
Abstract
Although interneurons in area CA1 of the hippocampus are less vulnerable to cerebral ischemia than CA1 pyramidal cells, it is not clear whether their relatively intact cellular morphology implies preservation of normal function. As maintenance of cellular excitability and firing properties is essential for interneurons to regulate neural networks, we investigated these aspects of interneuronal function after transient cerebral ischemia in rats. Cerebral ischemia in rats was induced for 8 mins by a combination of bilateral common carotid artery occlusion and hypovolemic hypotension, and whole cell patch clamp recordings were made in hippocampal slices prepared 24 h after reperfusion. Interneurons located within stratum pyramidale of area CA1 exhibited normal membrane properties and action potentials under these conditions. However, their excitability had declined, as evidenced by an increased action potential threshold and a rightward shift in the relationship between injected depolarizing current and firing rate. Voltage–clamp experiments revealed that transient cerebral ischemia reduced the peak Na+ current and shifted Na+ channel activation to more depolarized values, but did not alter steady-state inactivation of the channel. Double immunofluorescence cytochemistry showed that transient cerebral ischemia also reduced Nav1.1 subunit immunoreactivity in interneurons that coexpressed parvalbumin. We conclude that transient cerebral ischemia renders CA1 interneurons less excitable, that depressed excitability involves impaired Na+ channel activation and that Na+ channel dysfunction is explained, at least in part, by reduced expression of the Nav1.1 subunit. These changes may promote interneuron survival, but might also contribute to pyramidal cell death.
Keywords
Introduction
Inhibitory interneurons regulate the excitability of principal neurons, thus playing a critical role in neural network function. Morphological studies of both global and focal rodent brain ischemia models revealed that ischemia damages interneurons and principal neurons to different degrees; in several brain regions examined, interneurons were less vulnerable (Johansen et al, 1983, 1989; Nitsch et al, 1989; Johansen and Diemer, 1990; Ferrer et al, 1995; Katchanov et al, 2003; Frahm et al, 2004; Zhan et al, 2006). Multiple mechanisms, including lower expression of NMDA receptors (Nyíri et al, 2003; Avignone et al, 2005), greater calcium buffering capacity (Baimbridge et al, 1992), and better preservation of GABA transmission during the first few hours after reperfusion (Zhan et al, 2006), have been suggested to explain the lesser vulnerability of interneurons. However, it remains to be determined whether the relatively intact morphology of interneurons implies preservation of normal function. In fact, some evidence suggests altered interneuronal function after transient cerebral ischemia. For example, Inglefield et al (1997) found that dendrites of CA1 hippocampal interneurons lose immunoreactivity for the α1 subunit of the GABAA receptor after transient cerebral ischemia. Furthermore, interneurons become dysfunctional even earlier than principal neurons after a mild transient reduction in superfusion rate of rat neocortical slices combined with reduced glucose concentration (Wang, 2003).
Inhibitory interneurons differ from excitatory principal neurons in many respects, including morphology, neurochemical properties, firing pattern, distribution of excitatory amino-acid receptors, and calcium homeostasis (Miles, 2002). Interneurons typically fire action potentials at a higher frequency, as is appropriate for rapid action (Martina and Jonas, 1997; Martina et al, 1998). As the rate and pattern of cell firing are essential elements of normal interneuronal function, we investigated the possibility that transient cerebral ischemia alters these properties.
Materials and methods
All experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved in advance by the Duke University Institutional Animal Care and Use Committee.
Cerebral Ischemia
Adult male Sprague–Dawley rats (Charles River, Raleigh, NC, USA) that weighed 170 to 230 g (45 to 49 days old) were fasted for 7 to 10 h before surgery, but were allowed free access to water. Transient cerebral ischemia was induced for 8 mins by two-vessel occlusion with hypovolemic hypotension as described previously (Zhan et al, 2006). Rats were killed 24 h later for electrophysiological and immunofluorescence studies.
Electrophysiology in ex vivo Hippocampal Slices
Animals were re-anesthetized 24 h after reperfusion by inhalation of 2.5% halothane in O2 through a face mask. The animal was decapitated into oxygenated (95% O2/5% CO2) ice-cold artificial cerebrospinal fluid and the forebrain was removed. Transverse hippocampal slices (400 μm-thick) of the rostral hippocampus were prepared with a vibratome and transferred to artificial cerebrospinal fluid that contained 125 mmol/L NaCl, 2.5 mmol/L KCl, 2 mmol/L CaCl2, 1 mmol/L MgCl2, 1.25 mmol/L NaH2PO4, 26 mmol/L NaHCO3, 20 mmol/L d-glucose and 1 mmol/L ascorbic acid, pH 7.4 and saturated with 95% O2/5% CO2. Incubation continued at 34°C for 30 mins and then at room temperature thereafter.
A slice was transferred to a plexiglas recording chamber, held in place with curved platinum wires and superfused continuously (∼ 3 ml/mins) with oxygenated artificial cerebrospinal fluid at room temperature. Interneurons located within stratum pyramidale of area CA1 were visualized under a Nikon Eclipse E600-FN microscope equipped with infrared-differential interference contrast (IR-DIC) optics, a × 40 water immersion objective, and epifluorescence illumination (Nikon Inc., Melville, NY, USA). They were distinguished from the surrounding pyramidal cells based on the size and shape of the soma and the number of dendrites. Interneuron identity was confirmed by visualization of Alexa Fluor 488 that had diffused into the cell from the patch pipette (see below).
Signals were recorded with an Axopatch 200B amplifier (Axon Instruments, Union City, CA, USA). Patch electrodes were pulled from borosilicate glass (OD: 1.5 mm; ID: 1.1 mm) with a Flaming/Brown electrode puller (Sutter Instruments, Novato, CA, USA) to yield a tip resistance of 3 to 4 MΩ. Signals were low-pass filtered at 2 kHz and digitized at 20 kHz. Data were acquired and stored for analysis offline with PClamp 8.1 (Axon Instruments, Union City, CA, USA).
Resting membrane potential was measured in current clamp mode immediately after membrane rupture. Action potential threshold was taken as the membrane potential at which an action potential was first evoked as the cell was depolarized from resting Vm. Action potential amplitude was measured from onset to peak and action potential duration was measured at half the maximal amplitude. Amplitude of the postspike afterhyperpolarization (AHP) was measured from onset to peak. Input resistance (RN) was calculated from the steady state current evoked by a 100 ms, 10 mV hyperpolarization. Liquid junction potentials were estimated to be 4 mV for all internal solutions and were not corrected. Only cells with a resting membrane potential > −50 mV were studied.
