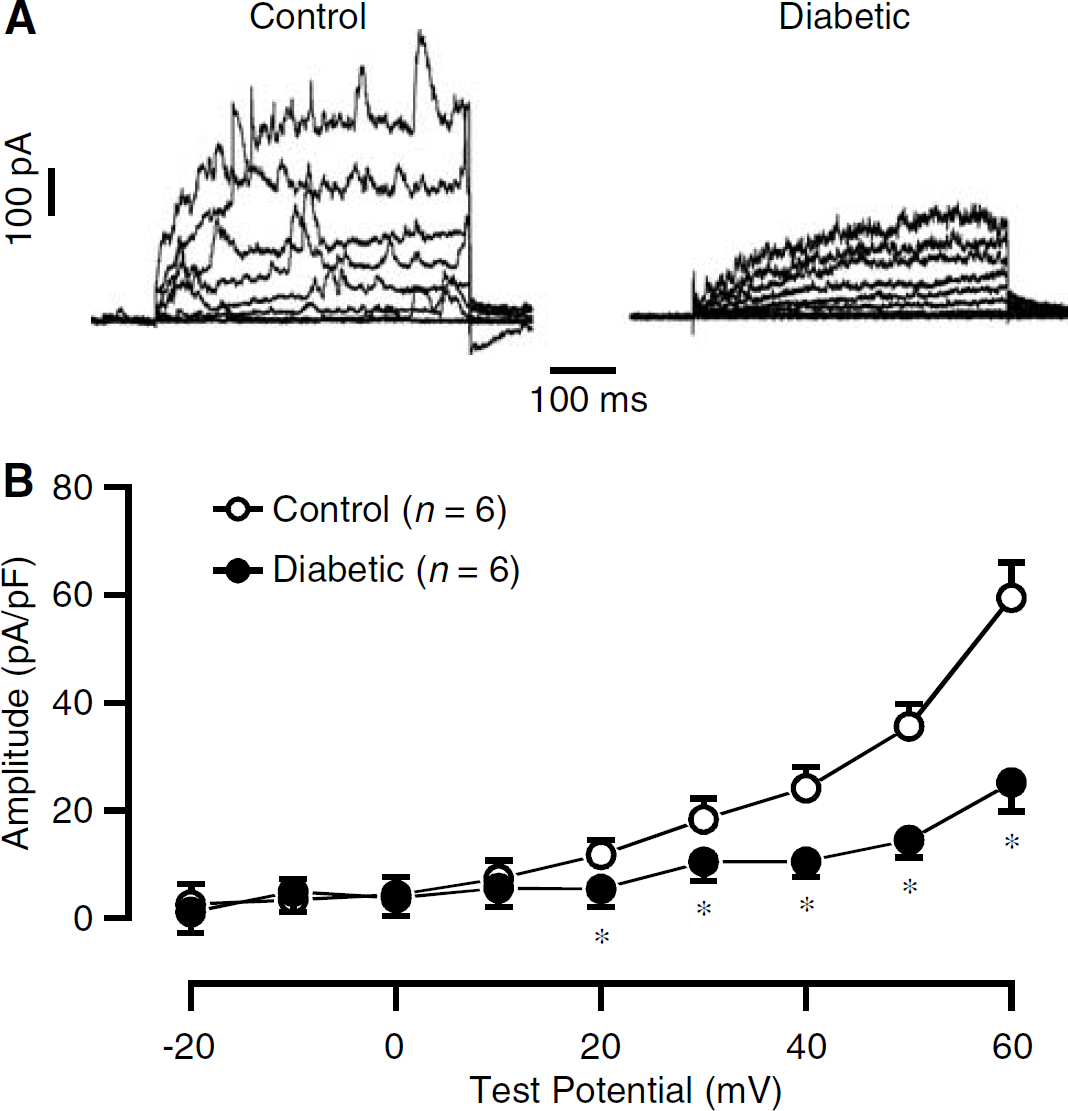
Whole-cell BK currents are reduced in freshly isolated CASMCs from mice with streptozotocin-induced type-1 diabetes for 5 weeks. (
The Sensitivity of Big-Conductance, Ca2+-Activated K+ Channels to the Specific Blocker Iberiotoxin is Attenuated in Type-1 Diabetic Mouse Cerebral Artery Smooth Muscle Cells
We also examined the effect of iberiotoxin, a specific BK channel inhibitor, on whole-cell outward K+ currents in control and diabetic CASMCs. Application of iberiotoxin (100 nmol/L) for 5 mins resulted in a smaller inhibition of outward K+ current in diabetic than control cells. The mean current inhibition by iberiotoxin was 63.7 ± 8.2% in control cells (n = 8 from five mice) and 38.5 ± 11.0% in diabetic cells (n = 6 from five mice), respectively (P < 0.05).
