
Abstract
Calcium toxicity remains the central focus of ischemic brain injury. Calcium channel antagonists have been reported to be neuroprotective in ischemic animal models but have failed in clinical trials. Rather than block the calcium channels, calbindin proteins can buffer excessive intracellular Ca2+ as a result, maintain the calcium homeostasis. In the present study, we investigated the effect of calbindin D 28k (CaBD) in ischemic brain using the novel technique protein transduction domain (PTD)-mediated protein transduction. We generated PTD-CaBD in Escherichia coli, tested its biologic activity in N-methyl-
Introduction
Intracellular calcium (Ca2+) overloading plays a critical role in ischemic brain injury. Normally, calcium is present at a ratio of 10,000:1 between the extracellular fluid and the intracellular cytosol (Berridge et al, 2003); however, when ischemia occurs, excessive Ca2+ accumulates in the cytosol and mitochondria because of the rapid exhaustion of ATP supply in the brain. The superfluous Ca2+ activates Ca2+-dependent catabolic enzymes such as phospholipases, protein kinases, proteases, and caspase cascades (White et al, 2000), which subsequently induce neuron necrosis and apoptosis. A number of calcium channel blockers and N-methyl-
Calbindin D 28k (CaBD) is a member of the superfamily of calcium-binding proteins. It is found in neurons and astrocytes throughout the nervous system (Baimbridge et al, 1992). Physiologically, CaBD can buffer and modulate intracellular calcium concentration ([Ca2+]i) to avoid neuronal injury; however, the buffering ability is greatly discounted in ischemic injury because of the large increase of [Ca2+]i. It has been reported that exogenous CaBD can reduce oxidative stress, preserve mitochondrial function (Guo et al, 1998), and protect neurons against glutamate- or ischemia-induced delayed excitotoxicity (D'Orlando et al, 2001, 2002; Meier et al, 1998). Therefore, increasing intracellular CaBD expression and buffering the excessive Ca2+ might provide a unique approach to reduce [Ca2+]i cytotoxicity.
In the present study, we hypothesized that buffering the excessive Ca2+ in the cytosol using CaBD, rather than blocking Ca2+ influx, would provide neuroprotection during cerebral ischemia. We firstly generated a novel fusion protein PTD-CaBD (PTD, protein transduction domain) in Escherichia coli; then, we further examined (1) whether PTD-CaBD could cross the cell membrane into cells; (2) whether the PTD-CaBD had biologic activity ex vivo; and (3) whether PTD-CaBD attenuated ischemic brain injury in the rat transient middle cerebral artery occlusion (tMCAO) model in vivo.
Materials and methods
Generation of Recombinant Fusion Proteins
The PTD-CaBD was generated according to the method described previously (Fan et al, 2006). Calbindin D 28k encoding sequence was PCR amplified using the primers 5′ ata gga tcc gca gaa tcc cac ctg cag tc 3′ (sense, modified by BamHI at 5′) and 5′ ata aga ata agc ggc cgc cta gtt gtc ccc agc aga gag aa 3′ (antisense, modified by NotI at 3′) from rat brain cDNA. The purified PCR fragment was cloned into the BamHI/NotI site of the pTransvector (qBiogene, St Louis, MO, USA), and into the pET32a (Novagen, Madison, WI, USA) as a control. PTD-CaBD and thioredoxin (Trx)-CaBD were expressed in E coli BL21 (DE3). The fusion proteins were extracted and purified under native conditions. Briefly, the bacteria pellet was suspended in a lysis buffer (50 mmol/L NaH2PO4, 500 mmol/L NaCl, 10% glycerol, 10 mmol/L imidazole and 1 mmol/L PMSF, pH 8.0). After ultrasonication and centrifugation, the supernatant was subjected to metal-affinity chromatography using a Ni-NTA resin column (Novagen). The column was washed with 20 mmol/L imidazole and finally eluted in elute buffer (50 mmol/L NaH2PO4, 300 mmol/L NaCl, and 500 mmol/L imidazole, pH 8.0). Finally, excessive salt and imidazole were removed from the purified product through dialysis using the 7,000 MWCO dialysis cassettes (Pierce, Rockford, IL, USA) in phosphate-buffered saline (PBS).
Some of the two recombinant proteins were labeled with fluorescein isothiocyanate (FITC) (Mao, 1994). Briefly, the protein solution was dialyzed overnight in 500 mmol/L carbonate, pH 9.5. Then, FITC dissolved in dimethyl sulfoxide (10 mg/mL) was added at a ratio of 40 to 80 μg FITC per mg protein. The solution was incubated with gentle shaking at room temperature for 1 h. Free FITC was removed thoroughly by dialysis in PBS. The PBS from the last time dialysis was collected and used as a control as described below.
PTD-CaBD Transduction
Cos7 cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (Invitrogen, San Diego, CA, USA). To observe the transduction ability, Cos7 cells were cultured up to 75% confluence and incubated with 1.0 μmol/L PTD-CaBD or Trx-PTD for 1 h at 37°C. The cells were washed and images were taken through a fluorescent microscope (Olympus, Melville, NY, USA). The time course and dose-dependent analysis were performed using flow cytometry. Different concentrations of PTD-CaBD (0 to 2.0 μmol/L) were used for the dose-dependent study, and time points of 0 to 120 mins were used for the time-course study. The cells were washed, trypsinized, and resuspended in PBS at 1 × 106 cells/mL for flow cytometry.