The excitability of recorded cells was assessed in current clamp recordings from their response to stepped current injections. For these experiments, the internal solution consisted of 136 mmol/L KCl, 1 mmol/L MgCl2, 0.1 mmol/L CaCl2, 0.5 mmol/L EGTA, 10 mmol/L HEPES, 2 mmol/L ATP tris salt and 0.4 mmol/L GTP tris salt, pH 7.30 (adjusted with KOH) and 292 to 295 mosm. Current steps from −0.5 to 1.5 nA in 0.1 nA increments were applied for 1 sec each.
Gramicidin-based perforated patch recordings were used to determine whether GABA transmission could affect responses triggered by current injection. This recording method does not alter the distribution of chloride ions across the plasma membrane (Akaike, 1996). The protocol was identical to that described previously (Zhan et al, 2006).
The activation and inactivation of voltage-gated Na+ channels were studied in voltage clamp mode. The internal solution contained 136 mmol/L CsCl, 1 mmol/L MgCl2, 0.1 mmol/L CaCl2, 1 mmol/L EGTA, 10 mmol/L HEPES, 2 mmol/L ATP tris salt, and 0.4 mmol/L GTP tris salt, pH 7.30 (adjusted with CsOH) and 292 to 295 mosm. To block voltage-gated K+ channels, the superfusion medium contained 20 mmol/L tetraethylammonium chloride and 2 mmol/L 4-aminopyridine; the NaCl concentration was reduced to 116 mmol/L to maintain constant osmolality. To block voltage-gated Ca2+ channels, the superfusion medium also contained 0.1 mmol/L CdCl2. To activate Na+ channels, the cell was held at a holding potential of −90 mV for 15 ms, hyperpolarized to −130 mV for 500 ms and then tested initially by depolarization to −90 mV for 15 ms. This protocol was repeated 16 times, incrementing the test potential by 10 mV each time until it reached + 60 mV. This procedure was then repeated in the presence of 1 μmol/L tetrodotoxin (TTX). Corresponding currents recorded in the absence and presence of TTX were subtracted to yield TTX-sensitive currents. To study Na+ channel inactivation, the cell was held at a holding potential of −90 mV for 100 ms, then stepped initially to a prepotential of −110 mV for 100 ms followed by a uniform test step to +30mV for 15 ms. This protocol was repeated 11 times, incrementing the test prepotential by 10 mV each time until it reached −10 mV.
Visualization of Recorded Cells
Alexa Fluor 488 hydrazide (Molecular Probes, Eugene, OR, USA) was added to all internal solutions at a concentration of 0.003% (w/v). At the end of the experiment, the recorded cell was visualized at an emission wavelength of 535 nm and fluorescent images were captured with a CCD camera (Quantix Photometrics, Tucson, AZ, USA).
Immunocytochemical Detection of Na+ Channel Subunits
The expression of voltage-gated Na+ channel subunits Nav1.1. (brain subunit α1), Nav1.2 (brain subunit α2), Nav1.3 (brain subunit α3), and β2 were examined 24 h after reperfusion. Animals were subjected to either a sham operation (n = 4) or 8 min of cerebral ischemia (n = 4). The procedures used for transcardial perfusion and brain tissue preparation were described previously (Zhan et al, 2001). Coronal brain sections of 14-μm thickness that included hippocampus and neocortex were cut with a cryostat, mounted individually on slide glasses, and air-dried before being processed. After exposure to 100% acetone for 90 secs at 4°C, sections were washed with 0.1mol/L phosphate-buffered saline (PBS, pH 7.4) three times for 5 mins each and then incubated with 5% (w/v) IgG-free bovine serum albumin (Sigma, St Louis, MO, USA) in PBS for 90 mins. Then they were incubated overnight at 4°C with a combination of a mouse monoclonal antibody directed against parvalbumin (1:400 dilution; Sigma Chemical Co., St Louis, MO, USA) and a rabbit polyclonal antibody directed against one of the Na+ channel subunits. The dilutions of Na+ channel subunit antisera were 1:400 for anti-Nav1.1, 1:200 for anti-Nav1.2, 1:50 for anti-Nav1.3 (all from Sigma Chemical Co., St Louis, MO, USA) and 1:200 for anti-β2 (Alomone Laboratories, Jerusalem, Israel). After sections were washed with PBS three times (10 mins each), they were incubated with a mixture of Alexa Fluor 488-labeled goat anti-mouse IgG (1:400) and Alexa Fluor 594-labeled goat anti-rabbit IgG (1:400) (Invitrogen, Carlsbad, CA, USA) for 150 mins at room temperature. After washing again with PBS, the sections were covered with 75% (v/v) glycerol in PBS and coverslips were applied.
Images were captured into Metamorph (Universal Imaging Corporation Inc., Downingtown, PA, USA) with a CCD camera and digitizer board. For each animal and each Na+ channel subunit, cell bodies of 8 to 10 parvalbumin-immunoreactive neurons (interneurons) within striatum pyramidale of area CA1 were outlined and the average immunostaining intensity within that area was determined by the software and expressed as a gray level on a scale of 0 to 255. For comparison, pyramidal cell immunolabeling was quantitated similarly from four parvalbumin-immunonegative neurons adjacent to each of the parvalbumin-immunoreactive neurons. Values obtained from all interneurons and pyramidal cells in the same animal were averaged to obtain single values for each animal. The fluorescence intensity of the overlying CA1 stratum radiatum was taken as background and subtracted from the gray level values computed for interneurons and pyramidal cells in the same section.
Statistical Analyses
Quantitative data are expressed as means ±s.e.m. Between-group differences regarding basic membrane properties and action potentials were assessed with an unpaired Student's t-test. The relationship between current injection and firing frequency was analyzed by linear regression. The activation and inactivation of voltage-gated Na+ channels were analyzed according to methods described by Ketelaars et al (2001). Fluorescence data were analyzed with two-way ANOVA, followed by Bonferroni post hoc tests for individual comparisons. P ≤ 0.05 were considered significant.
Results
Reduced Excitability of CA1 Interneurons after Transient Cerebral Ischemia
Whole cell patch clamp recordings were made from interneurons located within stratum pyramidale of area CA1 24 h after reperfusion. Recorded cells were identified as interneurons by their non-pyramidal-shaped soma from which multiple dendrites emerged. At 24 h after transient cerebral ischemia, the basic membrane properties of these cells, including resting Vm, RN, membrane time constant and whole cell capacitance had not changed significantly (Table 1). In contrast, transient ischemia increased the action potential threshold, and fewer action potentials fired in response to injected currents (Figure 1, Table 1).
