
Abstract
We have studied the possible mechanisms underlying the decrease of excitatory transmission induced by glucose deprivation by using electrophysiological recordings in corticostriatal slices. Extracellular field potentials were recorded in the striatum after cortical stimulation; these potentials were progressively reduced by glucose deprivation. The reduction started 5 minutes after the onset of aglycemia. The field potential was fully suppressed after 40 minutes of glucose deprivation. After the washout of the aglycemic solution only a partial recovery was observed. Aglycemia also induced a delayed inward current during single-microelectrode voltage-clamp recordings from spiny neurons. This inward current was coupled with an increased membrane conductance. The A1 adenosine receptor antagonists, 8-cyclopentyl-1,3-dimethylxanthine (CPT, 1 μmol/L) and 1,3-dipropyl-8-cyclopentylxanthine (CPX, 300 nmol/L), significantly reduced the aglycemia-induced decrease of field potential amplitude. Moreover, in the presence of CPT and CPX, a full recovery of the field potential amplitude after the interruption of the aglycemic solution was observed. Conversely, these antagonists affected neither the inward current nor the underlying conductance increase produced by glucose deprivation. The ATP-sensitive potassium channel blockers glibenclamide (10 μmol/L) and glipizide (100 nmol/L) had no effect on the aglycemia-induced decrease of the field potential amplitude. We suggest that endogenous adenosine, but not ATP-dependent potassium channels, plays a significant role in the aglycemia-induced depression of excitatory transmission at corticostriatal synapses probably through a presynaptic mechanism. Moreover, adenosine is not involved in the postsynaptic changes induced by glucose deprivation in spiny striatal neurons.
Electrical activity within the brain is rapidly lost under conditions of glucose deprivation (Auer et al., 1984; Auer and Siesjo, 1988). There is also evidence that energy deprivation, as a result of aglycemia, leads to depression of central synaptic transmission (Bachelard et al., 1984; Burke and Nadler, 1989; Crepel et al., 1992; Shoji, 1992). However, the mechanisms underlying the aglycemia-induced depression of excitatory synaptic transmission are still a matter of debate. In particular, three major pathogenetic mechanisms might be involved in this depressant effect induced by glucose deprivation. First, it is possible that endogenous adenosine, which is released during aglycemia (Butcher et al., 1987), causes a presynaptic inhibitory action on nerve terminals releasing excitatory amino acids (Zhu and Krnjevic, 1993; Khazipov et al., 1995). Accordingly, the activation of adenosine A1 receptors has been reported to cause presynaptic inhibition of glutamatergic excitation in different brain areas (Greene and Haas, 1991; Thompson et al., 1992; Ulrich and Huguenard, 1995) including ventral (Uchimura and North, 1991) and dorsal striatum (Malenka and Kocsis, 1990). Alternatively, it has been postulated that the aglycemia-induced depression of synaptic transmission is caused by activation of presynaptic ATP-sensitive potassium channels (Ashcroft, 1988; Freedmann and Lin, 1996). A third major cause for this depression might be the postsynaptic effects produced by energy deprivation. In fact, aglycemia causes various membrane potential changes (hyperpolarization and/or depolarization) in different neuronal subtypes (Spuler et al., 1988; Shoji, 1992; Knoepfel et al., 1990; Martin et al., 1994). Because these changes are usually coupled with an increased postsynaptic membrane conductance, it is possible that this increase reduces the amplitude of the excitatory postsynaptic potentials. We have studied the possible involvement of these mechanisms in the aglycemia-induced depression of the excitatory transmission in the striatum by using electrophysiological recordings from a brain slice preparation. The striatum, in fact, is considered a brain area that is highly vulnerable to hypoglycemia (Kalimo et al., 1985) and the corticostriatal projection is considered one of the most important glutamatergic pathways in the brain (Reubi and Cuenod, 1979). In fact, the stimulation of corticostriatal fibers produces excitatory synaptic potentials that are mediated by the release of endogenous excitatory amino acids acting on glutamate receptors localized on spiny striatal neurons (Cherubini et al., 1989; Calabresi et al., 1996). Because the striatum is involved in both motor and nonmotor functions (Calabresi et al., 1997), it is possible that the aglycemia-induced depression of glutamatergic transmission is responsible for part of the behavioral changes induced by this condition.