Hippocampal Slice Culture
The institutional animal care and use committee at Shanghai Medical School, Fudan University, China approved all procedures for the use of laboratory animals. Hippocampal slices were prepared according to the method of Stoppini et al (1991). Briefly, hippocampus was isolated on day 1 from a neonatal Sprague—Dawley rat. Transverse hippocampal slices of 400 μm in thickness were planted on six-well Transwell insert membranes (Corning Costar, Cambridge, MA, USA) and cultured in Neuro-basal culture medium (Invitrogen). To determine PTD-CaBD transduction efficiency, 2 μmol/L of PTD-CaBD-FITC or Trx-CaBD-FITC was added into the culture media. The plates were washed after 1 h of incubation. Images were captured using an inverted fluorescent microscope.
NMDA and OGD Exposure of Hippocampal Slices
Hippocampal slices exposed to NMDA and oxygen—glucose deprivation (OGD) were used to evaluate the biologic activity of PTD-CaBD. The hippocampal slices were incubated in a medium with 100 μmol/L NMDA (Sigma, St Louis, MO, USA) for 1 h at 37°C to induce Ca2+ influx. For the OGD treatment, inserts with slices were washed and transferred into neurobasal culture medium without glucose (Invitrogen) in six-well plates. The plates were put into an airtight microincubator chamber that was sealed tightly after being flushed with 95% N2/5% CO2 for 10 mins. The slices in the tightly sealed chamber were incubated at 37°C for 20 mins and then submerged into normal culture condition.
Ca2+ Assay
[Ca2+]i in hippocampal cells was measured using the sensitive indicator Fluo 3-AM (Yang and Steele, 2005). Hippocampal slices were loaded with Fluo 3-AM (5 μmol/L) in D-Hanks and incubated at 37°C for 60 mins. The slices were washed to remove free Fluo 3 AM and incubated in a serum-free medium. A confocal microscope (Bio-Rad, Hercules, CA, USA, Microradiance 2000) was used to detect [Ca2+]i.
Cell Death Determination
Propidium iodide (PI, 2 μg/mL) was added to the culture medium before or after treatment. Propidium iodide fluorescence images were captured from each slice immediately after PI application as the baseline measurement (Fb). Additional measurements were performed at different time points after treatment (Ft). At the end of the experiment, the slices were lethally challenged with NMDA and Kainic acid (KA) (500 μmol/L in PBS) for 2 h at 37°C and 24 h at 4°C. Additional PI fluorescence images were taken as Fmax. The percentage of cell death in each slice was expressed as Death% = (Ft–Fb)/(Fmax–Fb) × 100%, where Ft is slice fluorescence at time t, Fb is the background florescence at the start of the experiment, and Fmax was the maximal fluorescence after complete cell killing (Abdel-Hamid and Tymianski, 1997).
Transduction of PTD-CaBD in the Brain
Adult male Kunming mice were anesthetized with 1.5% isoflurane, and then PTD-CaBD (50 μg/kg) or Trx-CaBD (50 μg/kg) was intraperitoneally injected into mice. Animals were killed 8 h after injection and the brains were perfused transcardially with heparinized saline followed by 4% paraformaldehyde/PBS. Brains were removed and postfixed in 4% paraformaldehyde/PBS overnight at 4°C. Frozen sections 10 μm in thickness were prepared for immunoassaying. Anti-polyhistidine antibody (1:500 dilution; Novagen) and FITC-conjugated IgG (1:2,000 dilution; Sigma) were used to detect the histidine-tagged PTD-CaBD.
tMCAO in Rats
Adult male Sprague—Dawley rats weighing 200 to 220 g were anesthetized with 1.5% isoflurance. The internal carotid artery was isolated and the pterygopalatine artery was ligated. Then, a 3 cm length of 4-0 nylon suture with a slightly larger tip was gently advanced from the external carotid artery to the beginning of the middle cerebral artery for a distance of 18.0 ± 0.5 mm. The suture was withdrawn from the internal carotid artery to the common carotid artery for reperfusion (Longa et al, 1989; Zhang et al, 2003). The animals underwent 60 mins of tMCAO and 72 h of reperfusion. Surface cerebral blood flow was measured before and during MCAO using laser-Doppler flowmetry (Peri Flux System 5000, Perimed, Sweden) to ensure the success of MCAO. The rats were divided into the following experimental groups (six rats per group): (1) a sham control group; (2) tMCAO rats with saline intraperitoneal injection; and (3) rats with PTD-CaBD intraperitoneal injection (5 μg/kg) 30 mins before tMCAO.
Neurologic Severity Scores
Modified neurologic severity scores were used to evaluate neurologic function of each animal (Chen et al, 2001); neurologic severity score is a composite of motor, sensory, reflex, and balance tests. Neurologic function was graded on a scale of 0 to 18 (normal, 0; maximal deficit score, 18). The rat was scored 0 if it could pass all the tests, and the higher the score, the more severe the injury. All animals were trained to be familiar with the testing environment before the MCAO surgery. Neurologic examination was conducted in surviving animals at 24 and 72 h by a maskedinvestigator.