Membrane properties of morphologically identified CA1 interneurons 24 h after sham operation or transient cerebral ischemia
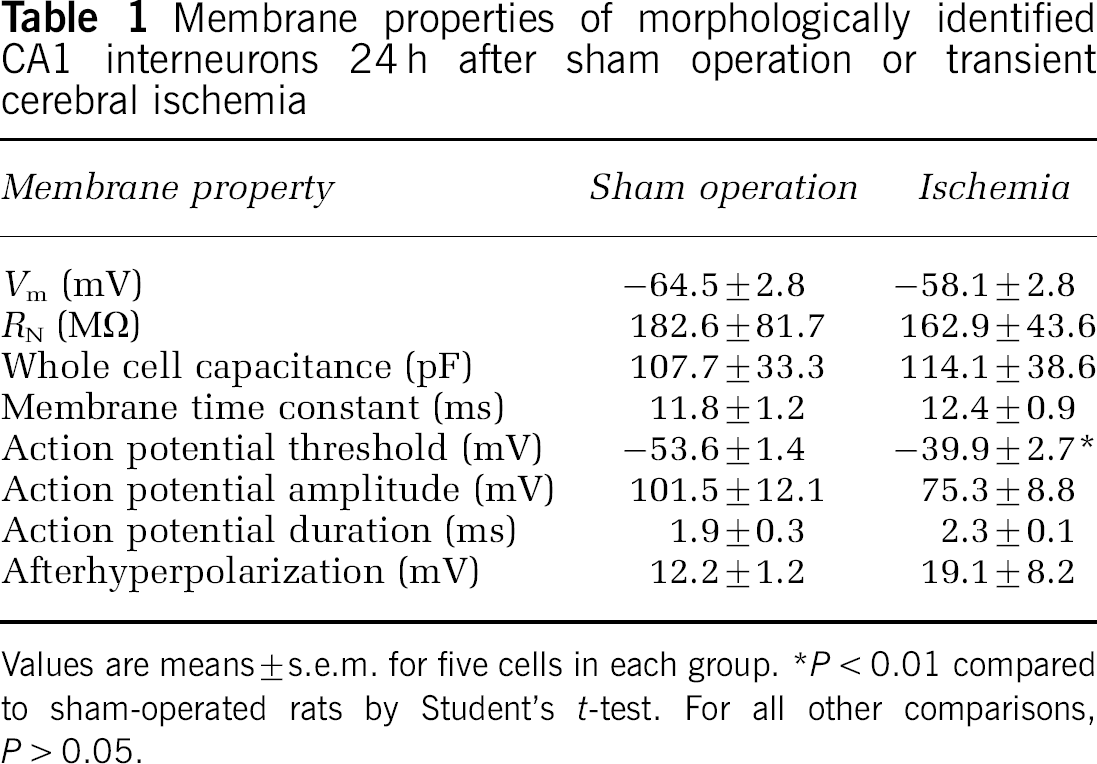
Values are means ± s.e.m. for five cells in each group. *P < 0.01 compared to sham-operated rats by Student's t-test. For all other comparisons, P > 0.05.
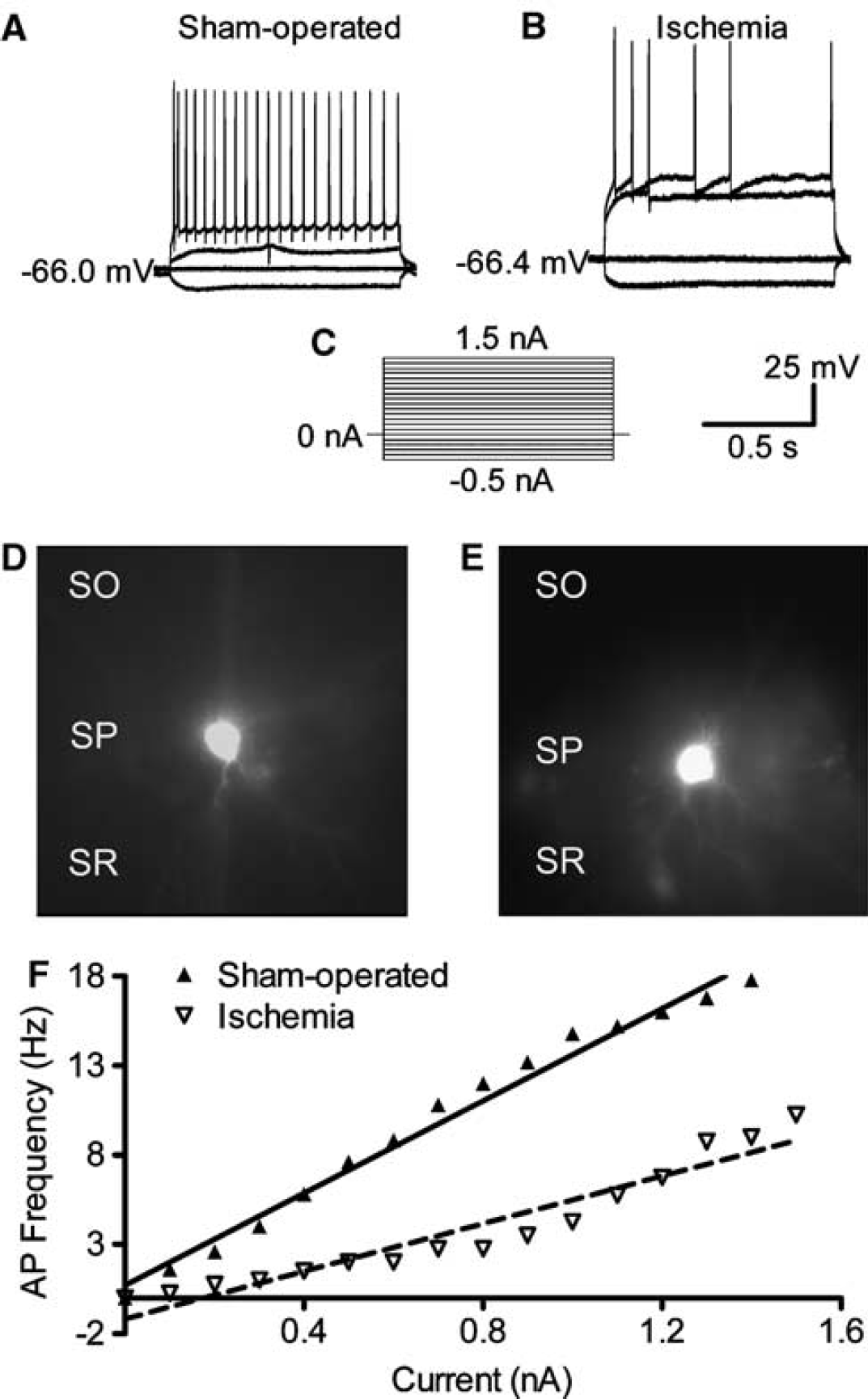
Transient cerebral ischemia reduces the excitability of area CA1 interneurons. (
Failure of GABAA Receptor Blockade to Restore Excitability of CA1 Interneurons
If the reduced interneuronal excitability was related to strengthened GABA inhibition, block of GABAA receptors should have restored it to normal. To test this possibility, we recorded firing rates in interneurons by using the gramicidin-based perforated patch technique. Although the application of 30 μmol/L bicuculline for 5 mins depolarized the cells by 5 to 10 mV (owing to diminished tonic inhibition), it did not change the relationship between firing rate and injected current (not shown). Thus, the reduced excitability of CA1 interneurons could not be explained by enhanced GABA inhibition.