METHODS
Wistar rats (150 to 250 g) were used. The preparation and maintenance of slices have been described previously (Calabresi et al., 1995a, b ). Briefly, corticostriatal coronal slices (200 to 300 m) were prepared from tissue blocks of the brain with the use of a vibratome. A single slice was transferred to a recording chamber and submerged in a continuously flowing Krebs solution (35°C, 2 to 3 mL/min) gassed with 95% O2 and 5% CO2. To study glucose metabolism in striatal neurons, slices were deprived of glucose by removing glucose totally from the perfusate and by adding saccharose to balance the osmolarity. In some experiments, the osmolarity was balanced by increasing the NaCl concentration (Jiang and Haddad, 1992). Because experiments performed by using these different procedures to replace glucose gave similar results, all the data were pooled together. Aglycemic solutions entered the recording chamber no later than 20 seconds after turning a 3-ways tap. Complete replacement of the medium in the chamber took 90 seconds. The composition of the control solution was (in m/mol/L): 126 NaCl, 2.5 KC1, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 Glucose, and 25 NaHCO3.
For the extracellular experiments, the electrodes were filled with 2 mol/L NaCl (5 to 10 MΩ). The intracellular recording electrodes were filled with 2M KC1 (30-60 MΩ) and located in the striatum close (1 to 3 mm) to the cortical areas. An Axoclamp 2A amplifier was used for recordings either in current-clamp or in voltage-clamp mode. In single-microelectrode voltage-clamp mode the switching frequency was 3 kHz. The head-stage signal was continuously monitored on a separate oscilloscope. Traces were displayed on an oscilloscope and stored on a digital system. For synaptic stimulation, bipolar electrodes were used (0.03 to 0.01 millisecond duration; 1 to 5 volts). These stimulating electrodes were located either in the cortical areas close to the recording electrode or in the white matter between the cortex and the striatum to activate corticostriatal fibers. The field potential amplitude was defined as the average of the amplitude from the peak of the early positivity to the peak negativity, and the amplitude from the peak negativity to peak late positivity (see Melchers et al., 1988). Quantitative data on modifications induced by aglycemia are expressed as a percentage of the controls, the latter representing the mean of responses recorded during a stable period (15 to 20 minutes) before the aglycemic phase. Values given in the text and in the figures are mean SD of changes in the respective cell populations. Student's t-test (for paired and unpaired observations) was used to compare the means. Drugs were applied by dissolving them to the desired final concentration in the saline and by switching the perfusion from control saline to drug-containing saline. 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) was from Tocris (Bristol, U.K.). D-2-amino-5-phosphonovalerate (D-APV) and tetrodotoxin were from Sigma (St. Louis, MO, U.S.A.). 8-cyclopentyl-1,3-dimethylxanhine (CPT), 1,3-dipropyl-8-cyclopentylxanthine (CPX), glipizide, and glibenclamide were from RBI (Amersham, Milano, Italia).
RESULTS
When extracellular recordings were performed in the striatum close to the stimulating electrode, a single activation of corticostriatal fibers produced a field potential. The amplitude of the field potential was related to the intensity of the synaptic stimulation. The corticostriatal field potential was blocked by tetrodotoxin (1 μmol/L, n = 4), low calcium (0.5 mmol/L)-high magnesium (10 mmol/L)–containing solutions (n = 4), and coadministration of 10 μmol/L CNQX plus 30 μmol/L APV (n = 5) (data not shown). These physiological and pharmacological characteristics are in agreement with previous findings indicating that cortically-evoked field potentials are mediated by the release of excitatory amino acids within the striatum (Calabresi et al., 1996). Glucose deprivation reduced the amplitude of these field potentials. As shown in Fig. 1, this inhibitory effect started 5 minutes after the onset of the perfusion of the slices with the aglycemic medium. The field potential was fully suppressed after 40 minutes of glucose deprivation. After the washout of the aglycemic medium, a partial recovery of the field potential amplitude was observed. Bath application of 1 μmol/L CPT produced a slight (13 ± 4%, n = 21), but significant (P < 0.01) increase of the field potential amplitude. For this reason, a slight decrease of the intensity of synaptic stimulation was necessary to restore the control field potential amplitude. A similar finding was obtained by using 300 nmol/L CPX (n = 12, data not shown). Incubation of the slices with either 1 μmol/L CPT (Fig. 1) or 300 nmol/L CPX (data not shown) 10 minutes before the onset of the glucose deprivation significantly reduced the amplitude of the aglycemia-induced depression of the field potential amplitude. Interestingly, in the presence of CPT or CPX a full recovery of the field potential amplitude was observed.
