
Abstract
The pathogenesis of cerebral ischaemia after subarachnoid haemorrhage (SAH) still remains elusive. The purpose of the present study was to examine whether specific protein kinas C (PKC) inhibition in rats could alter the transcriptional SAH induced Endothelin (ET) type B and 5-hydroxytryptamine type 1B (5-HT1B) receptor upregulation and prevent the associated cerebral blood flow (CBF) reduction. The PKC inhibitor RO-31-7549 or vehicle was injected intracisternally after the induced SAH in rats (n = 3 to 10 in each groups for each method). The involvement of the PKC isoforms was investigated with Western blot; only PKCδ and PKCα subtypes were increased after SAH RO-31-7549 treatment abolished this. At 2 days after the SAH basilar and middle cerebral arteries were harvested and the contractile response to endothelin-1 (ET-1; ETA and ETB receptor agonist) and 5-carboxamidotryptamine (5-CT; 5-HT1 receptor agonist) were investigated with a myograph. The contractile responses to ET-1 and 5-CT were increased (P < 0.05) after SAH compared with sham operated rats. In parallel, the ETB and 5-HT1B receptor mRNA and protein expression were significantly elevated after SAH, as analysed by quantitative real-time polymerase chain reaction and immunohistochemistry, respectively. Administration of RO-31-7549 prevented the upregulated contraction elicited by application of ET-1 and 5-CT in cerebral arteries and kept the ETB and 5-HT1B receptor mRNA and protein levels at pre-SAH levels. Regional and global CBF evaluated by an autoradiographic technique were reduced by 60%±4% after SAH (P < 0.05) and prevented by treatment with rO-31-7549. Our study suggests that PKC plays an important role in the pathogenesis of cerebral ischaemia after SAH.
Introduction
The cerebral ischaemia that occurs after subarachnoid haemorrhage (SAH) often results in death or severe disability. Several theories have appeared to explain the mechanism responsible for the cerebral ischaemia and vasospasm that occur after SAH; however, it still remains that no specific treatment exists. Through many years 5-hydroxytryptamine (5-HT) was thought of as a factor and several studies have shown involvement of 5-HT in SAH (Zervas, 1979; Zervas et al, 1975) with an increase in CSF concentration (Allen et al, 1974). There has not been any clinical efficacy of 5-HT antagonists in treating the cerebrovascular effects of SAH, however this does not eliminate an involvement of 5-HT, but suggest a multifactor pathogenesis. It is well known that 5-HT induces strong contraction of cerebral arteries, but it was only recently observed that the main cerebrovascular 5-HT receptor subtype present in man is the 5-HT1B receptor (Nilsson et al, 1999). Consequently, previous studies had no specific antagonist in there analysis. Increased levels of endothelin-1 (ET-1) have been shown in patients suffering from cerebral ischaemia (Zimmermann and Seifert, 1998). Endothelin-1 is a potent vasoconstrictor produced in the endothelium of blood vessels, which mediates its effects through endothe-lin A (ETA) and endothelin B (ETB) receptors (Masaki et al, 1994).
We have earlier shown upregulation of the ETB and 5-HT1B receptors in the major cerebral arteries from rat after SAH, an upregulation that imparts increased sensitivity of the arteries towards their endogenous agonists (Hansen-Schwartz and Edvinsson, 2000; Hansen-Schwartz et al, 2003a, b). The studies, however, were not designed to show whether these findings had any relevance to the reduction in cerebral blood flow (CBF).