Infarct Area Determination
TTC (2,3,5-triphenyl-tetrazolium chloride; Sigma) staining was performed to detect the infarct volume according to Wang et al (2004). The brains were removed 72 h after reperfusion, and a series of 2 mm coronal brain slices were stained in 1% TTC in 0.1 mol/L PBS for 30 mins at 37°C. Infarct volumes were measured by sampling stained sections with a digital camera (Nikon Corporation, Tokyo, Japan). The ischemic area was calculated as the difference between the area of the nonischemic hemisphere and the normal area of the ischemic hemisphere. The infarct volume was calculated by multiplying the infarct areas by the thickness of sections (Cao et al, 2002).
TUNEL Staining
The terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) assay kit (Oncogene, Boston, MA, USA) was used to detect DNA fragmentation. The brain sections were fixed in 4% paraformaldehyde/PBS for 15 mins, and incubated in 20 μg/ml proteinase K/PBS at room temperature for 10 mins. Then, 3% H2O2 was used to quench the endogenous horseradish peroxidase. The sections were then incubated in the labeling reaction mixture for 90 mins at 37°C. Anti-BrdU-biotin (1:20 dilution) was applied for 60 mins. After incubating in conjugate buffer, sections were stained with DAB-H2O2 (DAB, diaminobenzidine tetrahydrochloride), and counterstained with Harri's hematoxylin.
Immunostaining
The frozen brain sections were incubated in 1% Triton X-100 for 15 mins, in 3% H2O2 for 5 mins, and then blocked using 1% bovine serum albumin (Sigma) for 30 mins. The sections were incubated in primary antibody (anti-cleaved caspase-3 antibody, 1:100 dilution, Cell Signaling Tech, Beverly, MA, USA; anti-polyhistidine antibody, 1:500 dilution, Novagen) overnight at 4°C and in FITC- or horseradish peroxidase-coupled IgG antibody (1:100 dilution; Hua-Mei Biotech, Shanghai, China) for 1 h at room temperature. Finally, the sections were detected by fluorescent microscopy or by using a DAB reagent (Sigma).
To analyze TUNEL and cleaved caspase-3-positive staining, two coronal sections (20 μm thickness) 2 mm anterior and 1 mm posterior to the Bregma were chosen. We randomly selected five fields of view around the boundary zone of the ischemic core in each section. We then counted the total number of positive cells in each field. Finally, we calculated the average cell number per field of view. The average was used to deduce positive cells in the whole boundary zone.
Western Blot
Protein was extracted from the ischemic zone and the concentration was determined using bicinchoninic acid assay. An equal volume of proteins (80 μg) with sample buffer was boiled and loaded onto 15% polyacrylamide gels. Proteins were transferred onto a Hybond N nylon membrane (Amersham, Piscataway, NJ, USA). The membrane was incubated in 5% milk/PBS blocking solution, and immersed in anti-cleaved caspase-3 (1:250 dilution; Cell Signaling) or anti-polyhistidine antibody (1:500 dilution; Novagen) overnight. Then horseradish peroxidase-labeled anti-rabbit second antibody (1:100 dilution; Hua-Mei Biotech) was used to detect the immunoreactivity. Lastly, the membrane was developed in color with an enhanced ECL kit (Amersham) and exposed to Kodak film.
Statistical Analysis
All data were presented as mean ± standard deviation (s.d.). Student's t-test was used to compare the difference between the treated or untreated group. A probability of less than 1% or 5% was considered to be statistically significant.
Results
PTD-CaBD Generation
The fusion proteins were tagged 6 histidines for purification (Figure 1A). To generate the proteins, full-length CaBD-encoding cDNA was amplified. The fragment was purified by gel extraction and subsequently was cloned into pTransVector containing the PTD peptide, and into pET32a containing Trx but not PTD as a control. The recombinant plasmids pTV-CaBD and pET32-CaBD were verified by enzyme digestion and PCR amplification, indicating the insert of 786 bp fragments (Figure 1B).
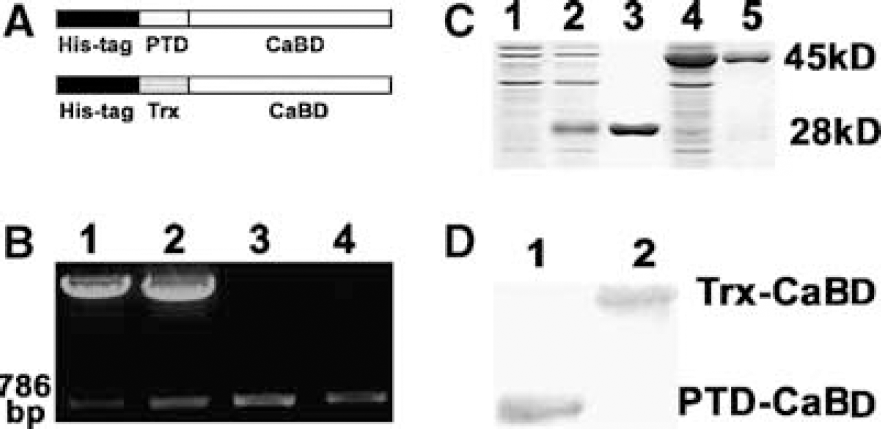
Generation of the fusion proteins PTD-CaBD and Trx-CaBD. (
The sequencing analysis showed that the fragment matched exactly with rat brain CaBD cDNA (GI: 203236). Finally, the proteins PTD-CaBD and Trx-CaBD were expressed in E coli and then purified to near homogeneity. The fusion proteins were assayed by SDS—polyacrylamide gel electrophoresis after Coomassie brilliant blue staining and further identified using Western blot analysis; the results showed that Trx-CaBD was 45 kDa and PTD-CaBD was 28 kDa (Figures 1C and 1D).