Impaired Activation and Reduced Peak Amplitude of Voltage-Gated Na+ Channels after Transient Cerebral Ischemia
The degree of voltage control required for accurate analysis of Na+ currents is best achieved in isolated cells. As it is difficult to isolate interneurons from area CA1 and, more importantly, because the isolation procedure changes cell structure and possibly also function, we recorded voltage-gated Na+ currents in intact hippocampal slices. Space clamp was likely to have been imperfect, but the data remain useful for between-group comparisons. In addition, interneurons accepted for inclusion in this study had to meet the following criteria. (1) Series resistance was ≤ 11 MΩ and could be compensated > 90% without destabilizing the recording. (2) Pipette capacitance was compensated completely and whole cell capacitance was compensated almost completely. (3) Recorded currents were confirmed to be voltage-dependent. (4) Currents exhibited a clear activation threshold and increased in parallel with the size of the depolarization. (5) Only TTX-sensitive currents were compared.
To assess Na+ channel activation, Na+ currents were evoked by stepping the resting membrane potential from −130 mV, a potential at which channel inactivation is removed, to test potentials from −90 mV to + 60 mV (Figure 2). In interneurons from sham-operated rats, Na+ channels were maximally activated around −50 mV (Figures 2A, 2D), corresponding to the action potential threshold (Table 1). The voltage-dependence of Na+ channel activation shifted significantly toward more depolarized potentials after transient ischemia (Figures 2B, 2D), in concert with the increased action potential threshold. Peak Na+ currents were also significantly smaller in interneurons from rats subjected to transient cerebral ischemia (Figure 2E).
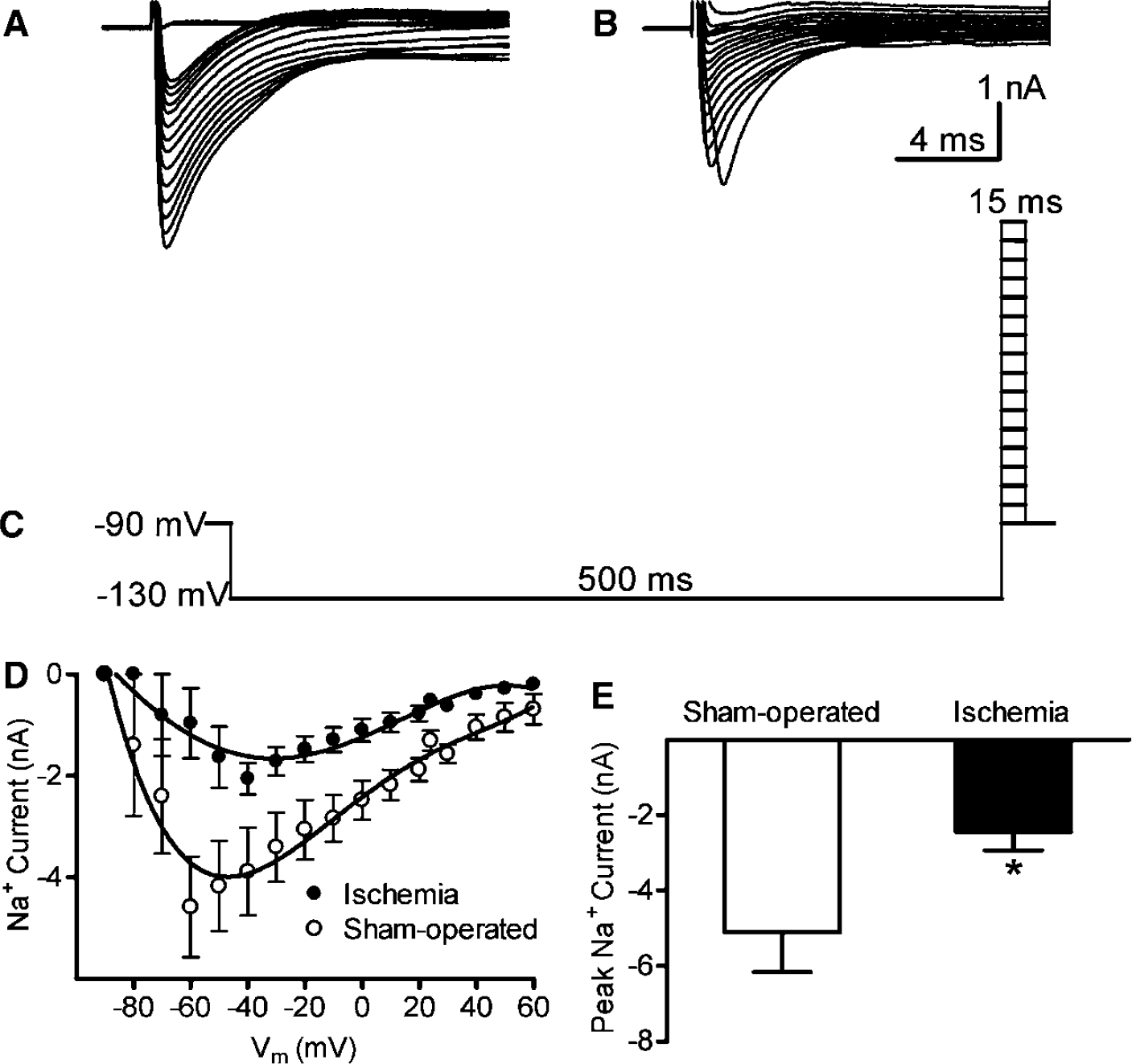
Transient cerebral ischemia altered the voltage-dependence of Na+ channel activation and reduced the magnitude of the current. TTX-sensitive Na+ currents were recorded 24 h after a sham operation (
Normal Na+ Channel Inactivation after Transient Cerebral Ischemia
The voltage dependence of steady-state inactivation is a property of Na+ channels that determines channel availability at specific membrane potentials. The protocol used to assess steady-state inactivation consisted of prepulses to varying membrane potentials from −110 to −10 mV followed by a constant test pulse to +30 mV. Transient cerebral ischemia had no observable effect on the voltage-dependence of channel inactivation (Figure 3).