Activity of protein kinase C (PKC) has in several studies been linked to cerebral ischaemia and cerebral vasospasm associated with SAH (Nishizawa et al, 2003). It has been shown in vitro that PKC activity is crucial to the upregulation of the ETB receptor (Hansen-Schwartz et al, 2002; Henriksson et al, 2003). Hypothetically, PKC may be a key intracellular mediator involved in the upregulation of ETB and 5-HT1B receptors in cerebral arteries. In the present study, we are testing the hypothesis that in vivo PKC inhibition alters the degree of ET and 5-HT1 receptor upregulation and in addition prevents the marked CBF reduction that is seen after SAH. A sensitive in vitro pharmacological method was used to measure the contractile responses to ET-1 (Endothelin ETA and ETB receptor agonist), sarafotoxin 6c (S6c; selective Endothelin ETB receptor agonist) and 5-carboxamidotryptamine (5-CT; specific 5-HT1 receptor agonist). To investigate if PKC inhibition had an influence on the ET and 5-HT1 receptor protein and mRNA levels, we used immunohistochemistry and quantitative real-time polymerase chain reaction (PCR). Western blot was used to examine which of the PKC subtypes that were involved in SAH and if the PKC inhibition had an influence on their activation. The regional CBF (rCBF) in the experiments were evaluated by a quantitative autoradiographic technique (Sakurada et al, 1978).
Materials and methods
All animal procedures were performed strictly within national laws and guidelines and approved by the Danish Animal Experimentation Inspectorate and the Ethical Committee for Laboratory Animal Experiments at the University of Lund.
Rat Subarachnoid Haemorrhage Model
Subarachnoid haemorrhage was induced by a model originally devised by Svendgaard et al (Prunell et al, 2002) and carefully described by Prunell et al (2003). The studies analysed the volume of blood injected, the time course, changes in CBF and cerebral metabolism associated as signs of vasospasm and neurologic outcome. In a previous study using the same SAH model, Delgado et al (1985) have in angiographic examinations of the arteries revealed a biphasic vasospasm with a maximal acute spasm at 10 mins and a late maximal spasm at two days after SAH. The acute phase is defined as within 10 mins after SAH.
Male Sprague–Dawley rats (350 to 400 g) were anaesthetized using 5% halothane (Halocarbon Laboratories, River Edge, NJ, USA) in N2O/O2 (30:70). The rat was intubated and artificially ventilated with inhalation of 0.5% to 1.5% halothane in N2O/O2 (70:30) during the surgical procedure. The depth of anaesthesia was carefully monitored and the respiration checked by regularly withdrawing arterial blood samples for blood gas analysis (Radiometer, Copenhagen, Denmark). An electrical temperature probe was inserted into the rectum of the rat to record the temperature, which was maintained at 37°C. An arterial catheter to measure blood pressure was placed in the tail artery and a catheter to monitor intracranial pressure (ICP) was placed in the subarachnoid space under the subocciptal membrane. At either side of the skull, 3 mm from the midline and 4 mm anteriorly from the bregma, holes were drilled through the skull bone down to dura mater (without perforation) allowing the placement of two laser-Doppler flow probes to measure cortical CBF. Finally, a 27G blunt canula with side hole was introduced 6.5 mm anterior to bregma in the midline at an angle of 30° to the vertical. With the aperture pointing to the right, the needle was lowered until the tip reached the skull base 2 to 3 mm anterior to the chiasma. After 30 mins of equilibration, 250 ml blood was withdrawn from the tail catheter and injected intracranially via this canula at a pressure equal to the mean arterial blood pressure (MABP) (80 to 100 mm Hg). Subsequently, the rat was kept under anaesthesia for another 60 mins to allow recovery from the cerebral insult after which catheters were removed and incisions closed with the exception of the subocciptal catheter, which was fixed by suture and used for injection of PKC inhibitor or vehicle. The rat was then revitalized and extubated. A subcutaneous injection of carprofen (4.0 mg/kg) (Pfizer, Denmark) was administered, and the rat was hydrated subcutaneously using 40 ml isotonic sodium chloride at the end of the operation and at day one. During the period, the rat was monitored regularly, and if showing severe distress the animal was prematurely killed. In addition, a series of sham-operated rats were prepared. They went through exactly the same procedure as described above with the exception that no blood was injected intracisternally. The reason for not giving saline as control is based on preliminary observations that both the ICP rise by the injection and the administered blood contribute to the SAH induced effects. All surviving animals were neurologically examined using an established scoring system (Bederson et al, 1986; Menzies et al, 1992). After 2 days, either harvesting of vessels or autoradiographic measurements were performed (see below for details). The ischaemic brain damage was examined by staining coronal slices of the brains with 1% 2.3.5-triphenyltetrazolium chloride dissolved in saline solution at 37°C for 20 mins. The slices were examined with a dissection microscope.
