
Abstract
Admission hyperglycemia complicates approximately one-third of acute ischemic strokes and is associated with a worse clinical outcome. Both human and animal studies have showed that hyperglycemia is particularly detrimental in ischemia/reperfusion. Decreased reperfusion blood flow has been observed after middle cerebral artery occlusion in acutely hyperglycemic animals, suggesting the vasculature as an important site of hyperglycemic reperfusion injury. This paper reviews biochemical and molecular pathways in the vasculature that are rapidly affected by hyperglycemia and concludes that these changes result in a pro-vasoconstrictive, pro-thrombotic and pro-inflammatory phenotype that renders the vasculature vulnerable to reperfusion injury. Understanding these pathways should lead to the development of rational therapies that reduce hyperglycemic reperfusion injury and thus improve outcome in this large subset of acute ischemic stroke patients.
Clinical Studies of Hyperglycemia in Acute Ischemic Stroke
Numerous observational studies have investigated the association between hyperglycemia and stroke outcome. The largest of these retrospective studies all found that admission hyperglycemia was associated with a worse clinical outcome (Moulin et al, 1997; Weir et al, 1997; Bruno et al, 1999). Reanalysis of the TOAST trial revealed an odds ratio of 0.82 for favorable outcome for each 5 mmol/L increase in admission glucose (Bruno et al, 1999). Diabetic and non-diabetic patients alike are worsened by hyperglycemia (Pulsinelli et al, 1983); however, ischemic stroke patients without a history of diabetes appear most affected by high admission glucose (Capes et al, 2001). Meta-analysis revealed a more than three-fold increase in 30-day mortality for non-diabetic hyperglycemics, compared with a two-fold increase for diabetic patients who were hyperglycemic on admission (Capes et al, 2001).
In patients receiving thrombolytic therapy, hyperglycemia independently predicts intracerebral hemorrhage, whether administered intravenously (OR 2.26) or intra-arterially (RR 4.2; Demchuk et al, 1999; Kase et al, 2001). In these studies, the symptomatic hemorrhage rates were 25% and 36% for patients with glucose levels >11.1 mmol/L (200 mg/dl). Hyperglycemia has effects beyond increasing the likelihood of hemorrhage, as it predicts poor outcome when patients with intracerebral hemorrhage are excluded. Alvarez-Sabin and co-workers found admission glucose >7.7 mmol/L (140 mg/dl) to be the only independent predictor of poor outcome at 3 months despite thrombolysis-induced recanalization of the affected vessel (OR: 8.4). Among patients who did recanalize, median infarct volume was approximately three times larger in hyperglycemic patients (96 versus 32.2 cm3). Hyperglycemia was not associated with a larger infarct size in patients who failed to recanalize within the 6-h monitoring period, suggesting that hyperglycemia was detrimental in tissue plasminogen activator (tPA)-treated patients only if early reperfusion occurred (Alvarez-Sabin et al, 2003).
Mechanistic Studies of Hyperglycemia in Acute Ischemic Stroke
Human studies have clearly showed that hyperglycemia is associated with poor outcome after acute ischemic stroke. Imaging studies suggest that metabolic derangements contribute to worse outcome (Parsons et al, 2002); however, human studies cannot distinguish cause and effect since they cannot control for the severity of the initial ischemic insult. Animal models can fill this void and assess the relative contributions of metabolic derangement and vascular injury to outcome. Using transient middle cerebral artery occlusion (tMCAO) models in rats made hyperglycemic, short-term ischemia (e.g. 30 mins) does not impair reperfusion blood flow and worsens infarct size predominantly through metabolic derangement (Gisselsson et al, 1999). However, prolonged ischemia before reperfusion (e.g. ≥90 mins) results in markedly poor reperfusion and increases the incidence of hemorrhagic transformation (Venables et al, 1985; Kawai et al, 1997; Quast et al, 1997).
Kawai and co-workers induced hyperglycemia by intraperitoneal glucose injection 20 mins before a 4-h tMCAO. After 2 h of reperfusion, cerebral blood flow (CBF) in the ischemic hemisphere of hyperglycemic rats was 43%±13% of the non-ischemic hemisphere, versus 106%±7% in normoglycemic controls. 2 h of ischemia followed by 2 h of reperfusion resulted in similarly reduced CBF levels in hyperglycemic rats (53%±14%) relative to controls (109%±18%). These reductions in CBF were paralleled by increases in infarct size: 157.6±37.5 mm3 in hyperglycemic rats versus 39.1±22.9 mm3 in controls (Kawai et al, 1997).
Other studies have identified the peri-infarct region as the primary site of reduced CBF. Venables et al (1985) occluded the MCA for 2 h in cats made acutely hyperglycemic by glucose infusion and found that, after 1 h of reperfusion, penumbral blood flow fell to 60% of pre-ischemic values in hyperglycemic cats, versus 89% in normoglycemic cats. Cerebral blood flow values in the ischemic core were not different from controls, likely reflecting loss of autoregulation in these severely damaged vessels. Quast et al found a similar reduction in penumbral CBF using perfusion MRI. Hyperglycemia was induced in rats by streptozotocin (STZ) 3 days before 2 h of tMCAO. After 2 h of reperfusion, penumbral CBF was reduced by 37% in hyperglycemic rats, matching an increase in acute ischemic territory as showed by diffusion imaging (Quast et al, 1997). Many of these animal studies also showed increased hemorrhage rates in hyperglycemic animals (Venables et al, 1985; Kawai et al, 1997), mimicking data from human studies.
Taken together, both animal and human studies suggest that the detrimental effects of hyperglycemia on stroke size and outcome are due in part to impaired effective reperfusion after recanalization of the affected vessel. We suggest that this impaired reperfusion results from hyperglycemia-induced exacerbation of reperfusion injury to the vessels themselves. These detrimental effects occur even when hyperglycemia is induced after ischemia (Venables et al, 1985). This rapid time course is consistent with human studies that find detrimental effects of hyperglycemia on short-term mortality, final infarct size and clinical outcome in non-diabetic patients who by definition have not had long-term hyperglycemia before the onset of ischemia (Capes et al, 2001; Parsons et al, 2002). Importantly, these studies implicate hyperglycemia—not diabetes per se—as a cause of poor outcome after hyperglycemic stroke.
Hyperglycemia in the Vasculature
Hyperglycemia induces a variety of biochemical changes within endothelial cells (Brownlee, 2001). Endothelial cells, including those in the cerebral vasculature, transport glucose via the GLUT-1 transporter (McEwen and Reagan, 2004). This facilitative transporter is not insulin-sensitive and continuously transports glucose; as a result extracellular hyperglycemia induces endothelial intracellular hyperglycemia (Mandarino et al, 1994). This intracellular hyperglycemia is the basis for many biochemical alterations involved in diabetic complications (Brownlee, 2001). These alterations have been best studied under conditions of chronic hyperglycemia and contribute to a number of diabetic complications, such as atherosclerosis and retinopathy (Beckman et al, 2002a; Brownlee, 2005). In this review, we discuss many of the same pathways, focusing on those changes that occur rapidly enough to explain the short time course of effect seen in the cerebral vasculature of hyperglycemic stroke models. We focus on the endothelium, both for its critical role in regulation of the vasculature and because its uptake of glucose is not insulin sensitive. Recent ultrastructural data shows that the hyperglycemic endothelium is severely damaged by ischemia as evidenced by abnormal mitochondrial morphology (Keep et al, 2005). Importantly, glucose uptake—not simply extracellular hyperglycemia—is required for impaired reperfusion in the rat hyperglycemic tMCAO model (Wei et al, 2003). This focus does not imply that the vascular smooth muscle is unaffected by hyperglycemia. Indeed, although decreased cerebrovascular tone can be seen with severe hyperglycemia (44 mmol/L; 792 mg/dL; Cipolla et al, 1997), less severely hyperglycemic pial arterioles (20 mmol/L; 360 mg/dL) denuded of endothelium show increased contractile tone (Ward et al, 2002), a finding consistent with the phenotype described here with intact endothelium. Interestingly, functional glucose transporters are essential for normal vascular smooth muscle function (Park et al, 2005); however, the relationship between this normal function and the pathological phenotype induced by hyperglycemia is not yet understood. Given the unique qualities of cerebral endothelium, we focus on studies performed in cerebral vessels whenever available, and specify the tissue source if studies were performed in peripheral vessels.