Single Big-Conductance, Ca2+-Activated K+ Channel Open Probability, Voltage Sensitivity, and Ca2+ Sensitivity are Decreased in Type-1 Diabetic Mouse Cerebral Artery Smooth Muscle Cells
Here, we first intended to determine whether the open probability of single BK channels was decreased in type-1 diabetic CASMCs. Using symmetric pipette and bath K+ concentration (140 mmol/L) and 1 μmol/L of free-bath Ca2+ solution, we found that single BK channel open probability was significantly decreased in diabetic cells. The mean open probability (NPo) was decreased from 0.16 ± 0.06 in control cells (n = 8 from three mice) to 0.04 ± 0.02 in diabetic cells (n = 8 from three mice, Figure 4A).
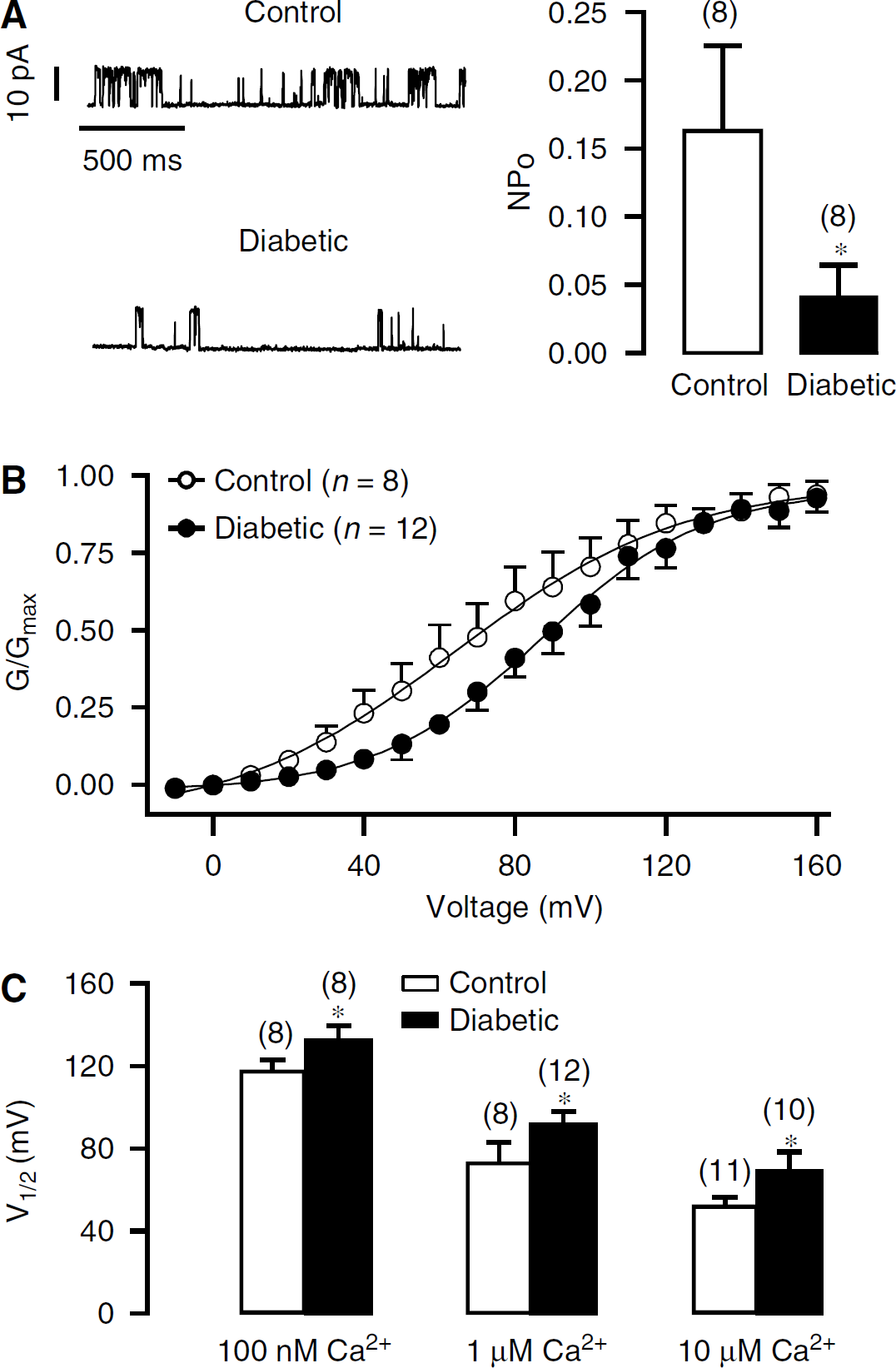
Single BK channel open probability, voltage sensitivity, and Ca2+ sensitivity are attenuated in freshly isolated CASMCs from mice with streptozotocin-induced type-1 diabetes for 5 weeks. (
Next, we examined the voltage sensitivity of BK channels in type-1 diabetic CASMCs. Macroscopic BK currents in inside-out patches were generated by step pulses from −10 to 160 mV in 10-mV increments for 500 ms. As shown in Figure 4B, the current—voltage curve was shifted to the right in diabetic cells. The V1/2 was increased from 72.5 ± 10.8 mV in control cells (n = 8 from four mice) to 92.3 ± 5.8mV in diabetic cells (n = 12 from three mice), indicating that the channel sensitivity to voltage is decreased in type-1 diabetic CASMCs.