Rat Subarachnoid Haemorrhage Model with Protein Kinas C Inhibition
This group of animals went through the same procedure as the above-mentioned SAH animals. In addition, they were treated with the PKC inhibitor RO-31-7549 (Calbiochem, Sweden) or vehicle in conjunction with the operation and after the induced SAH. All animals treated with RO-31-7549 received five injections intracisternally of RO-31-7549 or vehicle in similar volume. Thus, 50 ml 10−6mol/L of RO-31-7549 was injected intracisternally at 30 mins before the induced SAH and after the SAH 20 ml 10−6mol/L of RO-31-7549 was administered repeatedly after 3, 6, 24 and 32 h from the first RO-31-7549 injection. This dose was chosen on the basis of previous detailed work on isolated cerebral arteries (Hansen-Schwartz et al, 2002), the dose was chosen at near maximum inhibition and calculation of cerebrospinal fluid volume/turnover. RO-31-7549 is a selective PKC inhibitor with some isozyme specificity, primarily inhibiting classic PKCs (IC50 for PKCα = 10−7mol/L, PKCβI = 10−67mol/L, PKCβI = 10−68v, PKCγ = 10−67mol/L and PKCγ = 10−68mol/L) (Wilkinson et al, 1993).
Harvest of Cerebral Arteries
After 48 h of observation the sham, SAH treated with RO-31-7549 or SAH + vehicle operated rats (see above SAH model) were anaesthetized with CO2 and decapitated. The brains were quickly removed and chilled in ice-cold bicarbonate buffer solution (see composition below). Under a dissection microscope, the middle cerebral artery (MCA) and the basilar artery (BA) were carefully dissected free from the brain and cleared of connective tissue. The MCA and BA were immediately mounted in myographs for in vitro pharmacology or snap frozen at −80°C and examined by real-time PCR or immunohistochemistry. The brains were immediately snap frozen at −80°C and used for Western blot examination.
Autoradiographic Measurements of Regional Cerebral Blood Flow
Local CBF was measured by a model originally described by Sakurada et al (1978) and modified by Gjedde et al (1980).
In brief, after 48 h of observation rats in the various groups (sham, SAH and SAH treated with RO-31-7549) were anaesthetized using 5% halothane in N2O/O2 (30:70). The animal was intubated and artificially ventilated with inhalation of 0.5% to 1.5% halothane in N2O/O2 (70:30) during the surgical procedure. The anaesthesia and the respiration were monitored by regularly withdrawing arterial blood samples for blood gas analysis (Radiometer AS, Denmark). A catheter to measure MABP was placed in the right femoral artery and a catheter for blood sampling was placed in the left femoral artery. This catheter was connected to a constant velocity withdrawal pump (Harvard apparatus 22, USA) for mechanical integration of tracer concentration. In addition, a catheter was inserted in one femoral vein for injection of heparin and for infusion of the radioactive tracer. The MABP was continuously monitored with a Powerlab Unit (ADInstruments, UK). A temperature probe was inserted into the rectum of the rat to record the temperature, which was regularly maintained at 37°C. The haematocrit was measured by a haematocrit centrifuge (Beckman Microfuge 11, USA). After 30 mins of equilibration, a bolus injection of 50 μCi of 14C-iodoantipyrine 4[N-methyl-14C] (Perkin-Elmer, Boston, USA) was administered i.v. Arterial blood (122 μl) was withdrawn over 20 secs. Immediately after this the animal was decapitated, the brain removed and immersed in isopentane (JT Baker, Deventer, Netherlands) chilled to −50°C. The arterial blood sample was transferred to liquid scintillation counting vials containing 1 mL mixture of Soluene-350 (Perkin-Elmer, Boston, USA) and Isopropanol (JT Baker, Deventer, Netherlands) (1:1). After 2 h at 60°C, 0.2 mL of 30% hydrogen peroxide was added to the vials, and the samples were maintained at room temperature for 15 to 30 mins. Thereafter the samples were kept at 60°C for 30 mins and 10 to 15 mL Hionic-Fluor (Perkin-Elmer, Foster, CA, USA) was added. The β-radioactivity scintillation counting was performed on the samples with a program that included quench correction (Packard 2000 CA, Denmark). The 14C activity in the tissue was determined after sectioning the brain in 20 μm sections at −20°C in a cryostat (Wild Leitz A/S, Glostrup, Denmark). The sections were exposed to X-ray films (Kodak, Denmark) together with 14C methylmethacrylate standards (Amersham Life Science, England) and exposed the films for 20 days. Densities of the autoradiograms were measured with a Macintosh computer equipped with an analog CF 4/1 camera (Kaiser, Germany) and a transparency flat viewer (Color-Control 5000, Weilheim, Germany). The 14C content was determined in several brain regions (see Table 2). The CBF was calculated from the brain tissue 14C activity determined by autoradiography using Gjedde et al's equation (Gjedde et al, 1980):
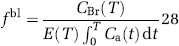
where fbl is the blood flow per unit mass, CBr (T) the isotope content, E (T) the next extraction fraction of the isotope in the time from t =0 to T, t the variable time, T the experimental time and Ca (t) is the arterial blood concentration of the isotope at time t.