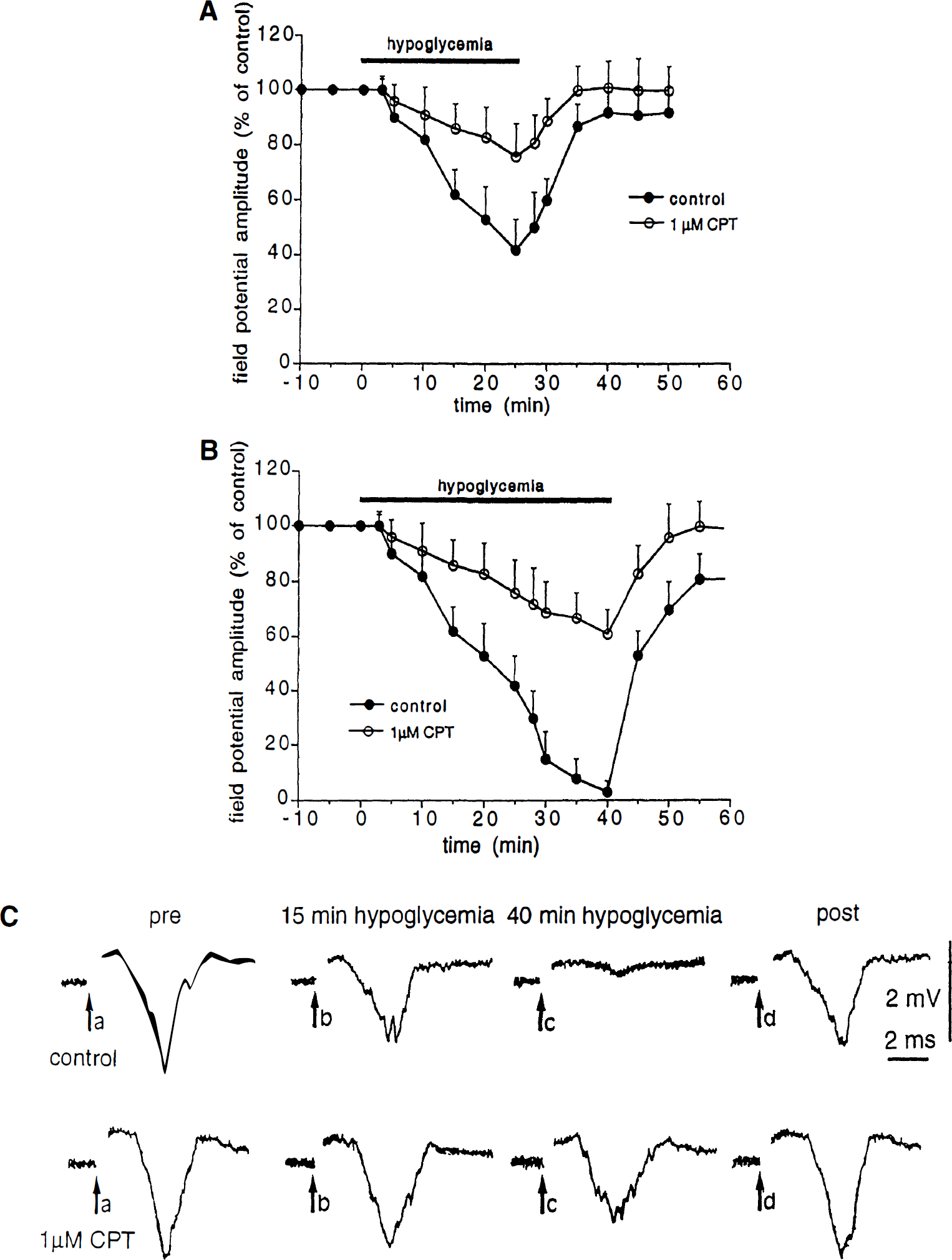
Effect of 8-cyclopentyl-1,3-dimethylxanhine (CPT) on the aglycemia-induced depression of corticostriatal synaptic transmission.
Incubation of the slices (15 minutes before the onset of aglycemia) in the presence of either 10 μmol/L glibenclamide (n = 7) or 100 nmol/L glipizide (n = 6) did not affect the field potentials amplitude. Moreover, the depression of the field potential amplitude caused by glucose deprivation was affected neither by glipizide (n = 6, Fig. 2) nor by glibenclamide (n = 7, data not shown).