To test whether the BK channel sensitivity to Ca2+ was also altered in type-1 diabetic CASMCs, we examined and compared V1/2 in inside-out patches from control and diabetic cells at different bath-free Ca2+ concentrations. As summarized in Figure 4C, V1/2 was greater in diabetic than control cells at 0.1, 1, and 10 μmol/L Ca2+ concentrations, indicating that the Ca2+ sensitivity of BK channels was reduced in type-1 diabetic CASMCs.
The BK channel β1-subunit appears to be necessary for the physiologic functions of the channel complex by regulating the Ca2+ and voltage sensitivity as well as conferring the actions of channel blockers in vascular SMCs (Lu et al, 2006; Ledoux et al, 2006). These findings, together with our results that BK channel sensitivity to voltage, Ca2+, and iberiotoxin are all reduced in diabetic cells, suggest that type-1 diabetes may impair the functions of the channel β1-subunit in CASMCs.
Big-Conductance, Ca2+-Activated K+ Channel β1, but not α-Subunit Expression is Decreased in Type-1 Diabetic Mouse Cerebral Arteries
The impaired activity of BK channels in type-1 diabetic CASMCs could result from decreased channel expression; therefore, we examined the BK channel α- and β1-subunit protein expression levels in cerebral arteries from control diabetic mice using Western blot analysis. The α- and β1-subunit proteins were immunoblotted with their specific antibodies and normalized to β-actin protein. As shown in Figure 5, subunit expression levels were similar in control and diabetic cerebral arteries; however, β1-subunit expression levels were significantly lower in diabetic than control cerebral arteries. These results are consistent with our electrophysiological findings that the BK channel β1-subunit functions are impaired in type-1 diabetic CASMCs.