Transduction Efficiency of PTD-CaBD
PTD-CaBD-FITC was efficiently delivered into cos7 cells after 1 h of incubation, whereas Trx-CaBD-FITC was not detected in cos7 cells (Figure 2A). Flow cytometer assay showed that PTD-CaBD was delivered into cos7 cells in a dose-dependent manner, and 0.05 μmol/L of FITC-PTD-CaBD could be delivered into approximately 80% cells (Figure 2B). FITC-PTD-CaBD-positive cells increased significantly after 10 mins of incubation, and peaked at 1 h (Figure 2C).
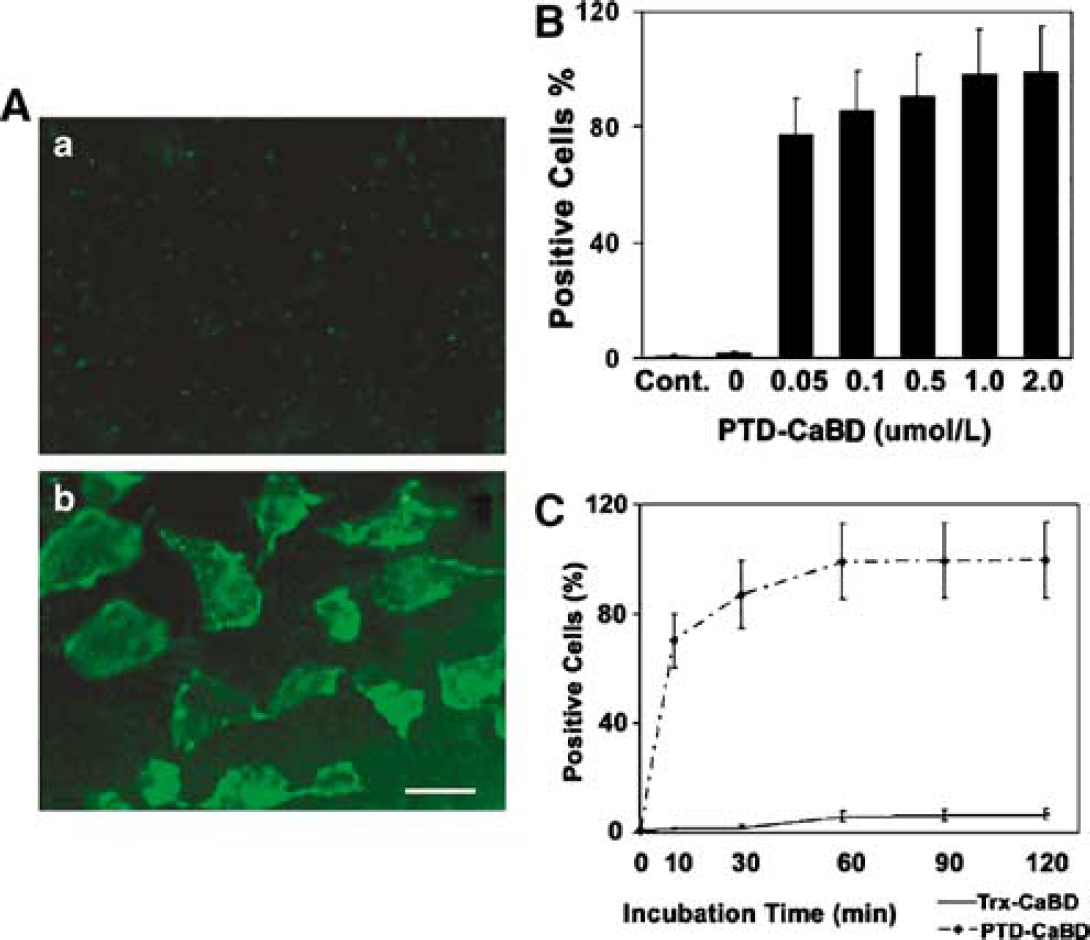
PTD-CaBD could be delivered into cos7 cells efficiently. (
Effect of PTD-CaBD on OGD or NMDA Exposure
Most cells in PTD-CaBD-FITC-treated slices showed strong fluorescence-positive staining after 1 h of incubation, whereas cells in Trx-CaBD-FITC-treated slices showed negative staining (Figure 3A). After pretreatment with 0.1 μmol/L PTD-CaBD for 1 h, intercellular Ca2+ in hippocampal slices subjected to OGD or 100 μmol/L NMDA significantly decreased compared with the control groups (P < 0.05; Figures 3B and 3C). Further analysis showed that pretreatment with different concentrations of PTD-CaBD (0, 10, 50, and 100 nmol/L) could attenuate hippocampal slice cell death induced by NMDA in a dose-dependent manner (Figures 3D and 3E).