Cerebral Microvessel Isolation
Isolated brains from the various groups were gently Dounce homogenized in ice-cold phosphate-buffered saline (PBS) (0.01 mol/L, pH 7.4) and centrifuged at 720g for 10 mins at 4°C. The supernatant was discarded, and pellets were resuspended in ice-cold PBS. The resuspended pellet was layered over a 15% dextran solution (35 to 40kDa), and centrifuged in a swinging bucket roter at 1300 g for 30 min × 2 at 4°C. The aqueous supernatant was discarded and the pellet containing cerebral blood vessels was collected and washed with ice-cold PBS over a nylon mesh (50 μm). The cerebral blood vessels were used for Western blot examination.
Tissue Lysis and Protein Content Determination
After cerebral microvessel isolation, the arteries were collected and placed on ice, homogenized in lysis-buffer with protease- and phosphatase inhibitors (10 mmol/LTris pH 7.4, 50 mmol/L β-Glycerophosphate, 100 μmol/L Na3VO4, 0.5% Deoxycholate, 1 mmol/L EGTA, 1 mmol/L EDTA, 1 mmol/L NaF, 20 mmol/L Na4P2O7, 1% Triton X-100, 1 mmol/L DTT, 20 μmol/L Pepstatin, 20 μmol/L Leupeptin, 0.1 U/ml Aprotinin, 1 nmol/L Calyculin and 1 mmol/L PMSF). After 20 mins incubation in lysis buffer on ice, homogenates were centrifuged at 4500 g for 10 mins at 4°C and the supernatant collected. Total protein concentration was determined using a BioRad DC kit (Hercules, CA, USA) and measuring absorbance at 750 nm on a Genesys 10 spectrophotometer (Thermo, Waltham, MA, USA). Lysates were used immediately for Western blot analysis or stored at −80°C.