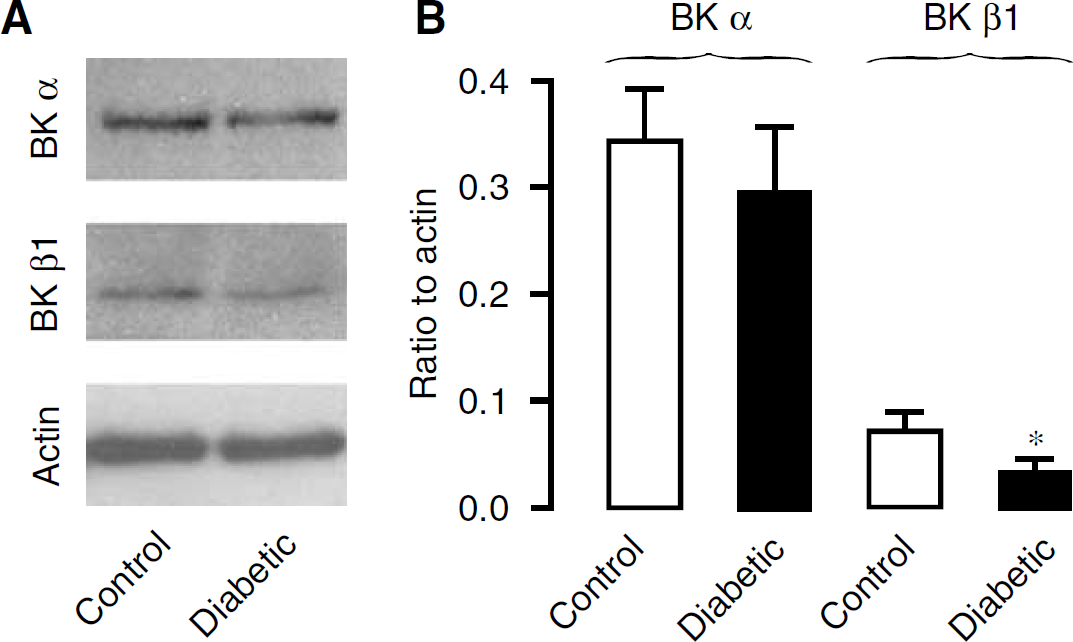
Big-conductance, Ca2+-activated K+ channel β1, but not α-subunit, protein expression levels are reduced in freshly isolated cerebral arteries from mice with diabetes for 5 weeks. (
Type-1 Diabetic Impairment of Big-Conductance, Ca2+-Activated K+ Channel Activity is Lost in β1–/–Mouse Cerebral Artery Smooth Muscle Cells
We further examined the potential importance of the β1-subunit in the type-1 diabetic inhibition of BK channel activity using β1−/− mice. The activity of BK channels, measured by the frequency and amplitude of STOCs, was similar in CASMCs from β1−/− mice with and without type-1 diabetes (Figure 6). The mean frequency and amplitude of STOCs were 4.4 ± 0.6 Hz and 80.3 ± 8.0 pA in cells from β1−/− mice without diabetes (nine cells from five mice) and 3.6 ± 0.7Hz and 86.1 ± 12.8pA in cells from β1−/− mice with diabetes (10 cells from five mice), respectively (P > 0.05). Thus, the diabetic effect on the BK channel activity is lost in β1−/− CASMCs.