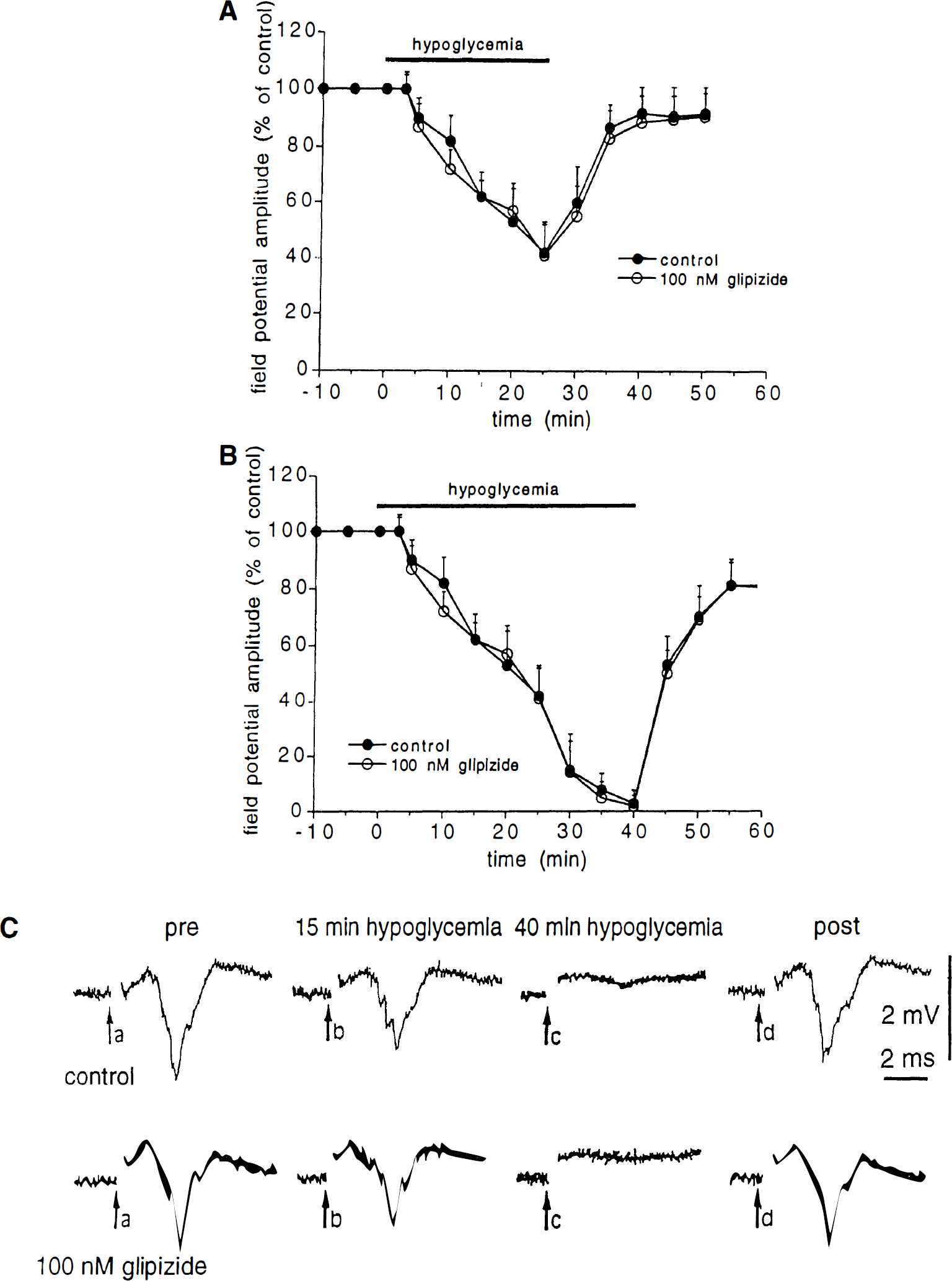
Effect of glipizide on the aglycemia-induced depression of corticostriatal synaptic transmission.
