
Abstract
Numerous studies in the last two decades have resulted in significant progress in our understanding of the role of inhibitors on axonal regeneration and conditions that influence mature neurons to regrow in an inhibitory environment. These studies have revealed putative therapeutic targets and strategies to interfere in the inhibitory signaling cascade and promote axonal regeneration. Some agents that were successful in animal models are now being tested in human patients. All of these advances have raised hope of a cure for an injury that was once thought to be ʻan ailment for which nothing is done' (Quote from Edwin Smith surgical papyrus, 1600BC).
Introduction
After injury, adult mammalian central nervous system (CNS) axons do not spontaneously regenerate, whereas their peripheral nervous system counterparts often do. In the early 1980s, David and Aguayo (1981) showed that a lesion in the CNS results in an inhibitory environment that prevents axon regrowth; however, when a piece of peripheral nervous system tissue is grafted into the lesion site, the injured CNS axons regrow, suggesting that adult CNS axons can regenerate when offered a permissive environment. Products of myelin breakdown released on injury in the adult CNS were thought to be responsible for the failure of axon regrowth (Berry, 1982). In seminal findings several years later, Caroni and Schwab (1988a) identified the axon growth inhibitory proteins present in the CNS myelin. Subsequently, they generated a monoclonal antibody IN-1, which blocked the inhibitory action of these proteins in cultures. Since then several myelin-associated inhibitory molecules have been identified (Chen et al 2000; GrandPre et al, 2000; Prinjha et al, 2000, see for a review Filbin, 2003). Nogo-A, myelin-associated glycoprotein (MAG) and oligodendrocyte myelin glycoprotein (OMgp) are the three major inhibitors associated with myelin. In addition to myelin inhibitors, the glial scar formed at the injury site is known to pose both a mechanical and biochemical barrier to the regenerating axons (McKeon et al, 1991; Asher et al, 2001, see for a review Silver and Miller, 2004). However, since it takes several weeks for the glial scar to mature, the myelin debris resulting from the injury poses the most immediate obstacle to the regenerating axons (Huang et al, 1999). Therefore, strategies aimed at blocking myelin inhibition should aid in axonal regrowth and functional recovery. This review will summarize the structure, function and signaling mechanisms of the major myelin-associated inhibitors and finally discuss various strategies to block their effects such that axons attempting to regenerate are no longer inhibited from doing so.
Inhibitors of myelin
Nogo-A
Nogo-A (also known as NI-220/250) is an antigen for monoclonal antibody IN-1, which was raised against inhibitory protein fractions of CNS myelin (Caroni and Schwab, 1988b). Application of IN-1 antibody blocked inhibition by myelin in culture as well as enhanced regrowth of injured corticospinal tract (CST) axons in vivo resulting in some functional recovery (Caroni and Schwab, 1988b; Bregman et al, 1995). The identification of the IN-1 antigen remained elusive for many years. In the year 2000, however, three groups independently cloned the IN-1 antigen (using a partial sequence of NI-250) and termed it Nogo (Chen et al, 2000; GrandPre et al, 2000; Prinjha et al, 2000). The Nogo gene encodes three distinct isoforms, Nogo-A, -B, and -C, generated via alternative splicing and promoter usage. Nogo-A is a 200 kDa membrane protein, mainly expressed by oligodendrocytes, motor neurons, and sensory ganglia neurons, but not astrocytes or Schwann cells. Nogo-B, however, is a 55 kDa protein expressed in many tissues and cell types including adult neurons. Nogo-C is a 25 kDa protein that is strongly expressed in skeletal muscle (GrandPre et al, 2000; Chen et al, 2000; Huber et al, 2002). All three Nogo isoforms share a common carboxy-terminal domain consisting of 188-amino-acid residues whose sequence is homologous to the reticulon protein family (GrandPre et al, 2000; Chen et al, 2000; Prinjha et al, 2000). Two long hydrophobic stretches, which could serve as transmembrane domains, are present within the carboxyl domain and a 66-amino-acid loop referred to as Nogo-66 is present in between the two hydrophobic stretches (Figure 1) (GrandPre et al, 2000). Nogo-66 is inhibitory to axon growth and is present in all the three isoforms, implying that all the three Nogo isoforms exert inhibitory properties. Nogo-A has an additional domain localized to a 195-amino-acid stretch near the N-terminus termed amino-Nogo that is absent in other isoforms (Figure 1). Amino-Nogo inhibits neurite outgrowth as well as abrogates 3T3 fibroblast spreading (Fournier et al, 2001). It is interesting to note that a high proportion of Nogo-A is found intracellularly associated with the endoplasmic reticulum and Golgi complex, while only about 1% of the total cellular Nogo-A is present on the cell surface (GrandPre et al, 2000; Oertle et al, 2003). The function of Nogo-A in the endoplasmic reticulum is not known. The large intracellular pool may represent an unprocessed precursor form and/or may have an intracellular function. In the adult CNS, Nogo-A is localized in the inner loop of the myelin sheath and in the outer myelin loop (Huber et al, 2002). Nogo-A is thought to have at least two different membrane topologies, one in which the N-terminal domain and Nogo-66 domain face the extracellular space and a second in which the N-terminal domain and Nogo-A-specific part are exposed to the cytoplasmic side of the membrane (GrandPre et al, 2000).