Biochemistry of Hyperglycemia in the Endothelium
Intracellular hyperglycemia increases glycolysis and thus pyruvate, which drives the tricarboxylic acid (TCA) cycle. Increased flux through the TCA cycle causes increased activity of the electron transport chain and consequently an increased proton gradient across the inner mitochondrial membrane. This increased proton gradient stabilizes superoxide-generating electron transport molecules such as ubisemiquinone (Brownlee, 2001). The resultant production of reactive oxygen species (ROS) activates poly(ADP-ribose) polymerase (PARP), which inhibits glyceraldehyde 3-phosphate dehydrogenase (GAPDH) via covalent modification (Du et al, 2003). Glyceraldehyde 3-phosphate dehydrogenase inhibition blocks a late step of glycolysis, limiting flux though the TCA cycle, and causing a buildup of upstream glycolytic intermediates. This altered availability of glycolytic intermediates affects biochemical and signaling pathways including the pentose phosphate pathway (PPP) for nicotinamide adenine dinucleotide phosphate (NADPH) production, the diacylglycerol (DAG) pathway for protein kinase C (PKC) activation, the hexosamine and methylglyoxal pathways for post-translational modification of intracellular proteins, and the polyol pathway that increases the NADH:NAD+ ratio (Figure 1; Brownlee, 2001). Evidence implicating several of these pathways in hyperglycemic cerebrovascular dysfunction will be discussed below.
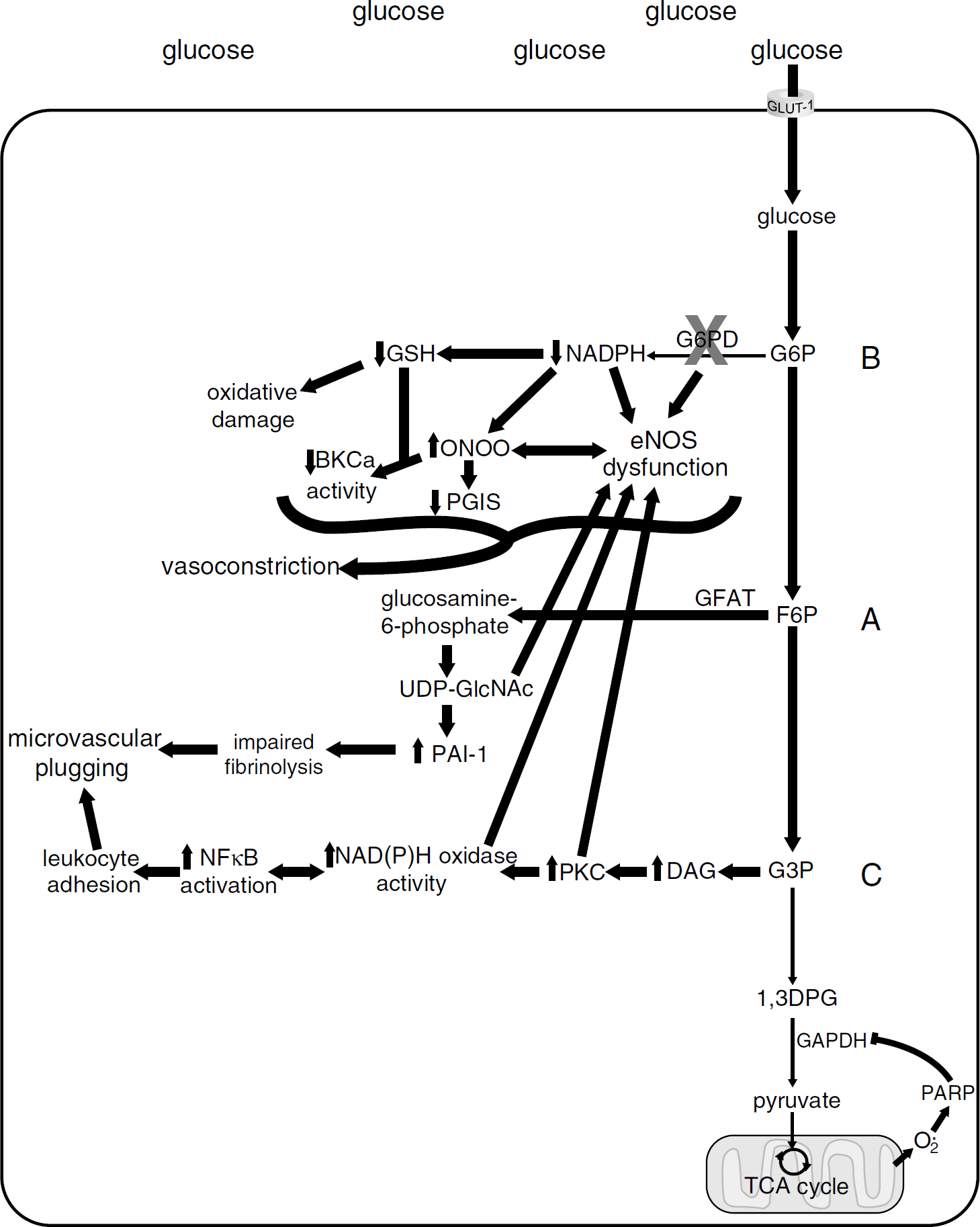
Endothelial biochemical pathways in hyperglycemia. Glucose enters the endothelial cell through the GLUT-1 facilitated transporter. Because GLUT-1 is not insulin sensitive, excessive extracellular glucose is transported into the cell, resulting in intracellular hyperglycemia. Increased intracellular glucose results in increased glycolysis, increased flux through the TCA cycle, and an increased proton gradient across the inner mitochondrial membrane. The increased proton gradient across the inner mitochondrial membrane stabilizes superoxide-generating components of the electron transport chain. The increased superoxide activates PARP, which causes covalent modification and partial inhibition of GAPDH. GAPDH inhibition limits production of pyruvate and flux through the TCA cycle, and by doing so increases levels of upstream glycolytic intermediates. The buildup of glycolytic intermediates alters a number of biochemical and cell signaling pathways (adapted from Brownlee, 2001). (
Hexosamine Pathway
The hexosamine pathway synthesizes uridine 5′-diphospho N-acetylglucosamine (UDP-GlcNAc)—used for dynamic post-translational O-acetylglucosamination of nucleocytoplasmic proteins—from glutamine and the glycolytic intermediate fructose-6-phosphate (Wells et al, 2003). Systemic endothelial cells exposed to high glucose produce excess UDP-GlcNAc (Wu et al, 2001). Cultured rat coronary microvascular endothelial cells exposed for 1 h to 15 mmol/L glucose contain five-fold more UDP-GlcNAc and those exposed to 30 mmol/L contain ∼9-fold more UDP-GlcNAc as compared with those exposed to 5 mmol/L glucose (Wu et al, 2001). Thus, hyperglycemia rapidly increases substrate availability for the post-translational modification of proteins through O-acetylglucosamination.