Western Blot Analysis
Proteins of interest were evaluated in cerebral arteries. Lysates were dissolved in Tris-glycine SDS sample buffer (Invitrogen A/S, Taastrup, Denmark) and boiled for 5 mins. Equal amounts of protein (10 μg/lane) were loaded on an 8% Tris-glycine gel (Invitrogen A/S, Taastrup, Denmark) and separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), molecular weight markers (New England BioLabs, Ipswich, MA, USA) were loaded on each gel for protein band identification. After separation, proteins were transferred onto a nitrocellulose membrane (BioRad, Hercules, CA, USA). Subsequently, the membrane was blocked with 6.5% non-fat milk in Tween-TBS (T-TBS) overnight 4°C. Membranes were then incubated with the primary antibody of interest: rabbit polyclonal anti-PKCα phosphospecific (1:1000 dilution; Biosource, Camarillo, CA, USA), rabbit polyclonal anti-PKCδ phosphospecific (1:1000 dilution; Biosource, Camarillo, CA, USA), rabbit polyclonal anti-PKCα phosphospecific (1:1000 dilution; Biosource, Camarillo, CA, USA), rabbit polyclonal anti-PKCbI phosphospecific (1:1000 dilution; Biosource, Camarillo, CA, USA), rabbit polyclonal anti-PKCbII phosphospecific (1:1000 dilution; Biosource, Camarillo, CA, USA), rabbit polyclonal anti-PKCg phosphospecific (1:1000 dilution; Biosource, Camarillo, CA, USA) rabbit polyclonal anti-PKCε phosphospecific (1:1000 dilution; Biosource, Camarillo, CA, USA) rabbit polyclonal anti-PKCθ phosphospecific (1:1000 dilution; Cell Signaling Technology, Beverly, MA, USA., or mouse polyclonal β-actin (Sigma, Saint Louis, USA) 1 h at 37°C, followed by 3 × 5 min wash with T-TBS. Subsequently, the membranes were incubated with the appropriate secondary antibody: goat antirabbit IgG-horseradish peroxidase or goat antimouse IgG-horseradish peroxidase (1:5000; Pierce, Rockford, IL, USA) for 1 h at room temperature, followed by 5 × 5 min wash with T-TBS. Levels of β-actin were used to confirm equal loading of the lanes. The membranes were developed using the Supersignal Dura kit (Pierce, Rockford, IL, USA) and visualized using a Fujifilm LAS-1000 Luminiscent Image Analyser (Stamford, CT, USA).
In vitro Pharmacology Myograph Experiments
For contractile experiments, a sensitive myograph was used for recording the isometric tension in isolated cerebral arteries (Hogestatt et al, 1983; Mulvany and Halpern 1977). The vessels were cut into 1 mm long cylindrical segments and mounted on two 40 μm in diameter stainless steel wires in a Myograph (Danish Myo Technology A/S, Denmark). One wire was connected to a force displacement transducer attached to an analogdigital converter unit (ADInstruments, Oxford, UK). The other wire was connected to a micrometer screw, allowing fine adjustments of vascular tone by varying the distance between the wires. Measurements were recorded on a computer by use of a PowerLab unit (ADInstruments). The segments were immersed in a temperature controlled buffer solution (37°C) of the following composition (mmol/L) NaCl 119, NaHCO3 15, KCl 4.6, MgCl2 1.2, NaH2PO4 1.2, CaCl2 1.5 and glucose 5.5. The buffer was continuously aerated with oxygen enriched with 5% CO2 resulting in a pH of 7.4. The vessels were stretched to an initial resting tone of 2 mN and then allowed to stabilize at this tone for 1 h. The contractile capacity was determined by exposing the vessels to an isotonic solution containing 63.5 mmol/L of K+, obtained by partial change of NaCl for KCl in the above buffer. The contraction induced by K+ was used as reference for the contractile capacity (Hogestatt et al, 1983). Only vessels responding by contraction of at least 2.0 mN to potassium for BA and 0.8 mN to potassium for MCA were included in the study. The presence of the endothelium was checked by precontracting the vessel using 5-HT (10−65 mol/L) (Sigma, St Louis, USA) and subsequently exposing the segments to carbachol (10−5mol/L) (Sigma, St Louis, USA). A relaxant response of the precontracted tension was considered indicative of a functional endothelium (Hansen-Schwartz et al, 2003a).
Concentration–response curves were obtained by cumulative application of 5-CT (Sigma, St Louis, USA) in the concentration range 10−12 to 10−5 mol/L, S6c (Sigma, St Louis, USA) in the concentration range 10−12 to 10−7mol/L, ET-1 (AnaSpec, San Jose, USA) in the concentration range 10−14 to 10−7 mol/L. ET-1 is investigated both without and with desensitized ETB receptors achieved by exposure to S6c.