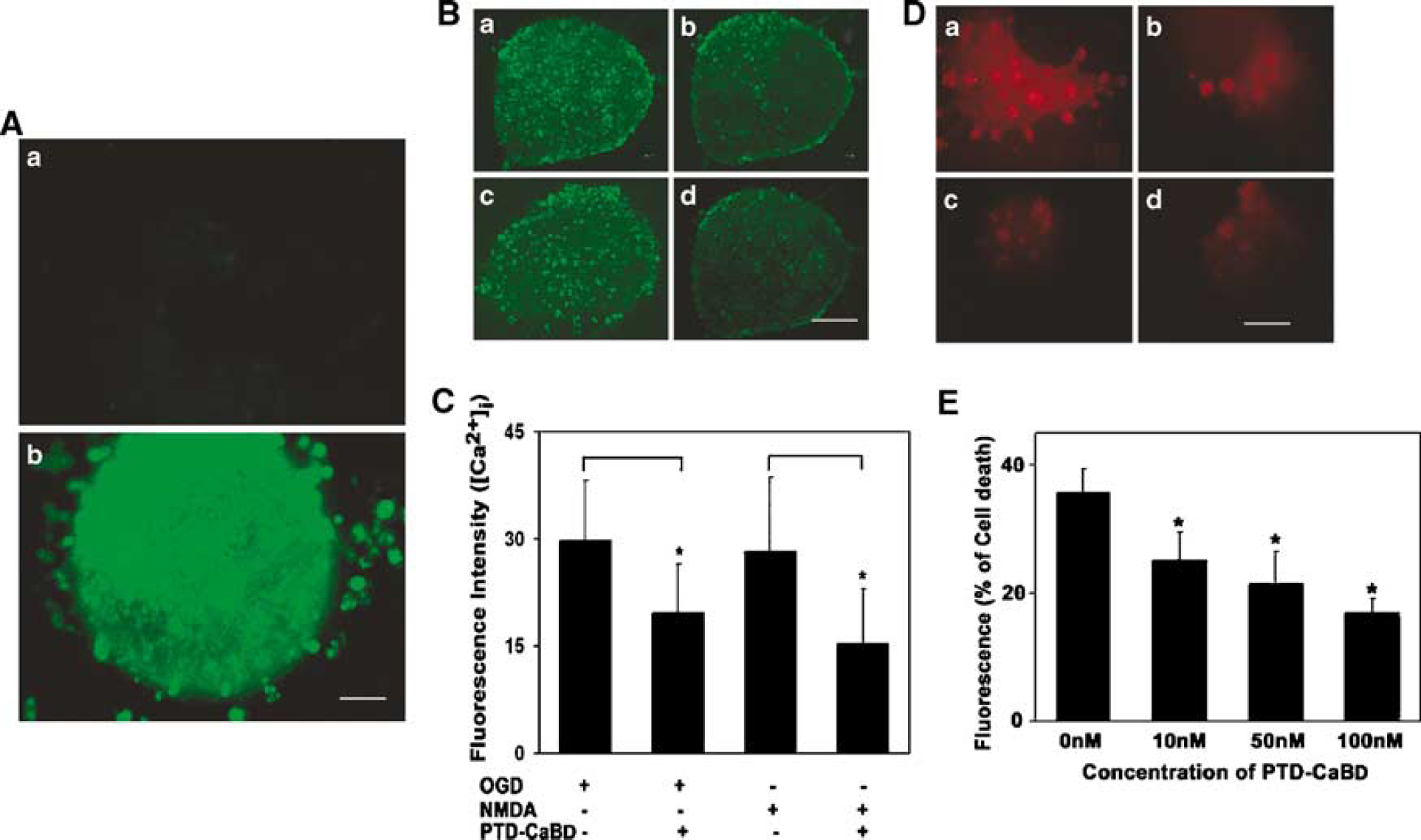
PTD-CaBD could buffer excessive Ca2+ in hippocampal slice cells. (
Effect of PTD-CaBD on Rat Brains after tMCAO
The surface cerebral blood flow was measured before and during MCAO operation to ensure the success of the model. The surface cerebral blood flow in the ipsilateral hemisphere of rats with tMCAO was reduced to 10% to 20% of baseline. The surface cerebral blood flow returned to at least 80% within 20 mins after the thread was withdrawn. There was no difference in physiologic parameters (e.g., arterial blood pressure, blood gases, body temperature) during tMCAO among the different experimental groups (data not shown).