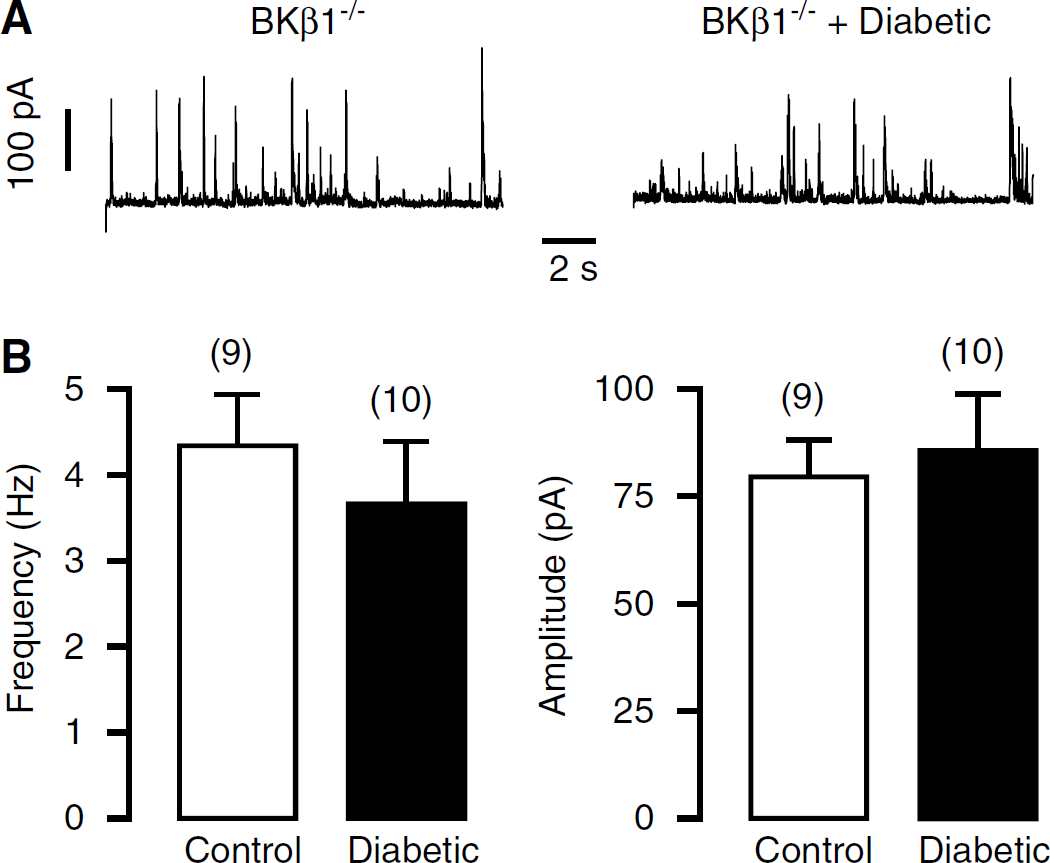
Activity of BK channels is similar in freshly isolated CASMCs from β1 gene deletion mice with and without streptozotocin-induced type-1 diabetes for 5 weeks. (
Myogenic Contraction is Increased in Type-1 Diabetic Mouse Cerebral Arteries
Considering that the activity of BK channels is important for the maintenance of normal vascular tone, we investigated whether myogenic contractions were altered in type-1 diabetic mouse cerebral arteries. As shown in Figure 7A, myogenic contraction, calculated as the percentage difference in diameters observed for Ca2+-containing versus Ca2+-free bath solution at each pressure step, was significantly increased in cerebral arteries from mice with type-1 diabetes than from control mice. The percentage difference in diameters at 100 mm Hg was increased from 5.8 ± 0.9% in control arteries (n = 11 from nine mice) to 9.6 ± 1.2% in diabetic arteries (n = 17 from eleven mice, P < 0.05).
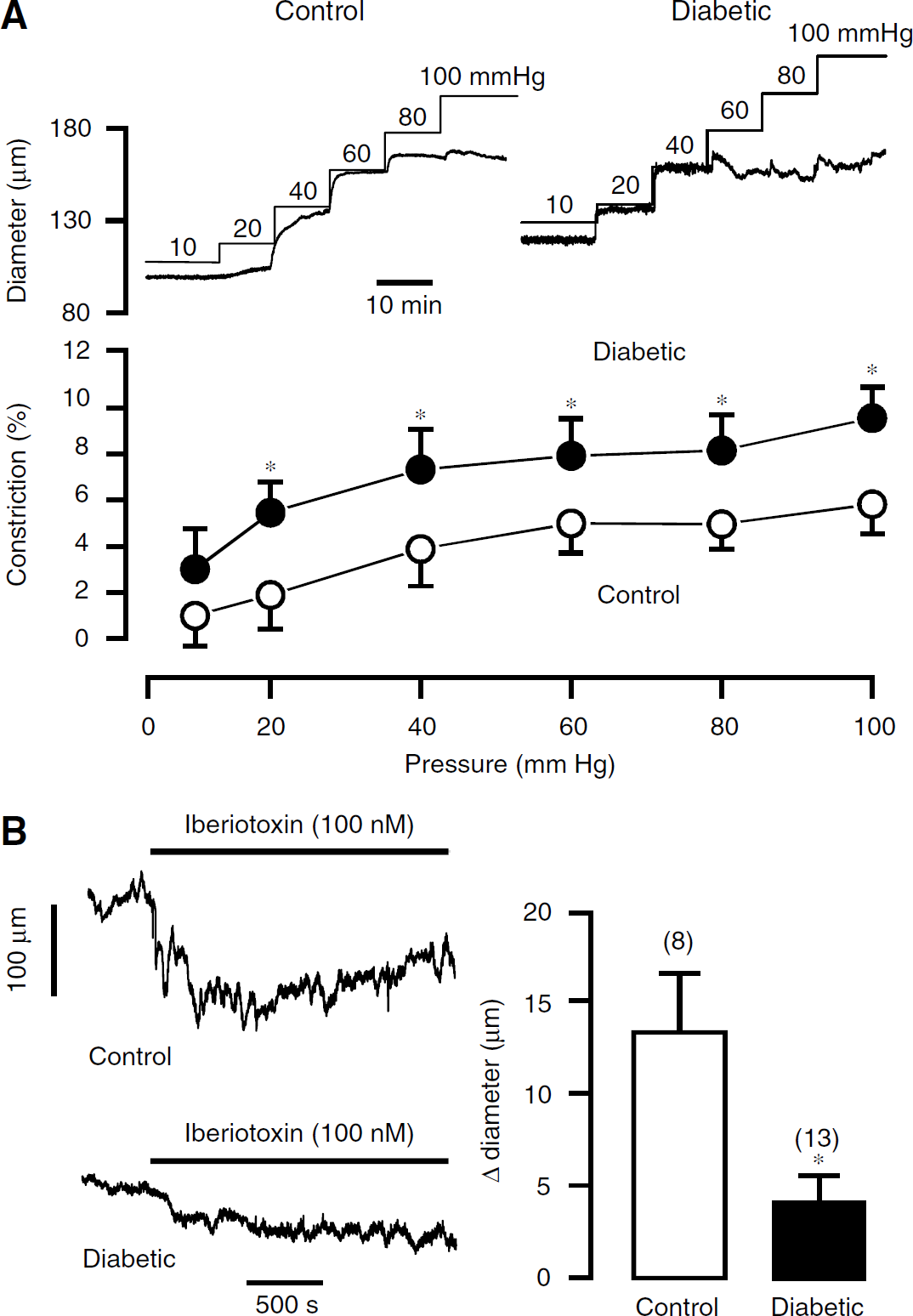
Myogenic and iberiotoxin-induced vasoconstrictions are enhanced in cerebral arteries from mice with diabetes for 5 weeks. (
Iberiotoxin-Induced Contraction is Diminished in Type-1 Diabetic Mouse Cerebral Arteries
Consistent with changes in myogenic contractions, the BK channel inhibitor iberiotoxin (100 nmol/L) caused a smaller vasoconstriction in diabetic arteries pressurized at 60 mm Hg than in control arteries. The mean vasoconstriction