Real-time Polymerase Chain Reaction
To quantify mRNA for the ETA, ETB and 5-HT1B receptors, RT-PCR and real-time detection monitoring the PCR products was used. BA or MCA were homogenized in 1 mL of the RNApro™ solution (Q-BIOgene, CA, USA) by using a FastPrep® instrument (Q-BIOgene, CA, USA). The total RNA was extracted after a protocol obtained from the FastRNA® Pro kit supplier. Reverse transcription of total RNA to cDNA was performed using the Gene Amp RT kit (PE Applied Biosystems, USA) in a Perkin-Elmer 2400 PCR machine at 42°C for 30 mins. The real-time quantitative PCR was performed with the GeneAmp SYBR Green PCR kit (PE Applied Biosystems) in a Perkin-Elmer realtime PCR machine (GeneAmp 5700 sequence detection system). The system automatically monitors the binding of a fluorescent dye to double-strand DNA by real-time detection of the fluorescence during each cycle of PCR amplification. Specific primers for the rat ETA, ETB and 5-HT1B receptor and house keeping gene elongation factor-1 (EF-1) were designed by using the Primer Express 2.0 software (PE Applied Biosystems) and synthesized by TAG Copenhagen A/S (Copenhagen, Denmark).
Receptor primers had the following sequences:

The housekeeping gene EF-1 is used as a reference, since it is continuously expressed to a constant amount in cells.
The EF-1 primers were designed as follows:
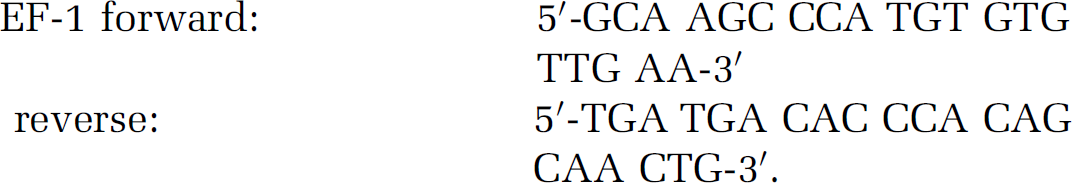
The PCR reaction was performed in a 50 μL volume and started at a temperature of 50°C for 2 mins, 95°C for 10 mins and the following 40 PCR cycles with 95°C for 15 secs and 60°C for 1 min. To verify that each primer-pair only generated one PCR product at the expected size, the real-time PCR products were separated electrophoretically on a gel. In addition, a dissociation analysis was performed after each real-time PCR run. A blank control (without template) was used in all experiments. The expected size of the amplification products were for the ETA receptor 51 base pairs, ETB receptor 86 base pairs, 5-HT1B receptor 51 base pairs and EF-1 96 base pairs. To prove that the cDNA of EF-1 and the ET and 5-HT1B receptors were amplified with a similar efficacy during real-time PCR, a standard curve were made in which the CT values were plotted against cDNA concentration on the basis of the following equation: CT = (log (1+ E))−1 log (concentration), where CT is the number of PCR cycles performed in one sample at a specific point of time, and E is the amplification efficiency with an optimal value of one. Standard curves for ETA, ETB, 5-HT1B and EF-1 were performed by dilution of cDNA sample (1:10, 1:100 and 1:1000) (data not shown).
Immunohistochemistry
The MCA and BA were dissected out and then placed onto Tissue TEK (Gibco, Invitrogen A/S Taastrup, Denmark) and frozen. They were then sectioned into 8 μm thick slices. The first antibodies used were rabbit antihuman ETB (IBL, 16207), diluted 1:400, goat anti-mouse 5-HT1B (Santa Cruz Biotechnologies, Santa Cruz, LA, USA, sc-1461), diluted 1:100, mouse anti-rat CD31 (Serotec, MCA1746), diluted 1:200, and mouse anti-rat smooth muscle actin (Serotec, MCA1905T) diluted 1:100. All dilutions were performed in PBS with 10% fetal calf serum. The second antibodies used were donkeyanti-mouse Cy™5 conjugated (JacksonImmunoResearch, 715-175-150) 1:100, donkeyantirabbit Cy™3 conjugated (Jack-sonImmunoResearch, 711-165-152) 1:100 in PBS with 10% fetal calf serum. The antibodies were detected at the appropriate wavelength in a confocal microscopy (Zeiss, USA). As control, only secondary antibodies were used. The images were analysed using ImageJ (http://rsb.info.nih.gov/ij/). This was performed by measuring the fluorescence in four to six different areas in each artery and the mean value for each vessel used (three animals in each group).