Immunofluorescence staining after 8 h of PTD-CaBD injection showed strong positive polyhistidine immunoreactivity in the subventricular zone, cerebral cortex, and caudate putamen injected with PTD-CaBD (Figure 4A). Interestingly, we did not find any detectable differences in the distribution of PTD-CaBD between the control group and PTD-CaBD injection plus MCAO group (data not shown), which indicated that PTD-mediated protein delivery into brain was not because of blood—brain barrier breakdown induced by ischemia. Infarct volume of PTD-CaBD-treated rats was significantly smaller than that of the control group (280 ± 47 versus 166 ± 70 mm3 P < 0.05; Figures 4B and 4C). Furthermore, behavioral outcome evaluated by neurologic severity scores had better scores in the PTD-CaBD-pretreated rat after 72 h of reperfusion compared with the controls (P < 0.05; Figure 4D).
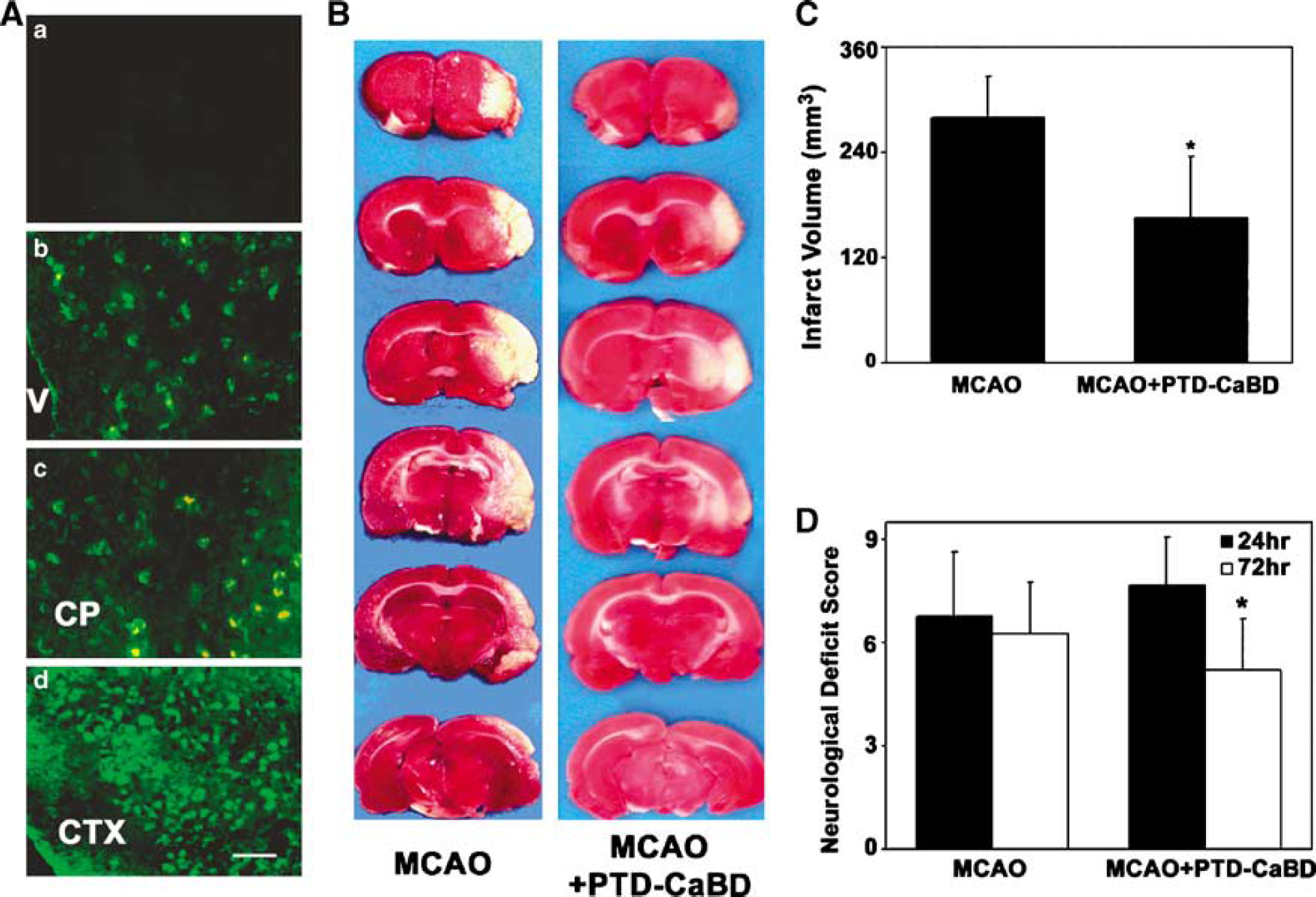
PTD-CaBD reduced brain infarct volume and improved neurologic outcomes in tMCAO rats. (
The Action of PTD-CaBD in Apoptosis Inhibition
TUNEL-positive cells in the ischemic boundary zone increased after ischemia, but were significantly reduced in the PTD-CaBD-treated group compared with the untreated group (58% ± 7% versus 29% ± 3%, P < 0.05; Figures 5A and 5C). Similarly, the cleaved caspase-3-positive cells significantly increased after ischemia/reperfusion compared with the controls. The positive cells were detected mainly in the ischemic perifocal region, although there were a few positive cells in the ischemic core areas. Importantly, the cleaved caspase-3-positive cells significantly decreased in the PTD-CaBD-treated rats compared with the tMCAO group (62 ± 4/field versus 31 ± 6/field, P < 0.05; Figures 5B and 5D). The Western blot result was parallel to the immunostaining findings (P < 0.05; Figures 5E and 5F).