was 13.3 ± 3.2 μm in control arteries (n = 8 from six mice) and 4.2 ± 1.3 μm in diabetic arteries (n = 13 from six mice), respectively (P < 0.05, Figure 7B). Similarly, vasoconstriction elicited by tetraethylammonium at a low concentration (2 mmol/L) to inhibit selectively BK channels was reduced from 21.3 ± 3.0 μm in control arteries (n = 8 from six mice) to 12.4 ± 2.9 μm in diabetic arteries (n = 8 from five mice, P < 0.05). These results further indicate that BK channels are impaired in type-1 diabetic CASMCs, which may play an important role in the increased vasoconstriction/diminished relaxation in vascular tissues.
Blood Pressure is Elevated in Type-1 Diabetic Mice
Hypertension often occurs in diabetes, and also is associated with the downregulation of BK channel activity (Amberg et al, 2003; Amberg and Santana, 2003; Bratz et al, 2005). Thus, we wondered whether blood pressure was elevated in type-1 diabetic mice. Blood pressure was measured using a noninvasive computerized tail—cuff system. The results indicate that blood pressure was considerably higher in diabetic mice; mean systolic and diastolic pressure were 108.0 ± 2.4 and 82.1 ± 2.5 mm Hg in control mice (n = 13) and 120.4 ± 2.7 and 90.3 ± 2.2 mm Hg in diabetic mice (n = 12), respectively (P < 0.05). Similarly, invasive measurement of blood pressure also showed higher systolic and diastolic arterial pressure in diabetic than control mice (data not shown).