We also tested whether the effect of A1 adenosine receptor antagonists on the aglycemia-induced depression of synaptic transmission was related to a possible interference with the postsynaptic membrane changes induced by glucose deprivation on spiny striatal neurons. For these reasons, the effects of aglycemia were studied in single neurons by using intracellular recordings in single-microelectrode voltage clamp mode. The intrinsic membrane properties of these neurons have previously been described in both current and voltage clamp modes (Calabresi et al., 1995a, b ). As shown in Fig. 3, aglycemia produced a time-related membrane depolarization in spiny neurons. This membrane depolarization developed slower than the aglycemia-induced synaptic inhibition. In fact, this depolarization started only 10 minutes after the onset of the aglycemic condition. In some experiments, the effects of aglycemia om membrane properties were measured by using single microelectrode voltage-clamp recordings (Fig. 3). The cells were clamped close to the resting membrane potential (–85 mV). The aglycemia-induced inward current was coupled with an increase of the membrane conductance as detected by the application of constant hyperpolarizing voltage steps (1 to 3 second duration, 5 to 15 mV amplitude). Both 1 μmol/L CPT (Fig. 3) and 300 nmol/L CPX (n = 4, data not shown) failed to alter the intrinsic membrane properties of the recorded neurons as well as the inward current induced by glucose deprivation. Moreover, these adenosine receptor antagonists did not affect the increase of the membrane conductance generated by aglycemia (Fig. 3).
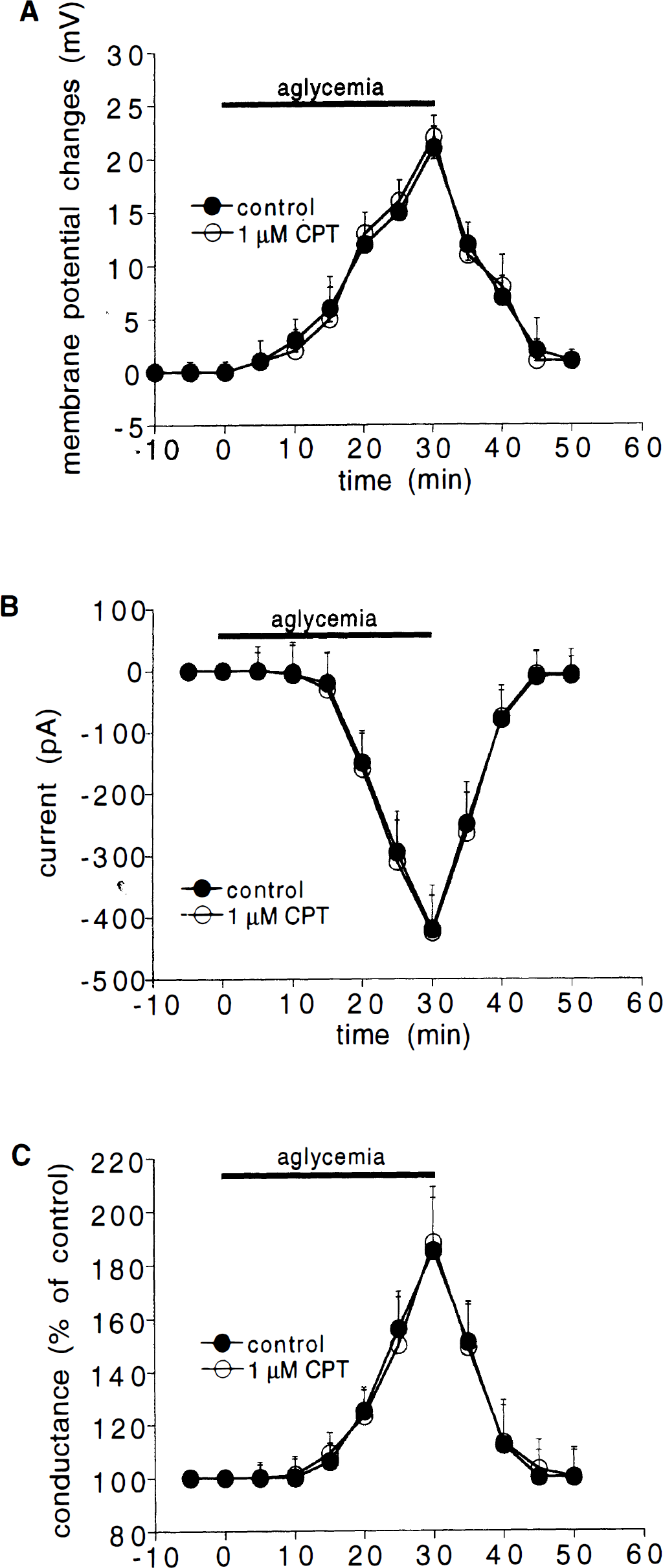
Effects of aglycemia on intrinsic membrane properties of striatal neurons.