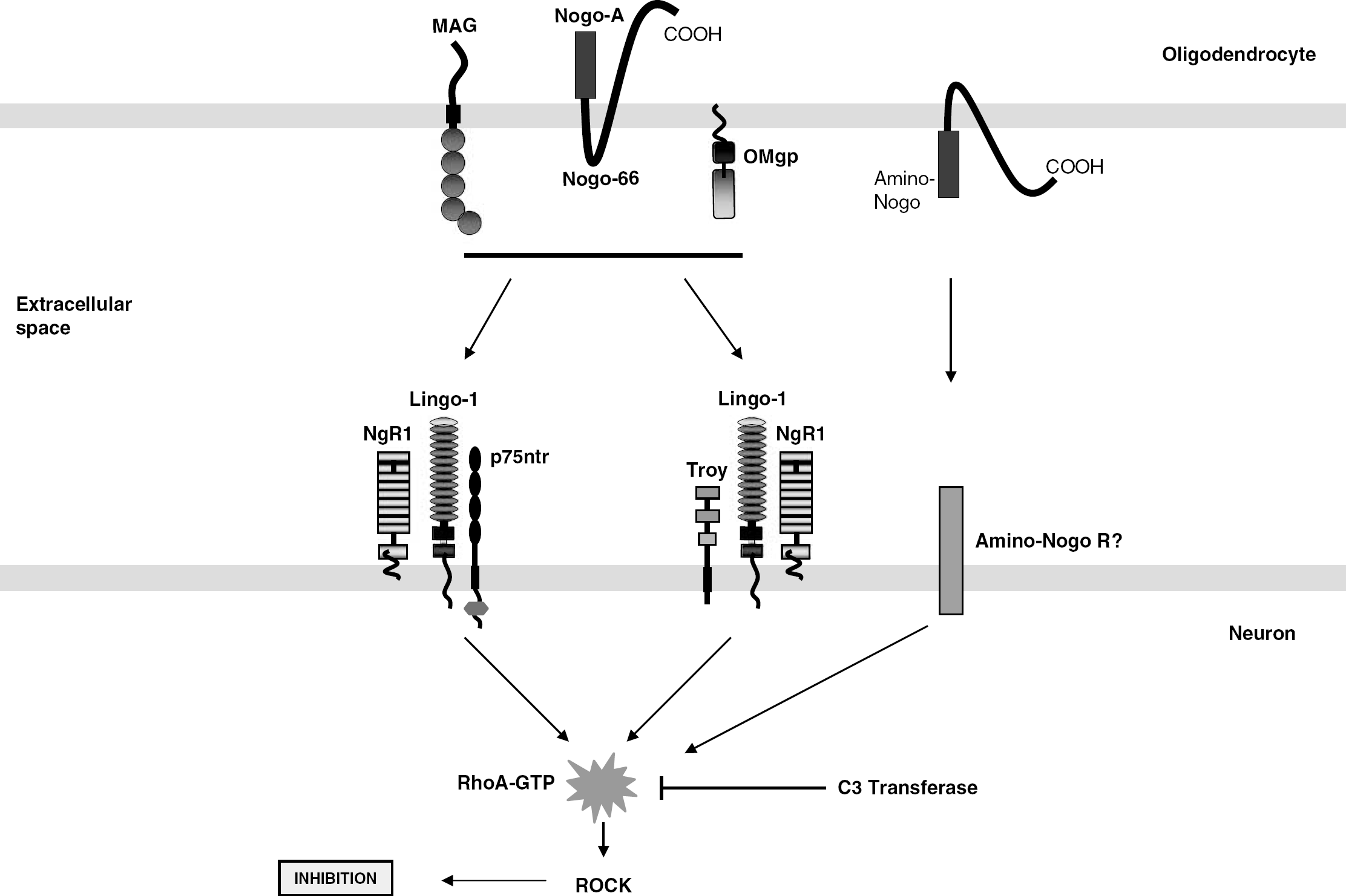
Inhibitors of axonal regeneration in the CNS. Nogo-A, MAG, and OMgp are the three major inhibitors present in CNS myelin. Nogo-A has two inhibitory domains, Nogo-66, a 66-amino-acid loop present in between the two transmembrane stretches, and amino-Nogo, present near the N-terminus that is unique to Nogo-A. Myelin-associated glycoprotein has five Ig-like domains in the extracellular region, a transmembrane, and a cytoplasmic domain. Oligodendrocyte myelin glycoprotein is a glycosyl phosphatidyl-inositol-linked protein that carries LRR in its extracellular region. Nogo-66, MAG, and OMgp bind to a common receptor complex consisting of the NgR, LINGO-1, and p75ntr. TROY, a tumor necrosis factor family receptor, can substitute for p75ntr in some neurons. Receptor for amino-Nogo is yet unidentified. On ligand binding, all the three inhibitors including amino-Nogo activate RhoA, followed by Rho kinase (a downstream effector of RhoA) and finally result in neurite outgrowth inhibition. Blocking RhoA activation using C3-transferase allows neurite outgrowth in the presence of inhibitors.
Myelin-Associated Glycoprotein
Myelin-associated glycoprotein was the first myelin inhibitor to be identified (Mukhopadhyay et al, 1994; McKerracher et al, 1994). Myelin-associated glycoprotein is a member of the immunoglobulin (Ig)-like family (Salzer et al, 1987, 1990). It contains five extracellular Ig-like domains, a single transmembrane domain, and a short cytoplasmic domain (Figure 1). Myelin-associated glycoprotein exists in two alternatively spliced forms, a large (L) and a small (S) form that differ in the length of their cytoplasmic domains. Myelin-associated glycoprotein is expressed in the myelin-forming cells: Schwann cells in the peripheral nervous system and oligodendrocytes in the CNS (Trapp et al, 1988, 1989). In the CNS, MAG is localized solely to the periaxonal membrane in the internodal segments of the myelin sheath, whereas in the peripheral nervous system, MAG is expressed in the paranodal region, Schmidt—Lanterman incisures, and outer mesaxon segments (Trapp et al, 1989). Myelin-associated glycoprotein is bifunctional, enhancing axonal growth from young neurons, but with a pronounced inhibitory action on older neurons (DeBellard et al, 1996; Mukhopadhyay et al, 1994; McKerracher et al, 1994; Cai et al, 2001; Turnley and Bartlett, 1998; Johnson et al, 1989). Furthermore, a soluble, proteolytic fragment of MAG consisting of the extracellular domain (ECD) secreted from damaged white matter in vivo is also inhibitory (Tang et al, 2001). Myelin-associated glycoprotein is also a sialic acid-binding protein (Kelm et al, 1994). It can bind to gangliosides, GT1b and GD1a, in a sialic acid-dependent manner. However, sialic acid binding is not necessary to exert inhibition (Tang et al, 1997).