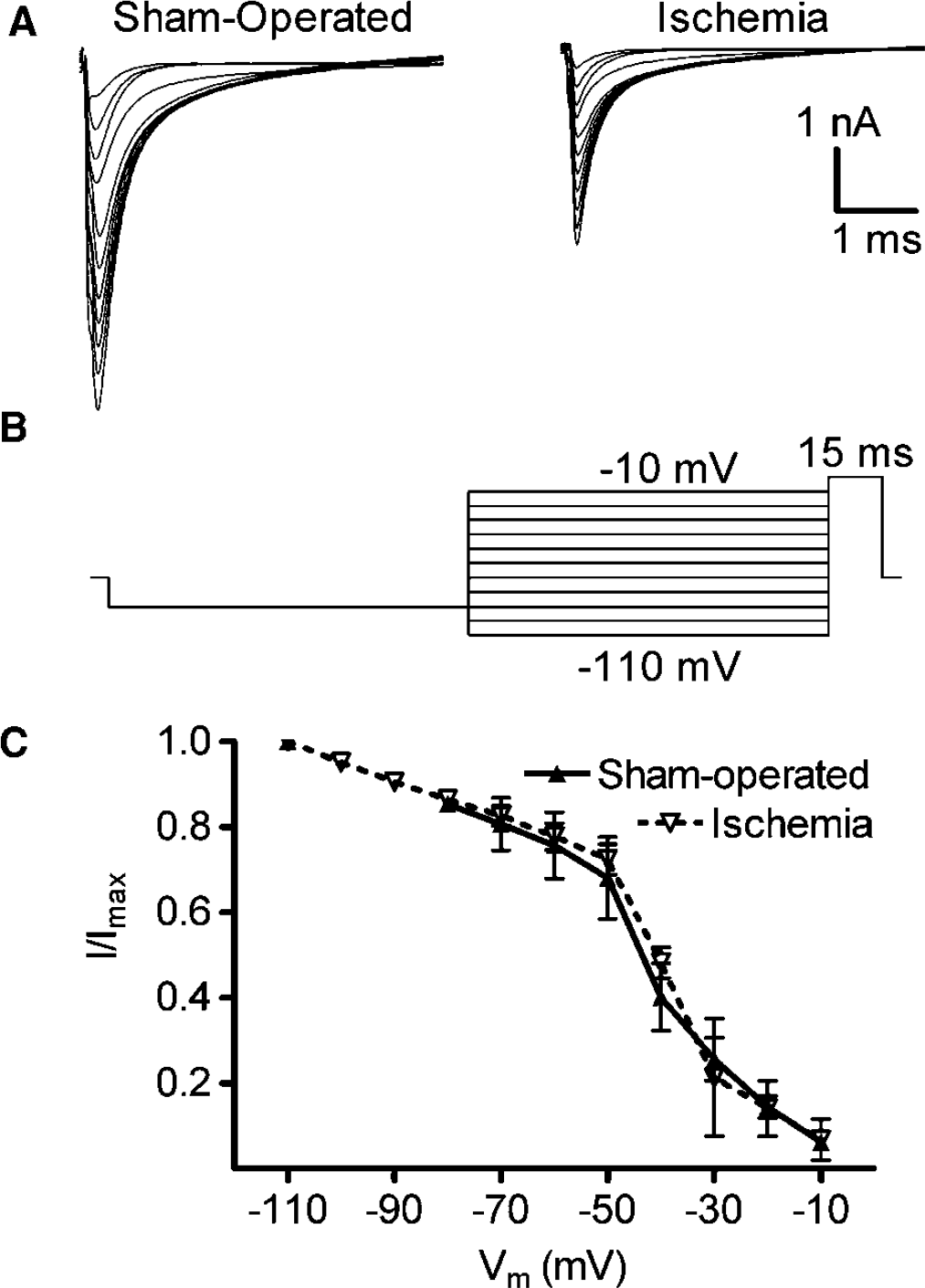
Transient cerebral ischemia did not affect the steady-state inactivation of voltage-gated Na+ channels. (
Reduced Nav1.1. Immunoreactivity in Parvulbumin-Immunoreactive CA1 Interneurons after Transient Cerebral Ischemia
To determine if the dysfunctional Na+ channel could be owing to an ischemia-induced alteration of Na+ channel structure, we assessed the expression of four Na+ channel subunits that comprise functional channels in the hippocampus. The Na+ channel subunits Nav1.1, Nav1.2, Nav1.3 and β2 were visualized in area CA1 by immunofluorescence. The four subunits were localized differentially within this region. Nav1.1 was expressed in both somata and proximal apical dendrites in association with neuronal plasma membranes (Figure 4). The most intense immunostaining was observed in neurons that also expressed parvalbumin and were therefore GABA interneurons. When visualized 24 h after transient cerebral ischemia, the intensity of Nav1.1 immunostaining had decreased in both parvalbumin-immmunoreactive and parvalbumin-immunonegative neurons. Neurons in stratum pyramidale were intensely Nav1.3-immunoreactive, without any noticeable difference between parvalbumin-immunoreactive and parvalbumin-immunonegative neurons (Figure 5). In contrast to Nav1.1, transient cerebral ischemia did not appear to change the intensity or distribution of Nav1.3 immunostaining (Figure 5). Stratum pyramidale exhibited only faint β2 immunoreactivity, whereas the dendritic layers of area CA1 were more intensely immunoreactive (Figure 6 left). White matter was also strongly immunoreactive. Transient cerebral ischemia had no obvious effect on β2 immunostaining (Figure 6 right). Expression of Nav1.2 in area CA1 closely resembled expression of β2 in both sham-operated rats and rats that had been subjected to transient cerebral ischemia (not shown). Quantitative comparisons of Nav1.1, Nav1.3 and β2 immunofluorescence in parvalbumin-immunoreactive and parvalbumin-immunonegative neurons within area CA1 are shown in Figure 7. Ischemia reduced Nav1.1 immunostaining of both interneuron and pyramidal cell somata by approximately 70%.