DISCUSSION
The present study shows that antagonists of A1 adenosine receptors reduce the depression of glutamatergic transmission induced by glucose deprivation. These antagonists affected neither the intrinsic membrane properties of the recorded cells nor the aglycemia-induced postsynaptic membrane changes. Thus, we argue that the depression of the glutamatergic transmission observed during glucose deprivation is probably caused by the release of endogenous adenosine acting on presynaptic A1 receptors located on glutamatergic corticostriatal terminals. It is possible that the increase of the postsynaptic membrane conductance generated after 10 minutes of glucose deprivation influences the decrease of synaptic transmission observed in the late phase of aglycemia. However, we presume that the early decrease of excitatory synaptic transmission (within the first 10 minutes) is mainly induced by presynaptic mechanisms. Nevertheless, it has to be stressed that further information about this issue will require intracellular experiments measuring postsynaptic sensitivity to exogenous glutamate during glucose deprivation and experiments dealing with the effect of aglycemia on paired-pulse facilitation to dissect between presynaptic and/or postsynaptic mechanisms.
It has been reported previously that adenosine release is the major cause of failure of synaptic transmission during glucose deprivation in rat hippocampal slices (Zhu and Krnjevic, 1993; Khazipov et al., 1995). In addition, our observation is fully in agreement with a variety of evidence that shows that at low concentration, adenosine blocks excitatory synaptic transmission by suppressing transmitter release (Lupica et al., 1992; Scholz and Miller, 1991). Interestingly, application of exogenous adenosine in the striatum blocks excitatory synaptic transmission, but does not affect intrinsic membrane properties of spiny neurons (Malenka and Kocsis, 1988; Uchimura and North, 1991). Moreover, our finding that blockers of ATP-dependent potassium channels failed to affect the aglycemia-induced decrease of excitatory transmission are in line with data obtained in other brain areas showing that these antagonists do not influence the synaptic changes induced by glucose deprivation (Shoji, 1992; Zhu and Krnjevic, 1993). The observation that aglycemia causes hyperpolarizing effects in the hippocampus caused by the activation of potassium conductances (Spuler et al., 1988), while it causes a late depolarization in spiny striatal neurons (present study), suggests that glucose deprivation seems to activate various ionic mechanisms in central neurons. These mechanisms might account for the differential vulnerability of neuronal subpopulations to energy deprivation in various brain areas.
The role of endogenous adenosine in the depression of synaptic transmission during energy metabolism failure is also supported by previous findings indicating that A1 receptor antagonism can delay the hypoxia-induced depression of synaptically evoked excitatory potentials in hippocampal slices (Fowler, 1989; Katchman and Hershkowitz, 1993). Accordingly, extracellular levels of adenosine can increase after a high metabolic demand, including hypoglycemia, hypoxia, and seizure activity (Geiger and Nagy, 1990; Greene and Haas, 1985; Snyder, 1985). This increase, most likely, is caused by the net breakdown of intracellular ATP to ADP and AMP. Adenosine, derived from intracellular AMP, is released from the cell by a nucleoside transporter (Martin et al., 1994). Adenosine can also be formed extracellularly from released nucleotides.
A cytoprotective role of adenosine and adenosine analogues has been shown in experimentally-induced cerebral ischemia (Ramkumar et al., 1995). Several mechanisms have been postulated to mediate this neuroprotection: interaction with antioxidant enzymes, activation of potassium channels, inhibition of calcium influx, and inhibition of neurotransmitters such as glutamate (for a review see Ramkumar et al., 1995). Our study suggests that the latter mechanism might play a role in the pathophysiology of corticostriatal transmission during metabolic stress. By using in vivo microdialysis in the freely moving rat, an increase of extracellular adenosine in the hippocampus and in the striatum during lights-off periods has recently been reported (Huston et al., 1996). These authors have suggested a possible role of this transmitter in regulation of sleep and in some motor and nonmotor behavioral activities related to these brain areas (Huston et al., 1996). Thus, the adenosine-mediated depression of glutamatergic transmission in the striatum during aglycemia might account for some of the behavioral changes observed after acute glucose deprivation.