Oligodendrocyte Myelin Glycoprotein
Oligodendrocyte myelin glycoprotein was first identified a decade ago and was found to be highly expressed in CNS myelin (Mikol and Stefansson, 1988; Mikol et al, 1990). Recently, it was reisolated and shown to cause growth cone collapse as well as neurite growth inhibition (Wang et al, 2002a). Oligodendrocyte myelin glycoprotein is a highly glycosylated extracellular membrane protein anchored to the surface through a glycosyl phosphatidyl-inositol lipid intermediate (Figure 1). Oligodendrocyte myelin glycoprotein consists of a highly conserved leucine-rich repeats (LRR) domain and a C-terminal LRR domain (Wang et al, 2002a). It is expressed not only in oligodendrocytes but also in many types of neurons and enriched near the nodes of Ranvier (Mikol and Stefansson, 1988). A recent study suggests that it may play a role in nodal architecture as well as prevent collateral sprouting (Huang et al, 2005). Oligodendrocyte myelin glycoprotein expression is developmentally regulated in rat CNS with an increase after birth to until postnatal week 6 (Vourc'h et al, 2003).
Receptors
Remarkably, all the three myelin-associated inhibitors (with the exception of amino-Nogo) bind to the same receptor complex consisting of the Nogo-66 receptor (NgR), p75ntr, and LINGO-1 (the
While attempting to identify a signal-transducing partner for NgR, Strittmatter's group discovered MAG to be a second ligand for NgR (Liu et al, 2002). Simultaneously, the Filbin group identified NgR as a receptor for MAG and showed that application of soluble NgR or dominant-negative forms of NgR can effectively block the inhibitory effects of MAG (Domeniconi et al, 2002). Both groups showed that the interaction between MAG and NgR is sialic acid-independent. It is still not clear if the two ligands bind to identical or overlapping sites on NgR. The evidence is contradictory as one study reported that excess soluble Nogo-66 can effectively block the binding of soluble MAG (Domeniconi et al, 2002) and a second study reported that neither excess Nogo-66 nor the Nogo inhibitory peptide Nogo extracellular peptide 1–40 significantly reduced MAG binding to NgR (Liu et al, 2002). During the same time He's group, who identified OMgp as a third myelin-associated inhibitor, showed that NgR is a functional receptor for OMgp. They further showed that Nogo-66 and OMgp bind to overlapping regions on NgR and hence compete for binding to NgR (Wang et al, 2002a).
Since NgR is a glycosyl phosphatidyl-inositol-anchored protein and lacks transmembrane or cytosolic domains, it was predicted to require a signal-transducing molecule to effect inhibition. By this time p75ntr, a transmembrane protein that binds to pro and mature neurotrophins had been implicated as a signal transducer for MAG-mediated inhibition. Using neurons from p75ntr null mice, Yamashita et al (2002) showed that MAG-dependent neurite inhibition and RhoA activation were reduced in the absence of p75ntr. This suggested that MAG-dependent inhibition requires p75ntr. But they did not show a direct interaction between NgR and p75ntr. He et al later established that p75ntr interacts with NgR to form a receptor complex and that this interaction is enhanced on ligand binding (Wang et al, 2002b). It appears that p75ntr interacts with NgR through its ECD. NgR, however, interacts with p75ntr via its N-terminal LRR, leucine-rich repeat C-terminal and C-terminal domains. Not surprisingly, neurons overexpressing truncated NgR (lacking the unique C-terminal domain) are not inhibited by MAG. Furthermore, the cytoplasmic domain in p75ntr is necessary to transduce inhibitory signals into the interior of the responding neurons. Accordingly, neurons overexpressing truncated p75ntr are not inhibited by myelin inhibitors (Wang et al, 2002b).
Recently, a third molecule, LINGO-1, has been found to be a necessary component of the NgR/p75ntr signaling complex (Mi et al, 2004). LINGO-1 is a nervous system-specific protein consisting of 12 LRR motifs in its ECD, an Ig-like domain, a transmembrane domain, and a short cytoplasmic tail. LINGO-1 is highly expressed in rat brain, with expression levels peaking at postnatal day 1 (P1) and decreasing thereafter into adulthood. LINGO-1 expression is also detected after injury in adult rat spinal cord. LINGO-1 binds to both NgR1 and p75ntr to constitute a functional receptor for myelin inhibitors. In the absence of LINGO-1, the inhibitory activity of myelin proteins is reduced. Coexpression of all the three components but not binary combinations of them is required to confer responsiveness to myelin components (Mi et al, 2004).
Although p75ntr is highly expressed in the developing nervous system (Roux and Barker, 2002; Chao, 2003), it is limited to certain types of neurons in the adult CNS. For example, adult cerebellar and cortical neurons do not express p75ntr (Park et al, 2005) and only 60% to 70% of adult dorsal root ganglion (DRG) neurons express p75ntr (McMahon et al, 1994; Wright and Snider, 1995; Park et al, 2005). Moreover, mice lacking p75ntr fail to undergo spontaneous regeneration after spinal cord injury (SCI) in vivo (Song et al, 2004; Zheng et al, 2005). Blockade of p75ntr function by administering soluble fusion protein consisting of the ECD from p75ntr (p75-Fc) does not improve regeneration (Song et al, 2004). All these observations led to the speculation that an alternate to p75ntr exists. Two research groups independently addressed this question and showed that TROY (also known as TAJ), a member of the tumor necrosis factor receptor family, as is p75ntr broadly expressed in postnatal and adult neurons, interacts with NgR and can substitute for p75ntr in the inhibitory receptor complex (Shao et al, 2005; Park et al, 2005). Like p75ntr, TROY can activate RhoA in response to inhibitor binding when both NgR and LINGO-1 are coexpressed. Neurons isolated from TAJ-deficient mice were resistant to the action of myelin inhibitors.