Discussion
A very recent and comprehensive report by McGahon et al (2007) have shown for the first time that caffeine-induced whole-cell BK currents, Ca2+ sensitivity of single BK channels, and mRNA expression of the BK channel β1-subunit are reduced in retinal arterial SMCs from streptozotocin-induced type-1 diabetic rats. In complement of these important findings, in this study, we have provided electrophysiological, functional, and molecular evidence that BK channels are impaired in CASMCs from mice with streptozotocin-induced diabetes. Our patch-clamp recordings indicate that the frequency and amplitude of STOCs are significantly decreased in diabetic CASMCs, and whole-cell BK currents are much smaller in diabetic than control cells. Single BK channel open probability is decreased in diabetic CASMCs as well. In support of our results, a previous meeting report has shown that whole-cell BK channel currents are decreased in CASMCs from spontaneously diabetic biobreeding (type-1 diabetic) rats (Gebremedhin et al, 1999). It has also been shown that the sensitivity of BK channels to carbon monoxide are attenuated in tail arteries from streptozotocin-induced diabetic rats (Wang et al, 2001). Although vascular diseases in type-2 diabetes are different from those in type-1 diabetes in many aspects from pathologic manifestations to molecular processes, dysfunctions of BK channels have been observed in coronary and mesenteric artery SMCs of Zucker diabetic fatty, type-2 diabetic rats (Erdos et al, 2002, 2004; Burnham et al, 2006). Moreover, BK channels are downregulated in CASMCs from rats with hypertension (Amberg et al, 2003; Amberg and Santana, 2003; Bratz et al, 2005). Thus, it is likely that BK channels are one of the most common cellular molecule targets for vascular diseases.
Big-conductance, Ca2+-activated K+ channels play an imperative role in the regulation of vascular tone by providing a negative feedback mechanism to inhibit membrane depolarization, Ca2+ influx, and cell contraction in vascular SMCs (Lu et al, 2006; Ledoux et al, 2006). As such, the impairment of BK channels would be involved in the vascular dysfunction in type-1 diabetes. In agreement with this view, we have shown that myogenic contraction is increased in streptozotocin-induced diabetic posterior cerebral arteries, and that the specific BK channel blocker iberiotoxin causes a much smaller contraction in posterior cerebral arteries in diabetic than control mice. It is unclear whether our observations in posterior cerebral arteries are reflective of other cerebral arteries owing to the potential regional vascular differences. However, a previous study has shown that contractile responses are similar in basilar and other cerebral arteries (Toyoda et al, 1997). Besides, the enhanced myogenic contraction is observed in posterior cerebral and gracilis skeletal muscle arteries from streptozotocin-induced diabetic rats (Zimmermann et al, 1997; Ungvari et al, 1999). We have also found that blood pressure is significantly elevated in type-1 diabetic mice. These findings, together with previous studies, report that Ca2+ influx through VDCCs is significantly augmented in various vascular SMCs from type-1 diabetic animals and patients (Agrawal and McNeill, 1987; Agrawal et al, 1987; White and Carrier, 1988, 1990; Kamata et al, 1988; Inazu et al, 1991; Ungvari et al, 1999; Fleischhacker et al, 1999; Chow et al, 2001; Burnham et al, 2006), suggest that in type-1 diabetes, BK channels are impaired, resulting in abnormal membrane depolarization, VDCC opening, and Ca2+ influx, contributing to vascular dysfunctions and associated hypertension. Cerebrovascular dysfunctions in streptozotocin-induced diabetes may also be associated with other K+ channels. Indeed, a number of studies have reported that contractile responses to various ATP-sensitive K+ channel openers are diminished in streptozotocin-induced rat cerebral arteries (Mayhan and Faraci, 1993; Mayhan, 1994; Zimmermann et al, 1997), indicating that these channels may be inhibited and consequently contribute to the abnormal Ca2+ influx and contraction in type-1 diabetic CASMCs.