Increased flux through the hexosamine pathway affects human vessels, as carotid plaques from diabetics show a 6.6-fold increase in O-GlcNAc immunoreactivity (Federici et al, 2002). Dynamic O-acetylglucosamination of proteins is a post-translational modification that, similar to phosphorylation, can modulate protein function. As O-GlcNAc can inhibit phosphorylation—by either steric hindrance or competition for the same amino acid residues—it has been suggested that phosphorylation and O-acetylglucosamination reciprocally modulate protein function (Wells et al, 2003). Two such reciprocally modulated proteins particularly relevant to hyperglycemic vascular function are endothelial nitric oxide synthase (eNOS) and Sp1.
Endothelial nitric oxide synthase is a constitutively active enzyme and the predominant source of vascular nitric oxide (NO) under normal conditions. Nitric oxide serves many functions: it stimulates cGMP production to induce vasodilation in all types of blood vessels, inhibits adhesion of leukocytes and platelets to the vessel wall, and inhibits nuclear factor κB (NFκB) activation (Peng et al, 1995; Forstermann, 2006). Loss of NO bioavailability has been found under a number of pathological conditions, including hyperglycemia (Forstermann and Munzel, 2006). In response to 48 h of hyperglycemia, production of NO from eNOS decreased by 67% in bovine aortic endothelial cells (Du et al, 2001). This decreased activity is associated with a 1.85-fold increase in eNOS O-acetylglucosamination and a concomitant 45% decrease in eNOS phosphorylation at Ser1177. Similar changes in eNOS activity and post-translational modifications were observed in the aortae of diabetic rats and in human coronary endothelial cells cultured in 20 mmol/L glucose for 72 h (Federici et al, 2002). These hyperglycemia-induced changes were reversed by inhibition of the hexosamine pathway's rate-limiting enzyme, glutamine:fructose-6-phosphate amidotransferase (GFAT), revealing an important role for the hexosamine pathway in hyperglycemic eNOS inhibition (Du et al, 2001; Federici et al, 2002).
O-acetylglucosamination is also involved in transcriptional changes that occur as a result of hyperglycemia. Exposing bovine aortic endothelial cells to 30 mmol/L glucose for 48 h results in a 1.7-fold increase in O-acetylglucosamination of the transcription factor Sp1, accompanied by a 70% to 80% decrease in its phosphorylation (Du et al, 2000). These hyperglycemic conditions also induced two- to three-fold increases in expression from reporters containing promoters for the genes transforming growth factor beta 1 (TGF-β1) and plasminogen activator inhibitor-1 (PAI-1), the primary endogenous inhibitor of tPA. Both the hyperglycemia-induced post-translational modifications of Sp1 and the increases in PAI-1 and TGF-β1 reporter expression were prevented by treatments that decreased flux through the hexosamine pathway (Du et al, 2000). Studies in glomerular mesangial cells similarly showed that hyperglycemia induced Sp1-mediated increases in PAI-1 transcription, which were dependent on increased flux through the hexosamine pathway (Goldberg et al, 2002). Increased flux through the hexosamine pathway may similarly affect transcription in vascular smooth muscle, as Sp1 sites also mediate hyperglycemia-induced activation of PAI-1 in rat aortic smooth muscle cells (Chen et al, 1998). The cerebrovascular consequences of elevated PAI-1 levels are discussed in the PKC section.
O-acetylglucosamination in response to hyperglycemia affects the transcription of genes such as PAI-1, and the function of proteins such as eNOS. Although the time course of these effects has only been studied at 48 to 96 h (Chen et al, 1998; Du et al, 2000, 2001; Federici et al, 2002; Goldberg et al, 2002), hyperglycemia produces dramatically increased levels of the hexosamine pathway end product UDP-N-acetylglucosamine in as little as 1 h (Wu et al, 2001). It is thus possible that changes in protein function resulting from O-acetylglucosamination occur rapidly enough to contribute to the impaired reflow that occurs after hyperglycemic stroke (Figure 1A).
Pentose Phosphate Pathway
Endothelial cells exposed to high glucose become depleted of NADPH via inhibition of the PPP. Cultured bovine aortic endothelial cells exposed to 25 mmol/L glucose show inhibition of glucose-6-phosphate dehydrogenase (G6PD, the enzyme that shunts glucose-6-phosphate from glycolysis into the PPP) in as little as 15 mins, resulting in a 50% reduction of NADPH by 3 h (Zhang et al, 2000). Similarly, NADPH depletion has been attributed to impairment of the PPP in human umbilical vein endothelial cells exposed to 33 mmol/L glucose (Kashiwagi et al, 1997). Supporting the importance of this pathway, increasing flux through the PPP by transketolase stimulation prevented acute hyperglycemia-induced changes in cultured endothelial cells and prevented retinopathy after chronic hyperglycemia in rats with STZ-induced diabetes (Hammes, 2003). Nicotinamide adenine dinucleotide phosphate is a cofactor for a number of enzymes, including glutathione reductase, NAD(P)H oxidase, eNOS, and dihydrofolate reductase for tetrahydrobiopterin (BH4) biosynthesis (Stryer, 1995; Lassegue and Clempus, 2003). Further, NADPH is a powerful direct antioxidant in its own right, particularly because it binds enzymes or other macromolecules, protecting them from oxidative damage (Kirsch and de Groot, 2001).
Decreased NADPH results in depletion of reduced glutathione (GSH; Zhang et al, 2000), which is essential for redox signaling and detoxification/conjugation reactions (reviewed in Dickinson and Forman, 2002). Supporting the contention that the PPP is an important protection against oxidative stress, targeted disruption of the G6PD gene rendered murine embryonic stem cells susceptible to even mild oxidative stress (Pandolfi et al, 1995). Interestingly, this increased susceptibility to oxidative stress by G6PD deficiency is not a universal phenomenon. Glucose-6-phosphate dehydrogenase deficiency reduced vascular ROS production stimulated by angiotensin II (Matsui et al, 2005). However, ischemia/reperfusion—a particularly severe oxidative stress—depleted GSH stores and impaired contractile response in G6PD-deficient myocardium (Jain et al, 2004). An adequate supply of antioxidants/reducing agents limits the effects of ROS to the subcellular location in which they are produced, allowing cells to use ROS for intracellular signaling without altering their redox state (Lassegue and Clempus, 2003; Wolin, 2004). Hyperglycemia depletes human umbilical vein endothelial cells of GSH (Kashiwagi et al, 1994). We thus hypothesize that hyperglycemia is particularly detrimental because increases in vascular ROS (discussed below and in the PKC section) occur in the setting of NADPH and GSH depletion.