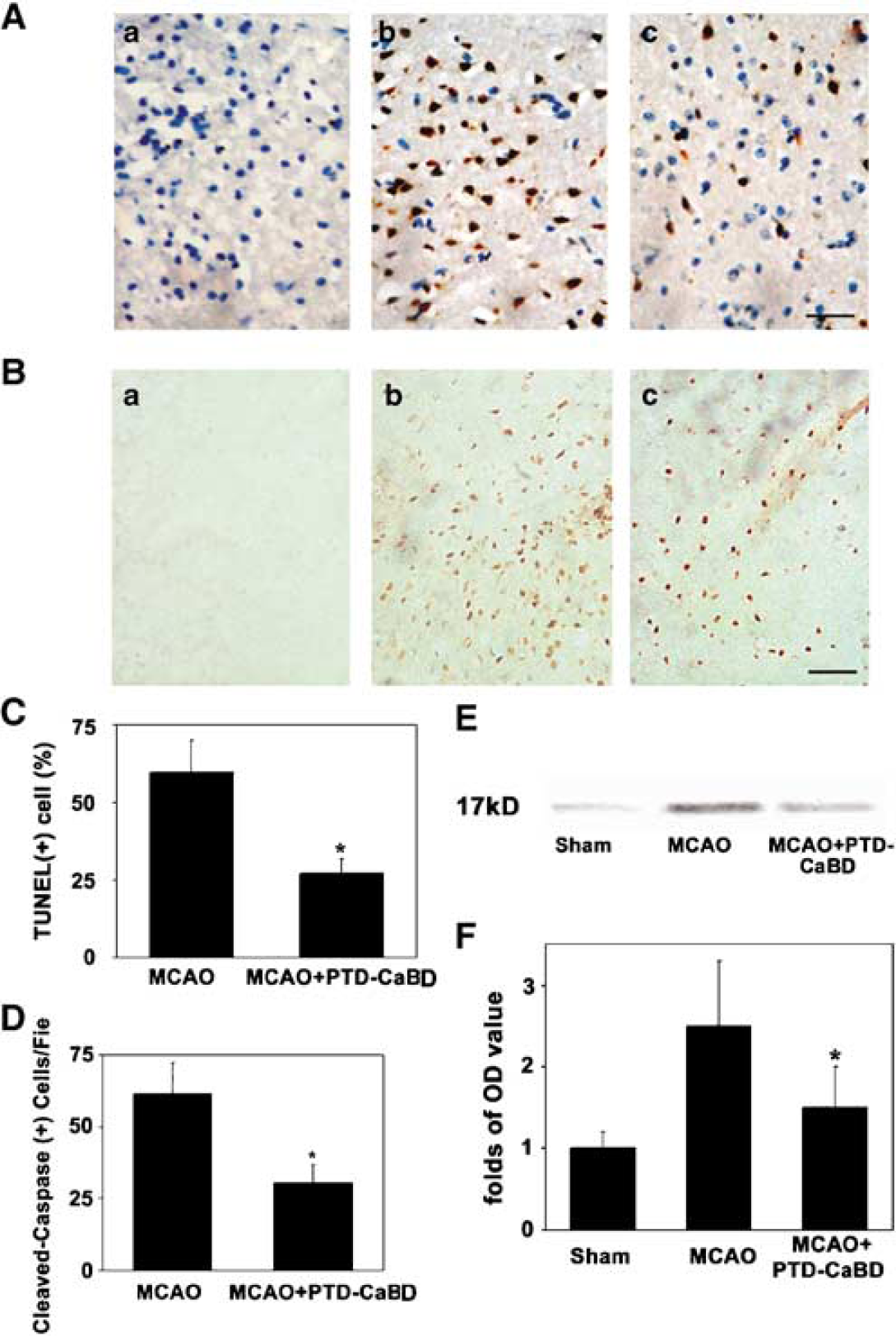
PTD-CaBD inhibited brain cell apoptosis. (
Discussion
For the first time, we used PTD-mediated protein transduction to explore the effect of CaBD in focal cerebral ischemic animal models. The results show that (1) PTD-CaBD could be delivered into cos7 cells or brain slices, and they buffer the excessive calcium, and therefore reduce NMDA- and OGD-induced neuronal injury, and (2) PTD-CaBD could be delivered into the rat brain and thus protect it from ischemia/reperfusion injury.
Protein transduction domains, also termed cell-permeable proteins or membrane translocating sequences, are small peptides that are able to ferry much larger molecules into cells independent of classical endocytosis (Beerens et al, 2003; Joliot and Prochiantz, 2004). This provides an exciting therapeutic opportunity for the treatment of brain ischemia and many other diseases. The PTD peptides can be found in several proteins such as TAT protein of the human immunodeficiency virus (HIV-tat) and antennapedia of Drosophila (Antp HD) (Leifert et al, 2002). Delivery of proteins or peptides fused to PTD into many kinds of cells in vitro or in vivo, even across the blood—brain barrier into brain parenchyma, has been reported (Dietz and Bahr, 2004). These proteins maintain their biologic activity and exert therapeutic action. Our previous studies showed that XIAP fused with PTD could inhibit apoptosis in the ischemic brain (Fan et al, 2006).