Big-conductance, Ca2+-activated K+ channels in the plasma membrane can be located very close to sarcolemmal ryanodine receptors/Ca2+ release channels (RyRs) (Lu et al, 2006; Ledoux et al, 2006). Through this colocalization design, BK channels are readily activated during Ca2+ release from RyRs. Presumably, the impaired activity of whole-cell BK channels could result from a decrease in RyR-mediated Ca2+ release; however, our data indicate that the frequency and amplitude of RyR-mediated Ca2+ sparks are both significantly increased in type-1 diabetic mouse CASMCs. Similarly, the amplitude of Ca2+ sparks is significantly larger in retinal arterial SMCs from streptozotocin-induced diabetic rats (McGahon et al, 2007). Consistent with the hyperfunctions of RyRs in type-1 diabetic CASMCs, previous studies have shown that the enhanced Ca2+ and contractile responses in tail and mesenteric artery SMCs from streptozotocin-induced diabetic rats are significantly decreased by inhibiting RyRs with ryanodine and procaine (Weber et al, 1996; Tam et al, 1997). Altogether, these data suggest that RyRs are likely to be involved in the enhanced Ca2+ and contractile responses in type-1 diabetic vascular SMCs by increasing Ca2+ release from the sarcoplasmic reticulum, but may not mediate the diminished functions of BK channels. Moreover, it can be reasonably inferred that the local BK channel and Ca2+ release signaling both play an important role in the development of vascular, particularly cerebral vascular artery diseases in type-1 diabetes.
Big-conductance, Ca2+-activated K+ channels are composed of α- and β1-subunit in the vasculature. The β1-subunit confers the Ca2+ and voltage sensitivity (Lu et al, 2006; Ledoux et al, 2006). In this study, we have found that the voltage for the half-maximum current activation is increased in inside-out patches from type-1 diabetic mouse CASMCs, whereas the maximal current amplitude is unaltered. Furthermore, the channel sensitivity to Ca2+ is also markedly decreased in diabetic cells. These electrophysiological results suggest that the β1-subunit is involved in the impaired functions of the BK channel complex in type-1 diabetic vascular SMCs. In harmony with our data, the sensitivity of BK channels to tamoxifen is decreased in diabetic rat retinal arterial SMCs (McGahon et al, 2007). Furthermore, previous studies have reported that sensitivity of BK channels to Ca2+ and voltage are decreased in Zucker diabetic fatty, type-2 diabetic rat mesenteric artery SMCs (Burnham et al, 2006), and hypertensive rat CASMCs (Amberg et al, 2003; Amberg and Santana, 2003; Bratz et al, 2005).
Our biochemical study further supports that the β1-subunit functions are impaired in type-1 diabetic vascular SMCs, in which the β1-subunit protein expression levels are much lower in streptozotocin-induced diabetic than control mouse cerebral arteries, whereas the α-subunit expression levels are similar between diabetic and control tissues. In complement of our results, using reverse transcriptase-polymerase chain reaction, McGahon et al (2007) have found that the β1, but not α-subunit, mRNA expression levels are reduced in streptozotocin-induced diabetic than control rat retinal arteries. Interestingly, similar findings were found in CASMCs from rats with hypertension (Amberg et al, 2003; Amberg and Santana, 2003; Bratz et al, 2005), whereas α and β1 proteins expression are both unchanged in mesenteric arteries from Zucker diabetic fatty, type-2 diabetic rats (Burnham et al, 2006). In addition, we have found that the frequency and amplitude of STOCs (as an indicator of the activity of BK channels) are similar in CASMCs from β1−/− mice with and without type-1 diabetes, indicating that the inhibitory effect of type-1 diabetes on functions of BK channels is vanished in β1−/− CASMCs. Thus, the differences in the changes in BK channel subunit expression in different vascular diseases may reflect the nature of distinct molecular pathogenesis.
Footnotes
Acknowledgements
We thank Mr Jeo Qiao and Ms Jodi Heim for their technical assistance, and Dr Richard W. Aldrich at University of Texas at Austin for kindly providing β1−/− mice.