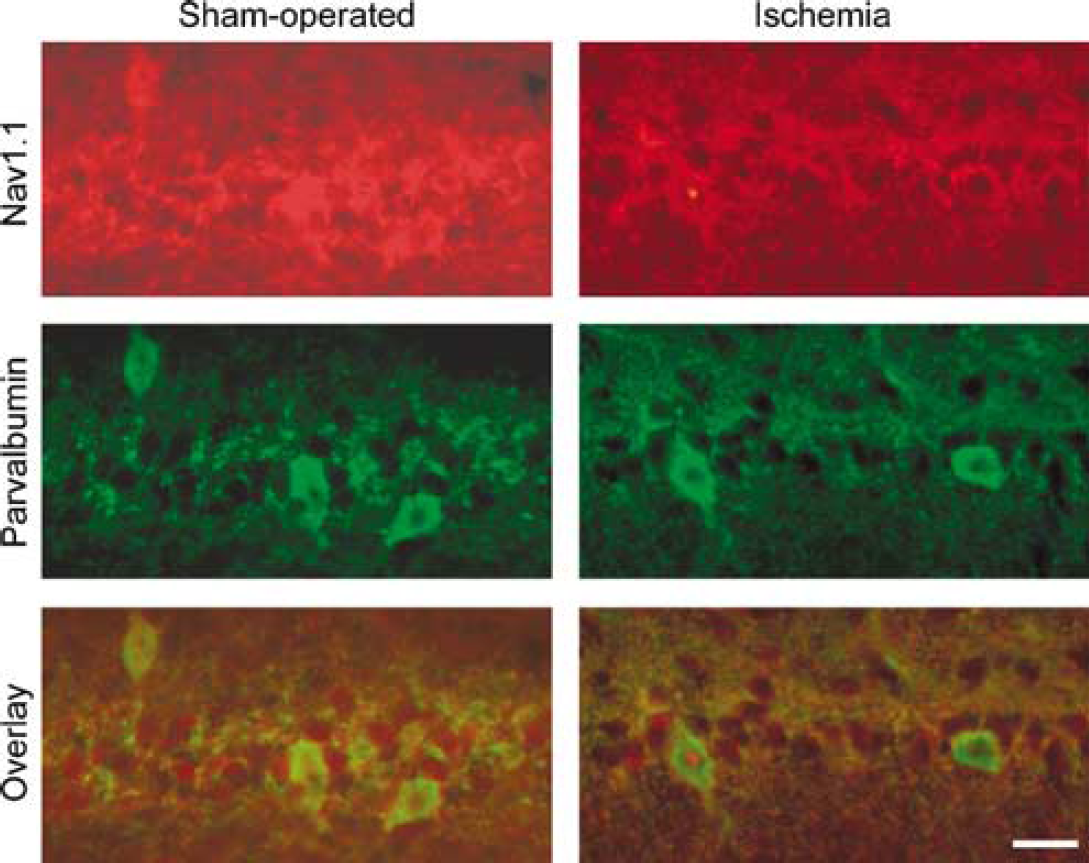
Transient cerebral ischemia reduced Nav1.1 Na+ channel subunit immunoreactivity in both parvalbumin-immunoreactive (interneurons) and parvalbumin-immunonegative (pyramidal cells) CA1 neurons 24 h after reperfusion. Scale bar, 20 μm.
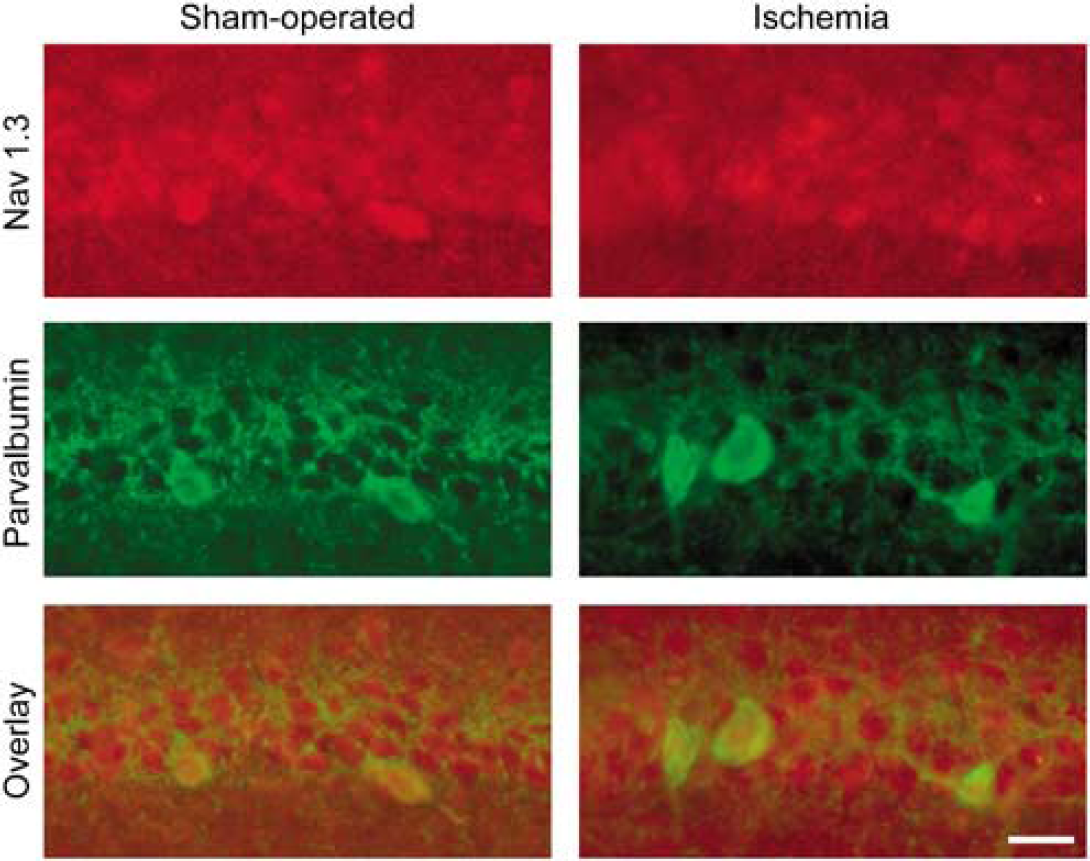
Transient cerebral ischemia did not change the pattern or intensity of Nav1.3 Na+ channel subunit immunostaining in CA1 neurons 24 h after reperfusion. Scale bar, 20 μm.