Hyperglycemia-induced oxidative stress in the systemic vasculature occurs in part through eNOS uncoupling (El Remessy et al, 2003; Ding et al, 2004; Cai et al, 2005). Uncoupling of eNOS refers to the enzyme's ability to produce superoxide instead of nitric oxide (Forstermann, 2006). Under optimal conditions, electron flow from NADPH to eNOS's heme center generates a ferrous-dioxygen intermediate that oxidizes
A number of studies have examined the relationship between hyperglycemia and eNOS uncoupling. Most studies using cultured aortic and retinal endothelial cells exposed to elevated glucose for 1 to 5 days showed 30% to 80% reductions in eNOS activity as reflected by NO production (Du et al, 2001; Cosentino et al, 2003; Ding et al, 2004; Cai et al, 2005), though others showed 12% to 30% increases in NO production (Cosentino et al, 1997; El Remessy et al, 2003). These decreases or slight increases in NO production occurred despite a doubling of eNOS expression (Cosentino et al, 1997, 2003; El Remessy et al, 2003; Ding et al, 2004; Cai et al, 2005), indicating that eNOS enzymatic activity for NO production is decreased in hyperglycemia. In response to hyperglycemia, superoxide production increased up to three-fold in some cases (Cosentino et al, 1997, 2003; El Remessy et al, 2003; Cai et al, 2005), the eNOS dimer:monomer ratio decreased by 30% to 70% (Zou et al, 2002; Cai et al, 2005), and BH4 levels decreased by 25% to 60% (Ding et al, 2004; Cai et al, 2005). This BH4 deficiency could be caused by either decreased production or increased degradation. Importantly, the decreased NO/increased superoxide production could be ameliorated with BH4 or NG-nitro-
One mechanism by which hyperglycemia could induce eNOS uncoupling is through PPP inhibition, as G6PD inhibition has been shown to promote eNOS uncoupling (Leopold et al, 2001). In bovine aortic endothelial cells, G6PD inhibition caused a marked increase in ROS in response to hydrogen peroxide (H2O2) that could be blocked by
Uncoupling of eNOS has been shown in many tissues and vascular beds (El Remessy et al, 2003; Bagi et al, 2004; Ding et al, 2004; Chu and Bohlen, 2004; Cai et al, 2005; Satoh et al, 2005). While direct evidence of eNOS uncoupling in the cerebral vasculature is lacking, several studies are consistent with this possibility. Kinoshita et al (1997) showed that BH4 depletion was associated with impaired NO-mediated dilations of the canine basilar artery, and that dilations could be restored to control values with superoxide dismutase. This study shows associations between BH4 depletion, eNOS dysfunction, and superoxide production consistent with eNOS uncoupling but does not identify eNOS as the source of superoxide. More evidence consistent with eNOS uncoupling comes from studies showing that
Based on these studies, it is likely that hyperglycemia-induced NADPH and GSH depletion would render the cerebral vasculature more susceptible to damage from the burst of ROS that occurs on reperfusion (Fabian et al, 1995). Hyperglycemia-induced PPP impairment might also promote eNOS uncoupling to impair vasodilation. Impaired NO-mediated vasodilation has been shown in the cerebral circulation in diabetes and acute hyperglycemia (Schwaninger et al, 2003; Sercombe et al, 2004; Didion et al, 2005). Although there are studies consistent with eNOS uncoupling in the cerebral vasculature (Quast et al, 1997; Kinoshita et al, 1997), eNOS uncoupling has yet to be conclusively showed in this vascular bed. However, the rapidity of PPP inhibition and its potential effects on the cerebral vasculature are consistent with hyperglycemia's particularly detrimental role in acute ischemic stroke with reperfusion (Figure 1B).
Activation of Protein Kinase C
Hyperglycemia results in increased DAG synthesis through reduction and progressive acylation of the glycolytic intermediate dihydroxyacetone phosphate (Brownlee, 2001). As DAG recruits PKC to the plasma membrane for activation, de novo DAG synthesis appears to be the mechanism by which hyperglycemia causes PKC activation. In the vasculature hyperglycemia primarily activates beta and delta isoforms (Brownlee, 2001), both of which are expressed prominently in cerebral microvessels (Krizbai et al, 1995).
Activation of PKC in response to hyperglycemia has been documented repeatedly in vascular cells and tissues after short term as well as prolonged glucose exposure, indicating an acute response as well as a sustained elevation (Brownlee, 2001). Pieper and Riaz (1997) saw PKC-dependent responses in bovine aortic endothelial cells as early as 1 h after culture in 35 mmol/L glucose. Others have found a similarly rapid time course: after 1 h of exposure to 22.2 mmol/L glucose, membrane-bound PKC activity of human aortic endothelial cells (Cosentino et al, 2003) and bovine aortic endothelial cells (Kouroedov et al, 2004) approximately doubled. Thus, activation of PKC occurs rapidly enough to account for hyperglycemic cerebrovascular reperfusion injury.
Protein kinase C activation in response to hyperglycemia has been linked to stimulation of ROS production by NAD(P)H oxidase, pro-inflammatory gene expression through NFκB activation, and decreased vasodilation linked to eNOS dysfunction (Brownlee, 2001). Hyperglycemia also increases production of PAI-1, TGF-β1 and vascular endothelial growth factor in a PKC-dependent manner (Figure 1C; Brownlee, 2001).
Protein Kinase C-Dependent Reactive Oxygen Species
Hyperglycemia induces PKC-dependent increases in ROS that are detrimental in hyperglycemic tMCAO (Hannah et al, 2005). Increased ROS in response to hyperglycemia have been attributed to NAD(P)H oxidase activity in the systemic (Inoguchi et al, 2000; Guzik et al, 2002; Cosentino et al, 2003; Sonta et al, 2004) as well as the cerebral circulations (Kusaka et al, 2004). Expression of certain NAD(P)H oxidase subunits was increased by 30% to 50% in rat brains after 8 weeks of STZ-induced diabetes and was similarly increased by 2 h tMCAO (Kusaka et al, 2004). Interestingly, 2 h tMCAO in diabetic rats increased NAD(P)H oxidase subunit expression to much higher levels than either diabetes or ischemia/reperfusion alone. These increases in NAD(P)H oxidase subunit expression were correlated with infarct sizes that were more than twice as large as controls, more severe neurological deficits, and more brain edema. These deficits as well as the increases in lipid peroxidation and NAD(P)H oxidase subunit expression were normalized by an angiotensin-receptor blocker (Kusaka et al, 2004), suggesting, based on interactions of the angiotensin receptor with NAD(P)H oxidase signaling, that NAD(P)H oxidase-mediated increases in ROS contributed to poor outcome in this model of hyperglycemic ischemia/reperfusion. Direct intervention with specific NAD(P)H oxidase inhibitors, however, is needed to establish causality.
The reaction of superoxide—such as that produced from NAD(P)H oxidase—with NO produces peroxynitrite, so the consequences of excessive NAD(P)H oxidase activation include both consumption of NO as well as peroxynitrite formation (Forstermann and Munzel, 2006). Consumption of NO by superoxide has been postulated as the main detrimental effect of NAD(P)H oxidase in hyperglycemia (Cosentino et al, 1997), and NAD(P)H oxidase activity does impair NO-mediated basilar artery vasodilation within a few hours of exposure to hyperglycemia (Sercombe et al, 2004). It is important to note, however, that BH4 is particularly sensitive to oxidation by peroxynitrite (Kuzkaya et al, 2003). Thus, excessive production of superoxide from NAD(P)H oxidase may initially decrease NO bioavailability by consumption, but as peroxynitrite levels rise and BH4 is depleted, eNOS may become uncoupled. In this way, an initial excess of ROS would lead to a more profound increase in ROS and decrease in NO (Forstermann and Munzel, 2006). Experiments establishing this mechanism in hypertension showed that aortic ROS were not increased in mice lacking either eNOS or the p47phox subunit of NAD(P)H oxidase (Landmesser et al, 2003).