Signaling by inhibitors: Even before the identification of the NgR complex, Rho family members had been implicated in myelin-induced inhibition (Yamashita et al, 2002). In its inactive state, RhoA remains bound to Rho guanine dissociation inhibitor, a Rho guanine dissociation inhibitor. On ligand binding, p75ntr associates with Rho guanine dissociation inhibitor and displaces Rho guanine dissociation inhibitor. This interaction appears to be particularly relevant to axon growth inhibition induced by myelin. Interference with p75ntr and Rho guanine dissociation inhibitor interaction disrupts p75ntr-dependent signaling (Yamashita and Tohyama, 2003). Treatment with C3-ADP-ribosyltransferase (C3), a bacterial exoenzyme and a specific inhibitor of Rho blocks inhibition and promotes neurite outgrowth on myelin inhibitors (Lehmann et al, 1999) (Figure 1). Treatment with Y-27632, a specific inhibitor of Rho kinase (Rho effector protein), also blocks inhibition (Dergham et al, 2002). Interestingly, both these inhibitors block inhibitory effects exerted by chondroitin sulfate proteoglycans (CSPGs) as well (Dergham et al, 2002; Monnier et al, 2003). Activation of RhoA is therefore a crucial event in the signal-transduction cascade activated by various myelin-associated inhibitors.
In an attempt to further unravel inhibitory signaling mechanisms, the Filbin group recently showed that binding of MAG to NgR induces regulated intramembrane proteolysis of its coreceptor p75ntr and this cleavage is necessary for RhoA activation and axon growth inhibition (Domeniconi et al, 2005). On ligand binding to NgR, p75ntr undergoes sequential cleavage first by α-secretase bound to the membrane to release the ECD and then cleavage within the transmembrane domain by γ-secretase to yield a 25 kDa intracellular domain (p75ICD). γ-Secretase activity is dependent on protein kinase C (PKC) activation, whereas α-secretase does not require PKC activation. The resulting p75ICD is believed to activate RhoA as inhibitors of γ-secretase interfere with RhoA activation (Figure 2) (Domeniconi et al, 2005; Ceni and Barker, 2005). These findings support an earlier report by He and his group that treatment with myelin-associated inhibitors induces PKC activation and blocking PKC activity attenuates RhoA activation and axon growth inhibition (Sivasankaran et al, 2004). Together, these two studies identify secretases and PKC as potential therapeutic targets for promoting regeneration in vivo. Accordingly, intrathecal delivery of PKC inhibitor allows robust regeneration of the ascending dorsal column axons (but not corticospinal axons) in vivo (Sivasankaran et al, 2004). The effects of blocking secretase activity on axon regeneration in an SCI model are yet to be tested.
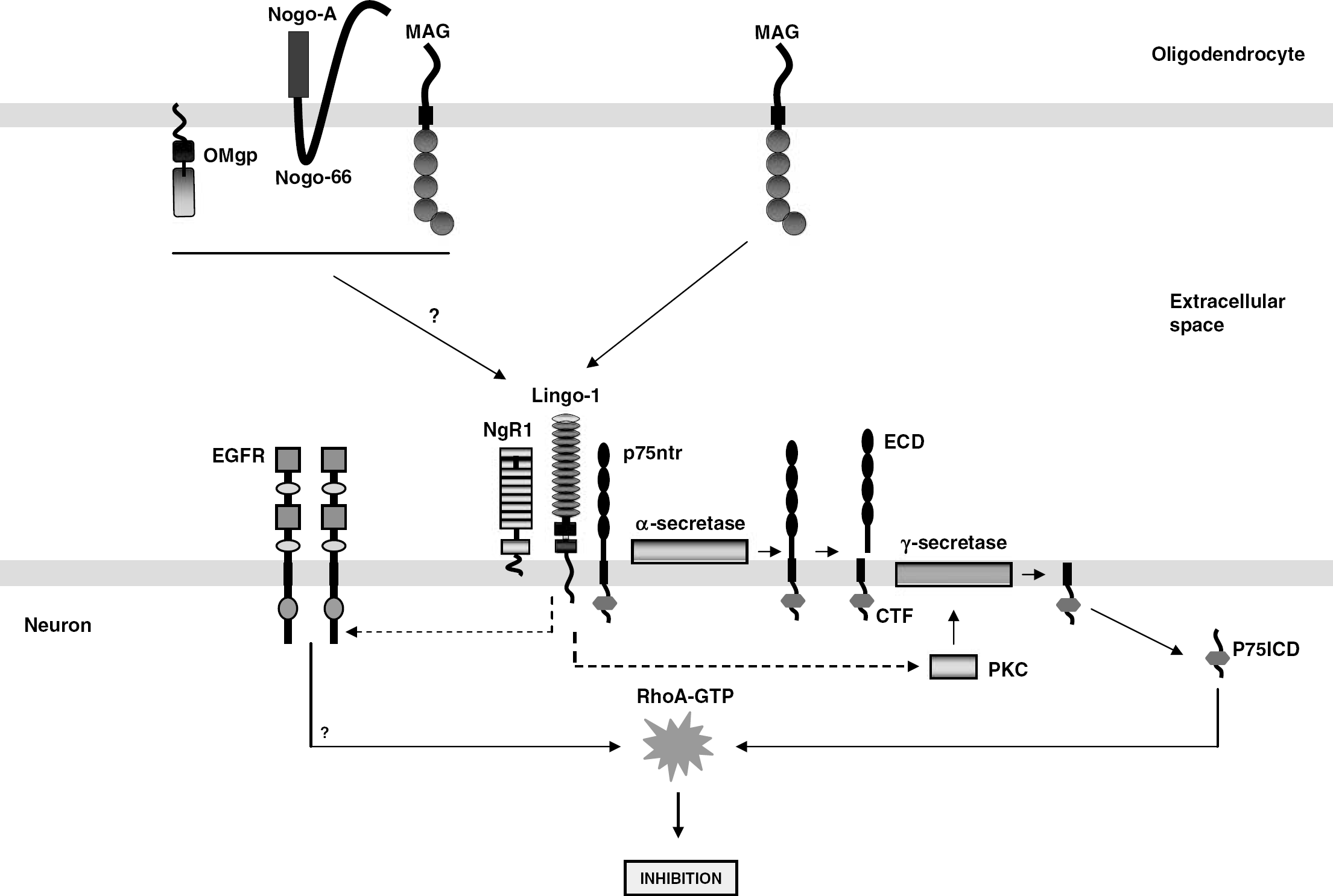
Signaling by myelin-associated inhibitors. Activation of NgR by myelin-associated inhibitors results in transactivation of the EGFR and through unknown sequence of events leads to RhoA activation and finally neurite outgrowth inhibition. Transactivation of EGFR requires Ca2+. Binding of MAG to NgR induces regulated intramembrane proteolysis of its coreceptor p75ntr. In a sequence of events, p75ntr is first cleaved by α-secretase bound to the membrane to release the ECD, followed by cleavage within the transmembrane by γ-secretase to yield intracellular domain (p75ICD). The released p75 intracellular domain activates RhoA. In addition, ligand binding to NgR complex is known to activate PKC and is essential for γ-secretase activity.