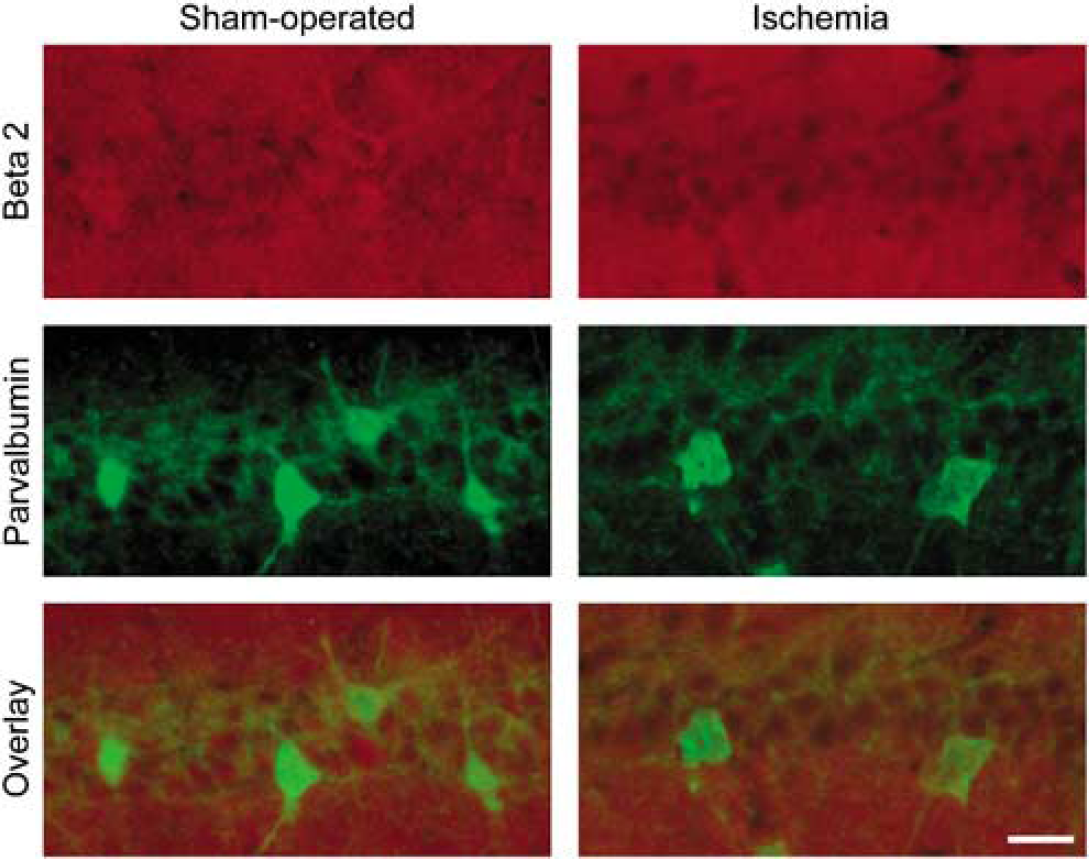
Transient cerebral ischemia did not change the pattern or intensity of β2 Na+ channel subunit immunostaining in CA1 neurons 24 h after reperfusion. Scale bar, 20 μm.
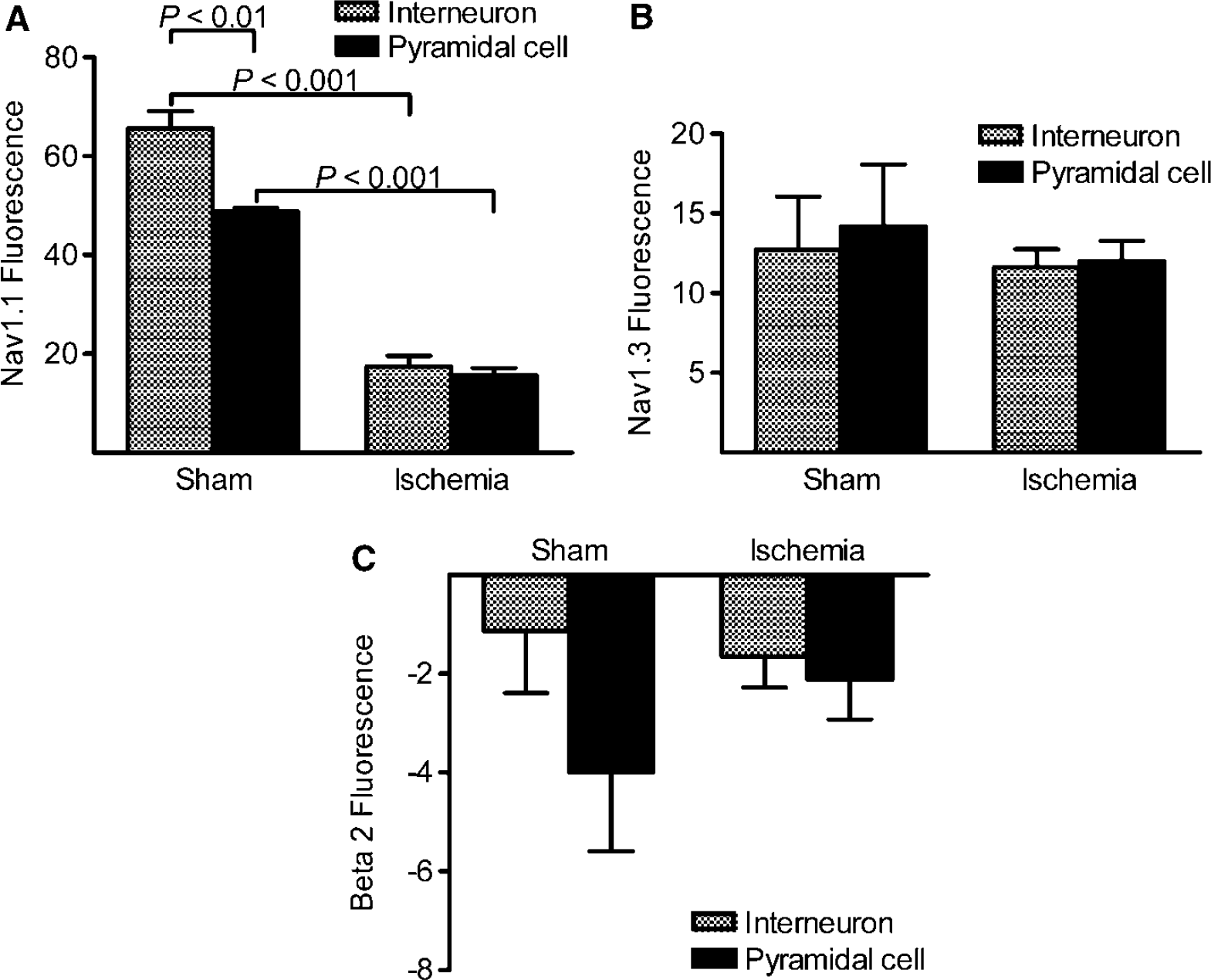
Transient cerebral ischemia reduced the Nav 1.1 Na+ channel subunit immunoreactivity of both CA1 interneurons and pyramidal cells. (
Discussion
Previous studies of hippocampal area CA1 after transient cerebral ischemia focused on the delayed death of pyramidal cells and the mechanisms that might explain this type of pathology. Inhibitory CA1 interneurons have drawn less attention, because they appear to survive the insult. As expected, CA1 interneurons appeared relatively normal, both structurally and functionally, 24 h after transient cerebral ischemia. In particular, passive membrane properties, which are sensitive to changes in the energetic state of the neuron, were similar to those of sham-operated rats. The low variability in passive membrane properties, again comparable to that observed in sham-operated controls, suggests further that the interneurons had all been exposed to a similar degree of energetic stress. In contrast, CA1 pyramidal cells exhibit evidence of dysfunction hours earlier, including reduced GABA-mediated responses (Zhan et al, 2006), release of cytochrome C into the cytoplasm (Sugawara et al, 1999; Zhao et al, 2005), induction of p53 mRNA (Tomasevic et al, 1999), and expression of caspase-3 and caspase inhibitor proteins (Siegelin et al, 2005; Zhao et al, 2005). However, overt morphological damage does not appear for another 2 to 3 days.
Although CA1 interneurons appeared undamaged, ischemia might have induced changes in the physiology of these neurons that altered circuit function and impacted the survival of pyramidal cells, the interneurons themselves, or both. Our results indeed revealed depression of interneuronal excitability. Transient cerebral ischemia increased action potential threshold, impaired Na+ channel activation, and reduced the peak amplitude of Na+ currents in CA1 interneurons. These changes may be explained, at least in part, by the reduced expression of Nav1.1 Na+ channel subunits.
Activation of voltage-gated Na+ channels is essential for the initiation and propagation of action potentials. Reduced Na+ channel activation in the absence of other changes increases action potential threshold and reduces the firing rate (Matzner and Devor, 1992). Voltage-gated Na+ channels consist of one pore-forming α subunit with one or more auxiliary β subunits. At least four α subunits (Nav1.1, Nav1.2, Nav1.3, Nav1.6) and two β subunits are expressed in the central nervous system (Goldin et al, 2000). Pathological conditions, such as hypoxia and carbon dioxide exposure, alter Na+ channel function (Banasiak et al, 2004; Gu et al, 2004). Mutations in Nav1.1 can alter both channel activation and inactivation, depending on the type of mutation (Rhodes et al, 2005). In the present study, transient cerebral ischemia reduced Nav1.1 immunoreactivity in CA1 interneurons that co-expressed parvalbumin. A similar result was reported after focal cerebral ischemia in the rat (Yao et