Peroxynitrite may also affect cerebrovascular reactivity by inhibiting prostacyclin synthase (PGIS; Schmidt et al, 2003; Bachschmid et al, 2005) and Calcium-dependent K+ (BKCa) channels (Elliott et al, 1998; Brzezinska et al, 2000). Prostacyclin synthase is particularly susceptible to tyrosine nitration and inactivation by peroxynitrite (Schmidt et al, 2003). Prostacyclin synthase is expressed in human cerebral vessels (Siegle et al, 2000), and its decreased transcription is associated with an increased risk of ischemic stroke (Nakayama et al, 2000). Further, PGIS gene transfer reduced infarct volume in a rat ischemia—reperfusion model (Lin et al, 2002; Fang et al, 2006), demonstrating prostacyclin's importance in the cerebral circulation. Studies in human aortic endothelial cells revealed that hyperglycemia causes PGIS inactivation via tyrosine nitration (Zou et al, 2002), leading to decreased prostacyclin production and increased thromboxane production (Cosentino et al, 2003). Prostacyclin synthase inhibition causes accumulation of PGH2, which is also a substrate for thromboxane A2 synthase, and increases synthesis of the potent vasoconstrictor thromboxane A2 (TxA2; Bachschmid et al, 2005). Importantly PGH2 can also activate the dual-specificity TxA2 receptor to cause vasoconstriction, platelet aggregation, and possibly promote opening of the endothelial barrier (Bachschmid et al, 2005). Low dose (10 μmol/L) peroxynitrite can also inhibit BKCa channels causing constriction of rat MCAs and shortening of MCA vascular smooth muscle cells, effects prevented by GSH (Elliott et al, 1998; Brzezinska et al, 2000). It would be interesting to investigate this mechanism in hyperglycemia, given the concomitant peroxynitrite generation and GSH depletion. We speculate that inactivation of PGIS and possibly BKCa channels would contribute to the pro-constrictive, pro-thrombotic, and pro-inflammatory phenotype seen with hyperglycemic cerebral ischemia/reperfusion injury.
Hyperglycemia-induced ROS also appear to increase the activity of matrix metalloproteinase 9 (MMP-9; Uemura et al, 2001). The mechanisms involved are unclear, but S-nitrosylation can activate members of the matrix metalloproteinase family (Gu et al, 2002), raising the intriguing possibility that hyperglycemia may promote MMP-9 activation via peroxynitrite. Matrix metalloproteinase 9 is a zinc endopeptidase that degrades components of the extracellular matrix. Matrix metalloproteinase 9 mRNA levels are increased in bovine retinal endothelial cells cultured in high glucose for 24 h as well as in the retinas of 12-week diabetic rats (Giebel et al, 2005). Bovine aortic endothelial cells exposed to hyperglycemic conditions for 2 weeks showed elevated MMP-9 activity, associated with an increase in ROS, which could be ameliorated by free radical scavengers (Uemura et al, 2001). In normal human subjects, an oral glucose load (with its attendant mild hyperglycemia) more than doubled plasma MMP-9 levels by 90 mins; however, MMP-9 levels were undetectable in the plasma of diabetic subjects both at baseline and after oral glucose challenge, perhaps suggesting compensatory regulation of MMP-9 activity with chronic hyperglycemia (Sampson et al, 2004). In rodent models, MMP-9 expression and activity levels in the cerebral vasculature are increased after ischemia/reperfusion, particularly in the presence of tPA (Tsuji et al, 2005). In tPA-treated stroke patients, elevated MMP-9 levels have been associated with hemorrhagic conversion (Montaner et al, 2003). Thus, in acute ischemic stroke the increased risk of hemorrhage seen with hyperglycemia might be explained in part by increased MMP9 activity, though much remains to be understood about these relationships including the time course and mechanism(s) of effect.
Protein Kinase C-Induced nuclear factor κB Activation
As NO inhibits NFκB activation (Peng et al, 1995), NO consumption by superoxide would lead to NFκB activation. Nuclear factor κB is rapidly and dramatically activated in vascular cells in response to hyperglycemia: studies of cultured endothelial cells exposed to high glucose show PKC-dependent increases in NFκB activation after as little as 1 h, peaking at 4 h (Pieper and Riaz, 1997; Morigi et al, 1998; Du et al, 1999). Morigi et al (1998) showed that NFκB activation in response to elevated glucose resulted in a subsequent 458% increase in leukocyte adhesion, mediated by intracellular adhesion molecule 1 (ICAM-1), vascular cellular adhesion molecule 1 (VCAM-1) and E-selectin. Antioxidants prevented hyperglycemia-induced NFκB activation (Du et al, 1999; Hattori et al, 2000), and hyperglycemia-mediated increases in VCAM-1 expression can be attenuated with isoform-specific PKC inhibitors (Kouroedov et al, 2004), implicating oxidative stress and PKC activation as causal mechanisms. Thus, NFκB activation in response to hyperglycemia results in a pro-inflammatory endothelium, and these changes occur rapidly enough to participate in hyperglycemia-induced cerebrovascular reperfusion injury.
Ischemia/reperfusion activates NFκB by 2 h of reperfusion after tMCAO (Han et al, 2003), and maximally by 30 to 45 mins after hypoxia/reoxygenation of human brain microvascular endothelial cells (Howard et al, 1998). In human cerebral endothelial cells, hypoxia-induced NFκB activation causes dramatic upregulation of ICAM-1, VCAM-1 and E-selectin (Howard et al, 1998; Manduteanu et al, 2003), suggesting that NFκB activation promotes leukocyte adhesivity in the cerebral endothelium just as it does in systemic endothelium. The leukostasis resulting from increased NFκB-directed expression of adhesion molecules would be expected to be detrimental to microvascular perfusion after stroke (del Zoppo and Mabuchi, 2003). Nuclear factor κB activation also participates in the impairment of NO-mediated vasodilation induced by NAD(P)H oxidase after tMCAO (Xie et al, 2005).
We are not aware of studies that have specifically examined NFκB activation in stroke models complicated by acute hyperglycemia. It seems likely that reperfusion with hyperglycemic blood would result in greater initial NFκB activation than reperfusion with normoglycemic blood; however, this remains to be tested. As the degree of initial NFκB activation correlates with the degree of ischemia/reperfusion injury in the liver (Fan et al, 2004), overzealous NFκB activation seems a promising mechanism by which hyperglycemia could exacerbate vascular ischemia/reperfusion injury in stroke.
Protein Kinase C-Dependent Plasminogen Activator Inhibitor-1 Expression
In cultured human brain-derived endothelial cells, PKC mediates enhanced expression of PAI-1 (Zidovetzki et al, 1999). Plasminogen activator inhibitor-1 binds tPA or urokinase plasminogen activator with 1:1 stoichiometry rendering them inactive (Horrevoets, 2004). Mice lacking PAI-1 achieve carotid recanalization more quickly than their wild-type littermates, both spontaneously and after tPA administration (Farrehi et al, 1998; Zhu et al, 1999; Konstantinides et al, 2001; Matsuno et al, 2002). These studies show a role for PAI-1 in stabilizing arterial clots and preventing their premature dissolution.