In another study, He and his co-workers identified a signaling event downstream of NgR activation (Koprivica et al, 2005). They showed that binding of myelin-associated inhibitors to NgR results in the activation of epidermal growth factor receptor (EGFR) (Figure 2) (Koprivica et al, 2005). This event requires transactivation by Ca2+. Inhibition of EGFR kinase activity using pharmacological inhibitors promotes neurite outgrowth on myelin inhibitory substrates in vitro and enhances axon regeneration in vivo in an optic nerve injury model. Moreover, CSPGs also activate EGFR in a Ca2+-dependent manner. Interestingly, Tarceva, a Food and Drug Administration-approved drug currently in clinical trial for treating human, non-small-cell lung cancer, is also an EGFR inhibitor. He's group showed that Tarceva can also block myelin-induced inhibition in vitro (Koprivica et al, 2005; Miller, 2005). Whether this drug is effective in blocking myelin-induced inhibition after SCI is yet to be determined.
Strategies to Overcome Central Nervous System Inhibition
Soon after the identification of the inhibitors present in myelin, attempts were made to ablate or neutralize the inhibitors. Moreover, binding of three major myelin-associated inhibitors to a single receptor complex suggests the existence of redundancy in the activity of these inhibitors. This provides yet another potential therapeutic target to block inhibition. In this regard, one strategy for therapeutic intervention is to deliver antibodies and peptides to block the binding of inhibitors to the receptor, so that the signaling is compromised.
Antibodies and peptides to block inhibition: Application of monoclonal antibody IN-1, raised against the IN-1 antigen, later identified as Nogo-A protein, induced regrowth of severed CST axons below the level of injury and recovery of locomotor function (Bregman et al, 1995). In another interesting study, mice were immunized against CNS myelin before SCI. Significant axonal regeneration and some functional recovery was observed in these mice in the absence of a cellular inflammatory response (Huang et al, 1999). Another approach was to use specific peptides to prevent inhibitors from binding. A peptide consisting of the first 40 amino-acid residues corresponding to the amino-terminus of the Nogo-66 domain (Nogo extracellular peptide 1–40) was shown to act as a competitive antagonist for NgR. Administration of Nogo extracellular peptide 1–40 to the rats with hemisected spinal cord resulted in ectopic sprouting of the severed CST axons and improved functional recovery (Grandpre et al, 2002). A truncated fragment of NgR (NgR ecto), Fc fusion proteins consisting of the ECD of p75ntr (p75-Fc) or LINGO-1 (LINGO-1-Fc) each have been shown to be effective in allowing neurite outgrowth on inhibitory substrates (Fournier et al, 2002; Wang et al, 2002b; Mi et al, 2004).
Interfering in downstream signaling events: In addition to blocking the inhibitors, it may be possible to manipulate downstream signaling pathways to promote axonal regeneration. Binding on myelin-associated inhibitors to NgR induces PKC activation. Blocking PKC activity using inhibitor Go6976 promotes regeneration of the dorsal column fibers (Sivasankaran et al, 2004). As described earlier, RhoA activation is a convergent point in the inhibitory signaling cascade. Inactivation of RhoA and its downstream effectors is thus a potential strategy to block inhibitory signaling and promote axon regeneration. Dergham et al (2002) compared the effects of RhoA inhibitor, C3-transferase, and the Rho kinase inhibitor, Y-27632, on axon regeneration in a SCI model. Both inhibitors induced long-distance axon regeneration and significant improvements in functional recovery. Fournier et al (2003), however, observed sprouting of the CST fibers and functional recovery with Y27632 treatment, but not with C3-transferase. Although the reasons for these divergent results are unclear, it is possible that the effective concentrations in the two studies were not the same since the two studies used different modes of delivery. The C3-transferase used in these studies has poor membrane permeability; however, chimeric-C3 with improved membrane permeability have been developed and are more effective in entering cells and blocking RhoA activation (Winton et al, 2002). One such membrane-permeable variant of C3, BA210, is now being tested in clinical trials of human SCI patients.