In our experiments, we found that 70% to 80% of cos7 cells were transduced even at a very low concentration of PTD-CaBD (0.05 μmol/L), or after a short incubation time (begins at 10 mins and peaks at 1 h), suggesting that PTD-mediated protein transduction was rapid and efficient. Moreover, the transduction could occur at 4°C and in the presence of known endocytosis inhibitors. The mechanism of PTD-mediated protein transduction, independent of energy and receptors binding, has not yet been fully elucidated. A nonspecific electrostatic interaction of basic amino acids PTD with cellular membrane might be the first crucial step (Bellet-Amalric et al, 2000); heparan sulfate in the cytoplasm (Rusnati et al, 1999; Ziegler and Seelig, 2004) and amphiphilic α-helices of PTD might be required during the transduction (Scheller et al, 1999). For the hippocampal slices, we obtained a similar result; after incubation with FITC-labeled PTD-CaBD, most of the cells showed FITC—positive staining.
After delivery into cells, the PTD fusion proteins in a partially denatured state are refolded and/or modified to perform their biologic functions (Schneider et al, 1996; Schwarze et al, 1999). In our experiments, when the hippocampal slices pretreated with PTD-CaBD were exposed to NMDA or OGD, [Ca2+]i significantly decreased and the cell death was much less than the control group. These results suggest that PTD-CaBD was not only delivered into cell cytosol, but also performed its biologic action through binding the overloaded [Ca2+]i and blocking the downstream lethal cascade, and consequently protecting the cells from OGD- or NMDA-induced injury. Previous reports also indicated that overexpression of CaBD protected glutamate-induced excitotoxicity in U937 cells (Jeon et al, 2004), neuroblastoma—retinal hybrid cells (D'Orlando et al, 2002), and pancreatic islet beta cells (Parkash et al, 2002; Rabinovitch et al, 2001).
Calbindin D 28k is a well-characterized Ca2+-binding protein; its major role is to buffer and transport Ca2+ to maintain calcium homeostasis (Heizmann and Braun, 1992). Expression of CaBD increases in many central nervous system diseases, such as Alzheimer's disease (Geula et al, 2003), transient forebrain ischemia (Hwang et al, 2003), and spinal ischemia (Lee et al, 2005), which may be associated with resistance to neuronal degeneration. Overexpression of CaBD could decrease neuronal death after transient focal ischemia (Yenari et al, 2001). In contrast to the low transfection, PTD can deliver its cargo into brain cells more efficiently across the blood—brain barrier into neurons and astrocytes in the cerebral cortex, striatum, and medulla oblongata (Asoh et al, 2005; Cao et al, 2002; Kilic et al, 2003). After injection of 50 μg/mL PTD-CaBD, we detected a number of anti-histidine-positive cells in the subventricular zone, cerebral cortex, and caudate putamen. The high efficiency of protein transduction makes it possible to apply systemically PTD fusion proteins in stroke therapy. Actually, we found that the infarct volume decreased and the neurologic recovery improved in the PTD-CaBD-treated rats compared with the non-treated control group. Although the neuroprotective effect of CaBDs appears controversial (Airaksinen et al, 1997; Bouilleret et al, 2000; Kiyama et al, 1990; Simic et al, 1999), our data indicate that the PTD-CaBD could be delivered into brain cells across the blood—brain barrier, and thus buffer ischemia-induced Ca2+ overload and attenuate the brain injury. Admittedly, organ-specific delivery remains a complex challenge with respect to the utility of this technology (Noguchi and Matsumoto, 2006) because systemic administration of PTD fusion proteins can be distributed in the liver, kidney, spleen, lung, bowel, and heart (Cai et al, 2006), which might cause unexpected side effects. Interestingly, in our experiments, we did not find detectable side effects after PTD-CaBD injection. This may result from the fact that the exogenous CaBD does not block the calcium channels but buffers the excessive intracellular calcium; therefore, normal calcium influx maintains the physiologic calcium homeostasis to avoid cell injury.
Brain ischemia not only leads to acute cell death in the ischemic core, but also initiates cell apoptosis, which is the major component of delayed neuronal injury in the boundary area (Kametsu et al, 2003; Nitatori et al, 1995). Ca2+ overload in mitochondria induced by ischemia activates caspase-9, which subsequently activates caspase-3, initiating the mitochondria-dependent apoptosis pathway (Chen et al, 2005; Miyamoto et al, 2005). Our results showed that delivery of PTD-CaBD significantly decreased TUNEL- and cleaved caspase-3-positive cells in the perifocal region, which suggests that PTD-CaBD could inhibit ischemia-induced apoptosis. This antiapoptotic effect results not only from its ability to chelate calcium and therefore inhibit caspase-9 activation, but also from its ability to directly bind and inhibit caspase-3 activity (Bellido et al, 2000).
Footnotes
Acknowledgements
We thank Huimin Ren, PhD, and neuroscientist in the Fudan University for experiment assistance. We also thank Jiahong Lu PhD, MD, and Qiang Dong, PhD, MD, Neurologists in the Fudan University, for the useful suggestions about the project.