Hyperglycemia increases PAI-1 expression in cultured vascular cells and in vivo. In bovine aortic endothelial cells exposed to 30 mM glucose for 48 h, PAI-1 transcription increased ∼3-fold (Du et al, 2000). Experiments with vascular smooth muscle cells indicate that upregulation of PAI-1 in response to acute hyperglycemia occurs rapidly: PKC-mediated PAI-1 mRNA expression in rat aortic vascular smooth muscle cells increased by 2 h, peaked at 4 h, then returned to baseline after 24 h of exposure to 27.5 mmol/L glucose (Suzuki et al, 2002). Similarly, free PAI-1 antigen levels in human umbilical vascular smooth muscle cells had more than tripled by 6 h after exposure to 20 mmol/L glucose (Pandolfi et al, 1996). In response to acute hyperglycemia, the vasculature increases PAI-1 expression, resulting in decreased fibrinolysis. In rats, 4 h of hyperglycemia reduced plasma fibrinolytic potential by more than 50% and increased PAI-1 activity more than four-fold (Pandolfi et al, 2001). Thus, hyperglycemia rapidly increases PAI-1 activity.
The vascular consequences of increased local PAI-1 activity encompass macrovascular effects such as impaired thrombolysis, as well as microvascular effects. In a study of 44 stroke patients, elevated PAI-1 levels independently predicted failure to recanalize the MCA despite thrombolysis within 3 h of symptom onset (Ribo et al, 2004). Conversely, lower PAI-1 levels correlated with vessel recanalization, dramatic clinical improvement at 12 h, and greater functional independence at 90 days. Admission glucose also independently predicts failure of the MCA to recanalize after thrombolytic therapy (Ribo et al, 2005). Microvascular effects of increased PAI-1 activity include plugging and fibrin deposition in smaller vessels, which would impair effective reperfusion (del Zoppo and Mabuchi, 2003). Within 4 h after permanent MCAO, PAI-1 expression increased in rat precapillary arterioles, capillaries, and post-capillary venules. This increased PAI-1 expression correlated with fibrin deposition and a microvascular plasma perfusion deficit, which expanded from the ischemic core at 1 h to include the cortex by 4 h (Zhang et al, 1999). Thus, increased PAI-1 seems to impair both macrovascular and microvascular reperfusion.
There is evidence that elevated PAI-1 levels contribute to hyperglycemia-induced exacerbation of ischemia/reperfusion injury in stroke models. One study evaluated expression and activity of PAI-1 and tPA after tMCAO in diabetic rats. Diabetic rats exhibited reduced fibrinolytic potential before ischemia that became more dramatic at 3 and 5 h of reperfusion, and infarct volume was five-fold larger in the diabetic rats (Liang et al, 2004). In rats rendered hyperglycemic shortly before tMCAO, a direct thrombin inhibitor reduced infarct size (Wei et al, 2004). Together, these studies show the deleterious effect of disturbed fibrinolytic balance in cerebral ischemia/reperfusion and suggest that hyperglycemia-induced reduction of fibrinolytic potential may exacerbate cerebral ischemia/reperfusion injury.
Protein Kinase C-Induced Changes in Vasomotor Tone
We previously discussed O-acetylglucosamination, PPP inhibition, and uncoupling as mechanisms by which eNOS activity might be decreased. Protein kinase C also plays a major role in loss of NO-mediated vasodilation (Bohlen and Nase, 2001). In vivo, human subjects exposed to hyperglycemia for 6 h exhibited impaired brachial artery vasodilation to methacholine, an effect ameliorated by pretreatment with a PKCβ2 inhibitor (Beckman et al, 2002b). Protein kinase C also affects vasomotor tone in the cerebral circulation, as pial arterioles exposed to 20 or 25 mmol/L glucose for 30 mins exhibited impaired endothelium-dependent relaxation via activation of PKC (Mayhan and Patel, 1995). The precise mechanisms by which PKC impairs NO-mediated vasodilation in the cerebral circulation are not fully understood but may include direct effects on eNOS post-translational modification: endothelial PKC activation can stimulate eNOS dephosphorylation at Ser1177 and phosphorylation at Thr495, both of which decrease NO production (Fleming and Busse, 2003). Alternatively, PKC may impair eNOS function indirectly through effects on NAD(P)H oxidase, as described above. It appears that multiple mechanisms act in concert to impair vascular NO bioavailability in hyperglycemia.
Differential Response of Hyperglycemic Vessels to Reperfusion after Acute Ischemic Stroke
The beneficial effect of
These observations and the data reviewed above indicate multiple mechanisms by which hyperglycemia leads to a pro-constrictive, pro-thrombotic and pro-inflammatory phenotype and suggests the following potential scenario by which hyperglycemia worsens reperfusion blood flow in transient cerebral ischemia: Ischemia initiates transcription of genes by hypoxia inducible factor 1α, such as PAI-1 and the potent cerebral vasoconstrictor endothelin 1 (ET-1) (Sharkey et al, 1993; Fink et al, 2002; Virley et al, 2004; Semenza, 2004). On reperfusion, oxygen-rich blood returns to the ischemic tissue, providing abundant oxygen substrate for production of vascular superoxide by NAD(P)H oxidase (Xie et al, 2005), and perhaps uncoupled eNOS as well as xanthine oxidase (Beetsch et al, 1998). Reactive oxygen species production increases in both normoglycemic and hyperglycemic animals, though the increase is more dramatic in hyperglycemic animals (Wei and Quast, 1998). In normoglycemic vessels, the benefits of restoring oxygen and glucose to the ischemic tissue, if performed rapidly enough, outweigh the detrimental effects of superoxide on NO-mediated vasodilation and NFκB activation. Coupled eNOS produces NO, helping counteract the vasoconstrictive effects of ET-1 and the inflammatory effects NFκB activation. In hyperglycemic vessels, however, eNOS may be uncoupled and—in the presence of oxygen—produces superoxide at the expense of nitric oxide. This excess superoxide, in combination with the lack of NO production due to eNOS uncoupling, would lead to an imbalance favoring vasoconstriction. Peroxynitrite may also promote impaired cerebral vasodilation due to PGIS and BKCa channel inactivation. NADPH depletion and inadequate GSH levels may render hyperglycemic vasculature particularly susceptible to peroxynitrite's damaging effects. In addition, ROS and perhaps peroxynitrite may promote MMP-9 activation and increase the risk of hemorrhage. Figure 2 illustrates our model of the detrimental effects of reperfusion after acute ischemic stroke complicated by hyperglycemia.