Gene knockout studies in spinal cord injury models: Three groups using three different strategies independently generated Nogo knockout mice to assess the contribution of Nogo to regeneration failure. Stephen Strittmatter's group generated Nogo mutants by disrupting both Nogo-A and -B expression, leaving Nogo-C expression unaltered (Kim et al, 2003). Marc Tessier-Levigne's group generated two different lines of mice, one lacking all three Nogo isoforms derived by deleting the C-terminal region common to all the three Nogo isoforms, and a second line lacking Nogo-A/B was generated by deleting the amino-terminal genomic fragment of exon 1 shared by Nogo-A and -B (Zheng et al, 2003). The latter line is similar to the one generated by Strittmatter's group and lacked the expression of only Nogo-A and -B. Finally, Martin Schwab's group generated knockout mice in which Nogo-A expression is specifically abolished (Simonen et al, 2003). This group, however, reported a compensatory increase in Nogo-B expression. All Nogo mutant mice are viable, fertile, and exhibit no obvious neurologic deficits. The central myelin extracted from all three mutant mice lines showed reduced axon growth inhibitory action in in vitro assays. Quite unexpectedly, each of these mice lines resulted in different regeneration phenotypes after SCI. A profound increase in CST axon sprouting was observed above the level of injury and a substantial number of axons caudal to the lesion site in the Nogo-A/B mutant mice generated by Strittmatter's group. This improved regeneration was, however, restricted to young adult mice that were <9 weeks old. In a subset of older mice (11 to 14 weeks), the extent of sprouting above the injury was significantly reduced and no regeneration was observed below the injury. In sharp contrast, Tessier-Levigne's group observed no axon regeneration in both Nogo-A/B and Nogo-A/B/C mutant mice. Schwab's group, however, observed a modest increase in axonal regrowth in their Nogo-A mutant mice. It is unclear at this point why each group obtained different results. Some possible explanations include differences in the genetic composition of the mice used in these studies. All three Nogo knockout studies used two different mouse strains (129X1/svj and C57/BL6) to generate chimeric mice. These mice have unknown proportions of genetic background. The observed differences in the capacity to regenerate might be attributed to these differences in the genetic composition. Martin Schwab's group addressed this issue in a recent study by backcrossing Nogo-A-specific knockout mice into 129X1/svj and C57BL/6 mouse strains to obtain pure strain backgrounds. The resulting mice exhibited remarkable differences in their regenerative response both in vitro and in vivo. In addition, the two mice lines showed different levels of growth-related gene expression, posttraumatic inflammation as well as endogenous neurite growth capacity, suggesting that genetic background may influence the capacity of these animals to regenerate (Dimou et al, 2006). Moreover, there are at least three inhibitors present in CNS myelin that might result in a potential redundancy in the myelin-mediated inhibitory signaling.
Alternatively, a null mutation in the ngr gene, whose product, NgR, binds to all three myelin-associated inhibitors (with the exception of amino-Nogo), was predicted to block myelin-induced axon growth inhibition more effectively and to achieve enhanced functional recovery. Two groups independently tested this hypothesis and obtained variable results. Strittmatter's group reported a block of myelin-mediated inhibition and significant improvements in locomotion as well as electrophysiologic measurements in ngr–/– mice (Kim et al, 2004), whereas Tessier-Levigne's group reported persistence of inhibition and lack of CST regeneration in their mice (Zheng et al, 2005). The extent of behavioral recovery obtained in Strittmatter's ngr–/– mice is similar to earlier studies that used IN-1 monoclonal antibody or Nogo extracellular peptide 1–40 peptide to block Nogo inhibition (Bregman et al, 1995; GrandPre et al, 2002). However, the anatomic results were different. While raphespinal and rubrospinal axon regeneration was present, CST regeneration observed in ngr–/– mice was insignificant. It is not clear why there is no CST regeneration in NgR–/– mice. In general, these results are inferior to those observed with NgR antagonist peptide (GrandPre et al, 2002). Is it the existence of NgR homologs, NgR2 and NgR3, that compensate for the loss of NgR1 in these mice? (Lauren et al, 2003). According to one report, none of the three major myelin-associated inhibitors bind to NgR2 and NgR3 (Barton et al, 2003). A recent report, however, showed MAG binding to NgR2 in a sialic acid-dependent manner (Venkatesh et al, 2005). Furthermore, since amino-Nogo can act independently of NgR, could this be responsible for the observed inhibition in ngr–/– mice? We do not have answers to any of these questions. These studies challenge the view that NgR plays a central role in myelin-mediated inhibitory signaling.
Axonal plasticity and recovery of function in stroke models: Although genetic studies have yielded inconclusive results about the role played by Nogo-A/B and NgR in SCI models, perturbation of Nogo-NgR function in stroke models has revealed some interesting results. Mice lacking Nogo-A/B and NgR showed improved functional recovery and enhanced neuronal plasticity after stroke (Lee et al, 2004). Significant axonal sprouting from the intact side to the stroke-denervated area was observed in the knockout mice. The enhanced neuronal plasticity in the absence of inhibitors is thought to be the underlying mechanism for stroke recovery in these mice. Treatment with monoclonal antibody IN-1 that neutralizes Nogo-A also revealed similar improvements in functional recovery (Papadopoulos et al, 2002; Wiessner et al, 2003; Seymour et al, 2005). Nogo-A neutralization resulted in up to 80% of forelimb functional recovery within 8 to 9 weeks after stroke. In all of these studies, neuroanatomic remodeling because of increased plasticity was observed. Therefore, treatments to perturb Nogo-NgR signaling may be a potential therapeutic strategy to promote neuronal plasticity and functional recovery after stroke and other neurodegenerative diseases.
Changing the intrinsic state of the mature neurons: The capacity to regenerate depends on the age of the animal at the time of injury. In contrast to mature neurons, young neurons show greater amount of axonal regeneration in vivo and are not inhibited by MAG/myelin in vitro (Bregman and Goldberger, 1982; Kunkel-Bagden et al, 1992; Bates and Stelzner, 1993; DeBellard et al, 1996). This indicates a differential growth state of embryonic neurons versus adult neurons. Embryonic neurons have cAMP levels significantly higher than that of their adult counterparts. The loss of regenerative ability in postnatal neurons indeed directly correlates with a precipitous decrease in endogenous cAMP levels (Cai et al, 2001). Thus, one way to enhance intrinsic growth capacity of mature neurons is to elevate levels of intracellular cAMP. This can be accomplished by priming mature neurons in culture with neurotrophins such as brain-derived neurotrophic factor, adding a membrane-permeable cAMP analog, dibutyryl cAMP, or by inhibiting phosphodiesterases, the enzymes that breakdown cAMP (Figure 3) (Cai et al, 1999; Song et al, 1998; Nikulina et al, 2004).