al, 2005). Some evidence suggests that Nav1.1 determines neuronal excitability. For example, increased Nav1.1 expression was associated with hyperexcitability of neocortical neurons in a model of epilepsy (Klein et al, 2004). In contrast, a loss-of-function mutation in Nav1.1 reduced Na+ current density, shifted the voltage dependence of channel activation toward more positive membrane potentials, and impaired action potential firing (Mantegazza et al, 2005). These effects closely resemble the changes observed in the present study. Recently, functional deletion of the gene that encodes Nav1.1 was shown to reduce Na+ current density in hippocampal interneurons, but not in pyramidal cells (Yu et al, 2006). We therefore suggest that transient cerebral ischemia reduces the excitability of CA1 interneurons, at least in part, through reduced expression of Nav1.1.
Although inhibitory interneurons constitute only approximately 10% of all hippocampal neurons (Woodson et al, 1989), each interneuron projects to hundreds of pyramidal cells and regulates their excitability (Buhl et al, 1994; Halasy et al, 1996). Transient cerebral ischemia produces a long-lasting membrane depolarization that favors activation of glutamate receptors. Overactivation of N-methyl-d-aspartate (NMDA) receptors and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors that lack a GluR2 subunit contributes to ischemic neuronal death (reviews by Rothman and Olney (1986); Weiss and Sensi (2000)). Considering that GABA still hyperpolarizes CA1 pyramidal cells after transient ischemia (Zhan et al, 2006), maintenance of normal GABA inhibition after ischemia could effectively counter the membrane depolarization and thus the overactivation of glutamate receptors. Our previous studies demonstrated that GABA inhibition, in fact, is not maintained during the postischemic period (Schwartz-Bloom and Sah, 2001; Zhan et al, 2006). GABA transmission onto CA1 pyramidal cells becomes impaired owing to reduced GABAA receptor sensitivity or clustering and possibly also to reduced GABA release. No such changes occur in CA1 interneurons. The present study demonstrates a further impairment of CA1 inhibition resulting from the loss of interneuronal excitability. Reduced GABA inhibition probably contributed to pyramidal cell death by enhancing the excitability of these cells and facilitating NMDA channel opening. Conversely, the same loss of interneuronal excitability, coupled with a markedly reduced excitatory drive from pyramidal cells, might enhance interneuron survival.
Ischemia-induced neuronal degeneration involves a complex cascade of cellular and molecular events. As impaired GABA inhibition promotes this cascade, drugs that improve GABA function are neuroprotective in animal models (Schwartz-Bloom and Sah, 2001). All these drugs enhance the action of GABA in a global fashion, but they would not be expected to normalize interneuronal firing. Thus, circuit function might remain abnormal in the presence of the drug. This consideration might explain, in part, the inability of GABA functional enhancement alone to improve neurological function significantly after a stroke (but see Lodder et al, 2005).