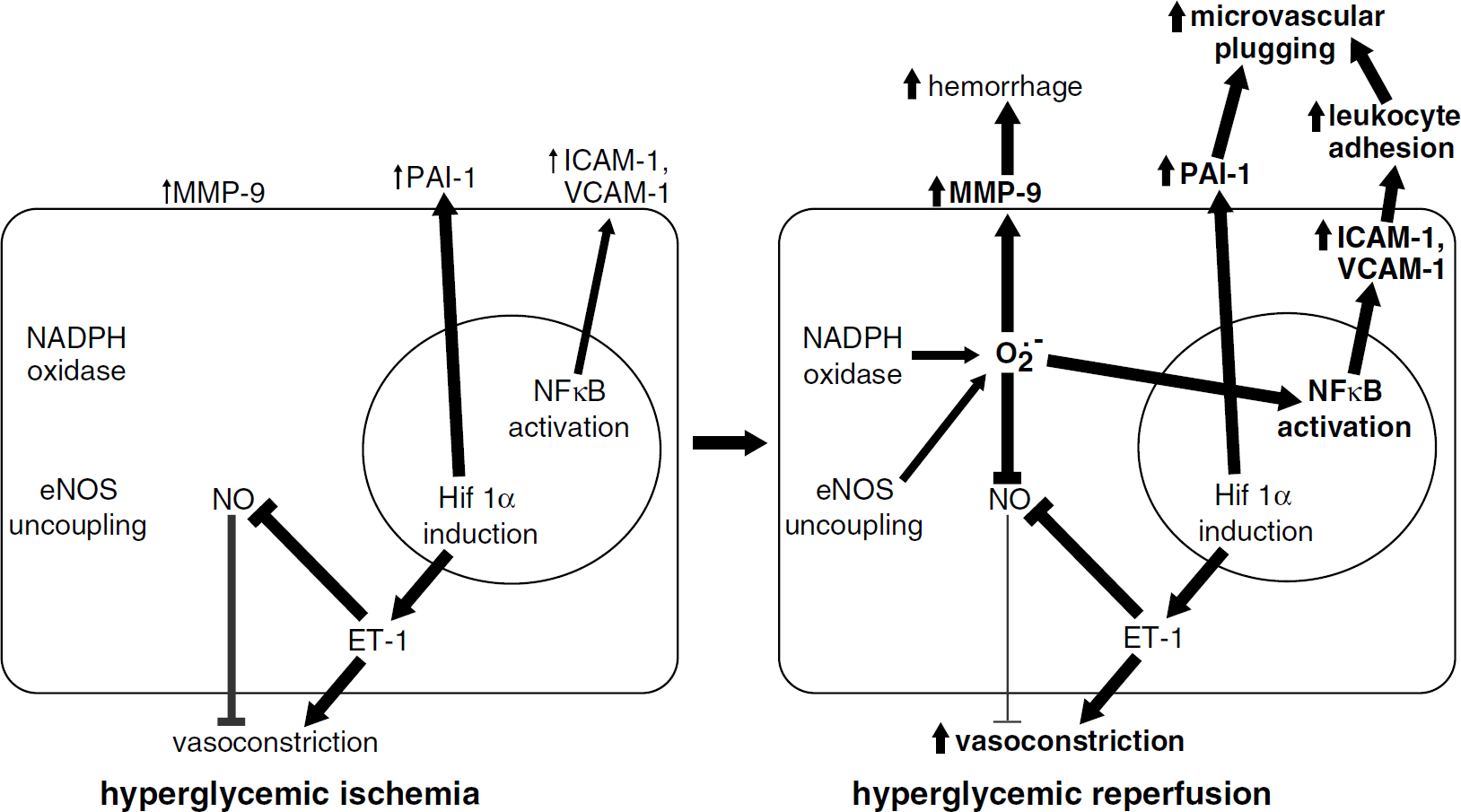
Detrimental effect of reperfusion under hyperglycemic conditions. (
Therapeutic Targets
In this review, we have summarized a number of biochemical effects of acute hyperglycemia on the vasculature. Acute hyperglycemia depletes endothelium and vascular smooth muscle of NADPH, reducing intracellular antioxidant potential. Protein kinase C activation increases ROS production, activates NFκB, and contributes to eNOS dysfunction. Acute hyperglycemia also results in post-translational modifications of proteins—including transcription factors—via the hexosamine pathway. The hexosamine and PKC pathways increase levels of PAI-1, promoting thrombus stabilization and microvascular plugging. These biochemical responses to hyperglycemia induce pro-inflammatory, pro-thrombotic and pro-vasoconstrictive changes in the vasculature in a matter of hours.
Perhaps the most obvious potential therapy for hyperglycemia in acute stroke is treatments that establish and maintain normoglycemia. Hyperglycemia is in general poorly controlled in acute stroke (Bruno et al, 1999). A recent retrospective study of acute ischemic stroke patients reported increased mortality in those with admission glucose >130; however, patients with admission hyperglycemia that normalized within the next 48 h (either spontaneously or with insulin and/or oral hypoglycemic agents) had a similar mortality rate as persistently normoglycemic patients (Gentile et al, 2006). These results suggest that control of hyperglycemia in acute stroke may indeed be beneficial. Alternatively, the results may reflect fundamental differences in stroke patients with easy-to-control versus hard-to-control hyperglycemia and thus must be confirmed prospectively.
There is increasing evidence that intensive insulin therapy benefits critically ill patients. A study of surgical ICU patients showed that intensive intravenous insulin therapy to maintain normoglycemia reduced the risk of organ failure and death (van den Berghe et al, 2001). Target blood glucose levels were 10.0 to 11.1 mmol/L (180 to 200 mg/dl) in the conventional treatment group and 4.4 to 6.1 mmol/L (80 to 110 mg/dl) in the intensive insulin therapy group. More than 1/3 were insulin-requiring diabetic patients. A preplanned subanalysis of this study revealed decreased NFκB activation and reduced levels of ICAM-1 and E-selectin (known NFκB targets) in subjects who received intensive insulin therapy (Langouche et al, 2005), indicating that intensive insulin therapy is capable of reversing at least some effects of hyperglycemia on the vascular endothelium. Beyond its ability to control blood glucose, insulin may exert additional protective effects on the endothelium. Insulin acutely increases aortic NO production via the PI3K/Akt pathway (Hartell et al, 2005). Akt has previously been shown to physically interact with and promote the activity of eNOS (Takahashi and Mendelsohn, 2003). In animal models, insulin therapy to normalize hyperglycemia reduces infarct volume after tMCAO (Yip et al, 1991; Hamilton et al, 1995). In later work this group examined the relationship between blood glucose level and cortical damage from tMCAO, and found that the least cortical damage occurred at blood glucose levels of 6 to 7 mmol/L (108 to 126 mg/dl). Importantly, both hypoglycemia and hyperglycemia increased infarct volume (Zhu and Auer, 2004). These results underscore the potential risks involved in overcorrecting elevated blood glucoses in acute ischemic stroke patients.
Insulin infusion protocols have been developed (Meijering et al, 2006) and are being increasingly utilized in the critical care setting. Clinically, intensive insulin therapy is highly resource-intensive and increases the potential for hypoglycemia. It remains to be seen if there is sufficient benefit to outweigh these risks and costs. Outcomes in van den Berghe et al (2001) did not show clear evidence for the benefit of tight glycemic control in stroke patients, but the number studied was small and their presence in the surgical ICU suggests they were not representative of all stroke patients (van den Berghe et al, 2001). Ongoing trials specifically in acute ischemic stroke are addressing the questions of safety as well as efficacy. The United Kingdom Glucose Insulin Stroke Trial (GIST-UK), including patients without a history of insulin-dependence, and Treatment of Hyperglycemia in Ischemic Stroke (THIS), including all hyperglycemic stroke patients, are multicenter randomized trials that seek to determine the feasibility and benefit of intensive insulin therapy in acute stroke.