The effect of increasing the intrinsic growth capacity of the neurons in vivo was first tested by Richardson and Issa (1984). They showed that spinal axons of L4 and L5 DRG neurons regenerated efficiently only if the ipsilateral sciatic nerve was cut. Neumann and Woolf (1999) later showed that transection of the ipsilateral sciatic nerve 1 or 2 weeks before performing a dorsal column lesion allowed spontaneous regeneration of injured dorsal column axons. This effect was later shown to be cAMP-dependent. One day after sciatic nerve transection, cAMP levels in DRG neurons are elevated about threefold and the levels drop to baseline by 7 days. The effect of a conditioning lesion can be mimicked by a single injection of dibutyryl cAMP into the DRG in vivo (Neumann et al, 2002; Qiu et al, 2002). Recently, it was shown that two priming lesions in the sciatic nerve, one made simultaneous to the dorsal column lesion and a second 1 week after the dorsal column lesion, promoted enhanced regeneration presumably via sustained intrinsic growth capacity (Neumann et al, 2005). Elevation of cAMP thus provides a promising strategy to overcome myelin-mediated inhibition of axonal regeneration. Three different laboratories recently tested the effect of cAMP elevation in combination with other therapeutic techniques in vivo (Lu et al, 2004; Pearse et al, 2004; Nikulina et al, 2004). The Tuszynski laboratory showed that injection of dibutyryl cAMP into DRG neurons when combined with neurotrophin application and bone marrow stromal cell transplantation improved regeneration after SCI (Lu et al, 2004). In the remaining studies, rolipram, a phosphodiesterase type 4 inhibitor, was injected to block cAMP degradation and elevate cAMP levels and then combined either with Schwann cell (Pearse et al, 2004) or embryonic tissue transplants at the lesion site (Nikulina et al, 2004). In both studies, significant improvement in axonal regeneration and functional recovery was reported.
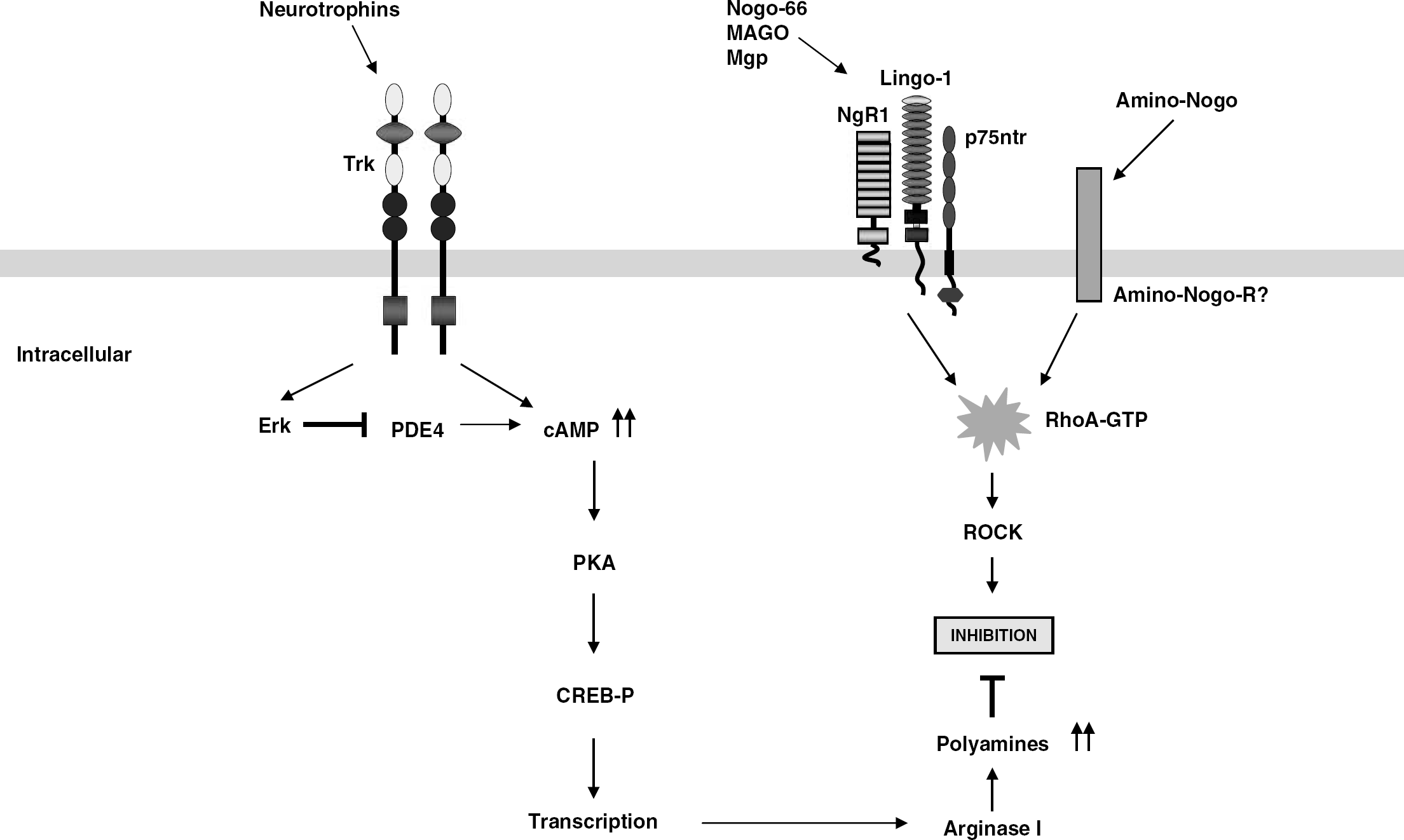
Overcoming myelin inhibitors to promote axonal regeneration. One approach to overcoming inhibitors in mature neurons is to elevate cAMP levels by priming with neurotrophins, which signal through tyrosine kinase receptors (trk) to inactivate cAMP hydrolyzing enzyme, phosphodiesterase type 4. In addition to priming, addition of membrane-permeable anolog, dibutyryl cAMP, and a peripheral conditioning lesion in vivo have been shown to elevate cAMP levels (not shown). Elevation of cAMP results in protein kinase A activation, which can inactivate RhoA directly and via transcription of genes such as Arg I, resulting in increased synthesis of polyamines in a parallel pathway that allows neurite outgrowth in the presence of myelin-associated inhibitors.