An agent that shows promise for establishing and maintaining normoglycemia is Glucagon-like peptide 1 (GLP-1). Glucagon-like peptide 1 is a short-lived endogenous peptide whose actions include glucose-dependent stimulation of insulin secretion/inhibition of glucagon secretion from the pancreas (D’Alessio et al, 2005). Glucagon-like peptide 1 is strictly glucose dependent, so it does not continue to decrease blood glucose once normoglycemia is attained (Nauck et al, 1993). In a small study of postoperative type II diabetic patients, GLP-1 infusion decreased blood glucose from 10 mmol/L (180 mg/dL) to ∼7 mmol/L (126 mg/dL) within 2 h and ∼6 mmol/L (108 mg/dl) within 3 h without hypoglycemic episodes (Meier et al., 2004). Glucagon-like peptide 1 infusion also lowered blood glucose in healthy subjects undergoing glucose infusion, indicating that the effects of GLP-1 are not limited to diabetic patients (Nauck et al, 2004). Similarly, a small pilot study found that GLP-1 infusion improved left ventricular function in both diabetic and non-diabetic patients after myocardial ischemia/reperfusion (Nikolaidis et al., 2004). Interestingly, in animal models GLP-1 can cross the blood—brain barrier (Kastin et al, 2002) and protect neurons against in vitro and in vivo excitotoxicity (Perry et al., 2002; During et al, 2003). Although not yet studied in stroke, the pleiotropic effects of GLP-1 may be of particular benefit in hyperglycemic stroke.
A novel approach that may benefit hyperglycemic acute ischemic stroke patients is exposing the ischemic territory to ultrasound. Our reanalysis of the multicenter CLOTBUST trial (Alexandrov et al, 2004) evaluating transcranial Doppler ultrasound as an adjunct to thrombolytic therapy suggests that ultrasound preferentially benefits 3-month outcome in hyperglycemic acute ischemic stroke patients, and that this benefit cannot be explained by improved recanalization of the affected vessel (Martini et al, 2006). A potential mechanism of this effect may involve actions of ultrasound on the endothelium to improve perfusion. In animal models, induction of permanent ischemia in heart and skeletal muscle decreases perfusion and causes tissue acidosis. In these settings, ultrasound increases capillary diameter, improves tissue perfusion, and normalizes acidosis through an NO-dependent mechanism (Suchkova et al, 2002; Siegel et al, 2004). In cultured endothelial cells, exposure to ultrasound increases NOS activity and NO production within seconds (Altland et al, 2004). This rapid time course suggests that ultrasound might act through signaling pathways that rapidly alter post-translational modifications (e.g., phosphorylation). Neither of these studies were performed in the presence of hyperglycemia; nevertheless, the detrimental effects of hyperglycemia on eNOS function—in conjunction with our results that hyperglycemic stroke patients may benefit from ultrasound therapy—have lead us to hypothesize that ultrasound might alter the phosphorylation state of eNOS in a manner beneficial to hyperglycemic stroke patients (Martini et al, 2006).
This review suggests that eNOS uncoupling might contribute to oxidative stress after hyperglycemic cerebral ischemia/reperfusion. If eNOS uncoupling contributes to the detrimental effect of hyperglycemia in ischemic stroke, then administration of agents that ameliorate eNOS uncoupling may be beneficial. The eNOS cofactor BH4 is one such agent. BH4 oxidation is increased and its biosynthesis is decreased in endothelial cells exposed to hyperglycemia (Ding et al, 2004; Cai et al, 2005), so hyperglycemic patients may have reduced levels of functional BH4. In cultured endothelial cells BH4 binding improves eNOS coupling, thus increasing production of NO and reducing production of superoxide (El Remessy et al, 2003). Improved NO production and decreased superoxide production would be expected to improve vasodilation, decrease platelet aggregation, and reduce oxidative stress. Indeed, BH4 supplementation has been shown to restore impaired NO-dependent vasodilation in normal human subjects after transient hyperglycemia (Ihlemann et al, 2003), suggesting that BH4 depletion and its effects on eNOS coupling may be physiologically relevant in humans. Acute BH4 treatment can also restore impaired NO-mediated vasodilation in diabetic patients and vessels (Heitzer et al, 2000; Guzik et al, 2002; Bagi and Koller, 2003). As such, BH4 may benefit both diabetic and non-diabetic hyperglycemics in the setting of acute stroke. Although BH4 is a potent antioxidant, at high levels it is also capable of auto-oxidation to produce superoxide and may induce contractions in the cerebral circulation (Kinoshita and Katusic, 1996). Further, there may be unintended effects on other BH4-dependent enzymes. Of note, an inhibitor of GTP-cyclohydrolase-1 (GTPCH1) and thus BH4 synthesis decreased iNOS activation and reduced infarct volume in a normoglycemic ischemia/reperfusion model (Kidd et al, 2005), raising the concern that BH4 treatment may not be benign, particularly for normoglycemic stroke patients. BH4 may be similar to
As endothelial dysfunction is a hallmark of hyperglycemia, statin therapy may benefit hyperglycemic acute ischemic stroke patients. In normoglycemic ischemic stroke models, both pre- and post-ischemia statin administration decrease infarct size and improve outcome (Sironi et al, 2003; Endres et al, 2004). Through pharmacological and molecular studies, this benefit of pre-ischemia statins has been attributed to upregulation of eNOS (Endres et al, 2004). It has been suggested that increased eNOS production might not be beneficial in situations where eNOS becomes uncoupled, such as hyperglycemia (Endres et al, 2004). A recent study indicates that this approach may indeed have promise. Using endothelial and vascular smooth muscle cell cultures as well as electromagnetic spin resonance measurements of oxidative stress in diabetic rats, Tsubouchi et al (2005) showed that statin therapy normalized the increased oxidative stress induced by hyperglycemia. Since oxidative stress is likely a key factor in hyperglycemic reperfusion injury after acute ischemic stroke, acute statin therapy may benefit hyperglycemic patients, but definitive conclusions await further study.
Conclusion
An approach to increasing the number of stroke patients that can benefit from treatment is to identify subgroups that respond poorly to existing treatments and to develop new or adjunctive therapies. We have termed this phenomenon ‘heterogeneity in acute ischemic stroke’ and identified hyperglycemic patients as a sizable subgroup that are deriving less benefit from present therapies (Kent et al, 2001). Through an understanding of the mechanisms by which hyperglycemia worsens outcome, rational therapies will likely emerge. Evidence reviewed here indicates that hyperglycemia generates a different vascular phenotype that is pro-constrictive, pro-inflammatory and pro-thrombotic. Within such a pro-constrictive phenotype, even successful recanalization may not result in parenchymal reperfusion. Despite the complexity of hyperglycemia's effects, there appear to be several potential key mechanisms. One of these is the production, through a variety of mechanisms, of ROS. The extent and rapidity of production of ROS after cerebral ischemia/reperfusion (Fabian et al, 1995) suggests that standard antioxidants, which scavenge free radicals with a 1:1 stoichiometry, are unlikely to be sufficiently beneficial (Brownlee, 2001). A more promising approach may include therapies that target the sources of ROS production, for example, NAD(P)H oxidase or uncoupled eNOS.
Although there is undoubtedly an involvement of metabolic derangement and neuronal injury in the detrimental effects of hyperglycemia in acute ischemic stroke, vascular injury appears to be an important factor that will limit the benefit derived from interventions that focus primarily on neuronal protection. In our view, it is unlikely that substantial progress will be made in the treatment of hyperglycemic stroke patients until the contribution of vascular injury to outcome is sufficiently appreciated.