Downstream effectors: Elevation of cAMP results in the activation of protein kinase A, a cAMP effector. Protein kinase A-specific inhibitors block cAMP effects in overcoming MAG/myelin inhibition (Cai et al, 1999). In addition to protein kinase A activation, priming with neurotrophins also leads to an accumulation of cAMP. This increase is known to occur via the activation of extracellular signal-related kinase, which in turn inhibits phosphodiesterase type 4, allowing cAMP to accumulate (Figure 3) (Gao et al, 2003). There are several downstream targets of protein kinase A and extracellular signal-related kinase, including the transcription factor, cAMP-response element-binding protein (Figure 3). cAMP-response element-binding protein plays a central role in cAMP-mediated effects. The Filbin lab has shown that activation of cAMP-response element-binding protein alone is sufficient to overcome MAG/myelin-mediated inhibition and allow spinal axon regeneration in vivo (Gao et al, 2004). Several genes are upregulated after cAMP-response element-binding protein activation. One such gene is arginase I (Arg I). Arginase I is a rate-limiting enzyme in the synthesis of polyamines, which include putrescine, spermidine, and spermine. Overexpression of Arg I or exogenous treatment with polyamines can overcome MAG/myelin-mediated inhibition in vitro (Figure 3). Blocking Arg I activity using a specific inhibitor can abrogate both Arg I and cAMP or brain-derived neurotrophic factor effects in overcoming inhibition (Cai et al, 2002). Interestingly, Arg I levels are high in young DRG neurons and low in older neurons. This decline correlates with the decrease in cAMP levels observed during the developmental switch in responsiveness to MAG. Overexpressing Arg I in older neurons blocks the switch (Cai et al, 2002). The mechanism by which polyamines function to alter the growth-inhibitory effect of MAG/myelin is not well understood. Arginase I and polyamines are by far the most specific targets for therapeutic intervention in the signaling cascade elicited by cAMP elevation. Further elucidation of the various signaling pathways might result in more specific therapeutic targets for treatment after injury and reduce the serious side effects that may occur as a result of modulating levels of a promiscuous molecule such as cAMP in neurons.
The glial scar and axon repulsion molecules
In addition to myelin-associated inhibitors, glial scar formation at the site of injury is known to pose both a physical and biochemical barrier to the regenerating axons (Silver and Miller, 2004). On injury, various extracellular matrix molecules are increasingly deposited in and around the scar by reactive astrocytes, inflammatory cells, and oligodendrocytes. One family of molecules of particular interest is CSPGs that include phosphacan, neurocan, versican, and NG2 (Levine, 1994; McKeon et al, 1999; Jones et al, 2002). Chondroitin sulfate proteoglycans are inhibitory to axonal growth both in vitro and in vivo (Rudge and Silver, 1990; McKeon et al, 1991, 1999; Asher et al, 2001). Altering CSPG levels using enzymes such as chondroitinase ABC, which hydrolyzes the glycosaminoglycan chains of proteoglycan (Bradbury et al, 2002) or DNA enzyme that interfere with glycosaminoglycan synthesis (Grimpe and Silver, 2004), improves regeneration and functional recovery. Although the receptors for CSPGs are unknown, CSPG binding results in the activation of RhoA and its downstream effector Rho kinase (Dergham et al, 2002; Monnier et al, 2003), an event that occurs after NgR activation. An implication of this finding is the existence of a common therapeutic target for blocking multiple inhibitors. Finally, a number of chemorepulsive guidance molecules such as semaphorins, ephrins, slits, netrins, and their receptors have been identified in the mature nervous system (Niclou et al, 2006). Studies investigating the roles for these molecules in axonal regeneration are currently gaining interest. In a recent study, Goldshmit et al (2004) reported regeneration of corticospinal and rubrospinal axons as well as functional recovery in EphA4 knockout mice after hemisection injury. In EphA4 knockout mice, astrocytic gliosis and glial scar were greatly reduced, whereas in wild-type mice, EphA4 expression was upregulated. These findings suggest a role for EphA4 signaling in astrogliosis. In another study, ephrin-B3 was shown to be present in oligodendrocytes and myelin extracts. Myelin substrates lacking ephrin-B3 allowed extension of slightly longer neurites from postnatal cortical neurons compared with wild-type myelin (Benson et al, 2005). Furthermore, semaphorin 3A expression increases in meningeal fibroblasts of the fibrotic scar in penetrating CNS injuries (Pasterkamp et al, 1999). Injured neurons retain the expression of semaphorin 3A receptor components and are repelled by semaphorin 3A (Pasterkamp et al, 2001). Although the role of slits and netrins in axon regeneration is not clear, netrin-1 is induced in Schwann cells after injury and may play a role in peripheral nerve regeneration (Madison et al, 2000). All these studies imply that in addition to restricting axon growth during development, axon repulsive molecules can inhibit axon growth in the adult nervous system.
Future perspectives
Significant advances have been made in identifying the inhibitors associated with myelin and the signaling molecules involved in inhibition. However, more remains to be done. Targeted deletion of Nogo and NgR was thought to improve regeneration of the injured CNS axons, but the results were variable. While some fiber pathways regenerated, others did not. It remains to be answered if amino-Nogo plays a compensatory role and if there are other unidentified inhibitors and receptors that are responsible for the inferior capacity of these mice to regenerate. The results from the stroke models, however, are promising. Perturbation of Nogo-NgR pathway using knockout mice or treatment with monoclonal antibody IN-1 resulted in enhanced recovery of function and neuroplasticty. Clearly understanding the mechanism of sprouting and treatments to enhance meaningful sprouting are highly desired. Studies employing cAMP elevation, however, have led to promising discoveries in therapeutic treatment. Further work is necessary to elucidate the crosstalk between various cAMP-induced signaling pathways and to identify new and specific targets for therapeutic intervention. Results from the combinatorial studies clearly show the need for combined therapeutic strategies to achieve complete reversal of the consequences of SCI.
Footnotes
Acknowledgements
We gratefully acknowledge Dr Tim Spencer, Dr Christine Cain and Dr Wilfredo Mellado for helpful discussions and preparation of this paper.