Experimental Design Section
The total number of rats used in this study was 47. Three different experimental groups was included sham (n = 15), SAH +vehicle (n = 16) and SAH treated with RO-31-7549 (n = 16). At 2 days after the surgery, the rats in the various groups were further used for autoradiographic measurements (n = 5 rats in each group), in vitro pharmacology study (n = 4 to 8 rats in each group), real-time PCR (n = 7 to 10 rats in each group), immunohistochemistry (n = 3 rats in each group) and Western blot (n = 6 rats in each group). Some rats were used for several experiments.
Calculations and Statistics: Data are expressed as mean ±standard error of the mean (s.e.m), and n refers to the number of rats. Statistical analyses were performed using the nonparametric Kruskal-Wallis test, where P < 0.05 was considered significant.
Western Blot
Cerebrovascular protein lysates from the different groups were compared. Cerebral arteries from two animals were pooled for each group of experiment, and each experiment was repeated three times. Quantitation of band density was performed with the electrophoresis computer analysis program Fujifilm Science Lab Image Gauge 4.0. The immunoblot optical density values were determined with repeated measurement and presented as percentage activity in the treated groups compared with the sham group, where the sham group was set to 100%.
In vitro Pharmacology
Contractile responses in each segment are expressed as percentage of the 63.5 mol/L K+ induced contraction. Emax value represents the maximum contractile response elicited by an agonist and the pEC50 the negative logarithm of the drug concentration that elicited half the maximum response. For biphasic responses, Emax(1) and pEC50(1) describes the high affinity phase and Emax(2) and pEC50(2) describes the low affinity phase.
Real-time Polymerase Chain Reaction
Real-time PCR experiments were performed on BA and MCA from sham rats, rats with induced SAH and rats with induced SAH treated with PKC inhibitor. Data were analysed with the comparative cycle threshold (CT) method (Hansen-Schwartz et al, 2003a).
The CT values of EF-1 mRNA were used as a reference to quantify the relative amount of ETA, ETB and 5-HT1B mRNA. The relative amount of mRNA was calculated with the CT values of ETA, ETB and 5-HT1B receptor mRNA in relation to the CT values of EF-1 mRNA in the sample by the formula X0/R0 = 2 CtR-CtX , where X0 is the original amount of target mRNA, R0 is the original amount of EF-1 mRNA, CtR is the CT value for EF-1 and CTX is the CT value for the target.
Results
Subarachnoid Haemorrhage Model
The total number of rats used in the study was 47; 16 in the SAH groups, 15 in the sham group and 16 was used in the SAH + treatment with RO-31-7549 group. The mortality rate of the animal model of SAH was 5% and there was no difference in the mortality rate between the groups. The rats did not show any distressed behaviour. They were moving around, eating, drinking and their fur was not sprawl. All surviving animals were neurologically examined using an established scoring system (Bederson et al, 1986; Menzies et al, 1992). All SAH animals received a score of 1, and the sham and PKC treated groups got a score of 0. However, staining of coronal slices of the brain with 1% 2,3,5-triphenyltetrazolium chloride (TTC) did not show any changes to support the neurologic outcome.
In all operated rats, MABP (101±3 mm Hg), partial pCO2 (40± 0.5 mm Hg), partial pO2 (103±2 mm Hg), haematocrit (42±1 mm Hg) values and temperature were within acceptable limits during the operation. No statistical difference was seen in physiologic parameters between the groups; sham, SAH + vehicle (henceforth only mentioned as SAH) and SAH treated with RO-31-7549. A result of injecting the blood the cortical blood flow dropped over both hemispheres to 20±3% of resting flow (there was no difference between the two Laser Doppler probe data) and the ICP increased from 10 ± 1 to 129±10 mm Hg. The Laser Doppler blood flow and the elevated ICP returned to the basal values within 1 h of postoperative monitoring. There was no difference in the ICP and cortical blood flow between the two groups SAH and SAH treated with RO-31-7549 or vehicle (Figure 1). Injection of RO-31-7549 or vehicle alone had no effect on the cortical blood flow measured with Laser Doppler.
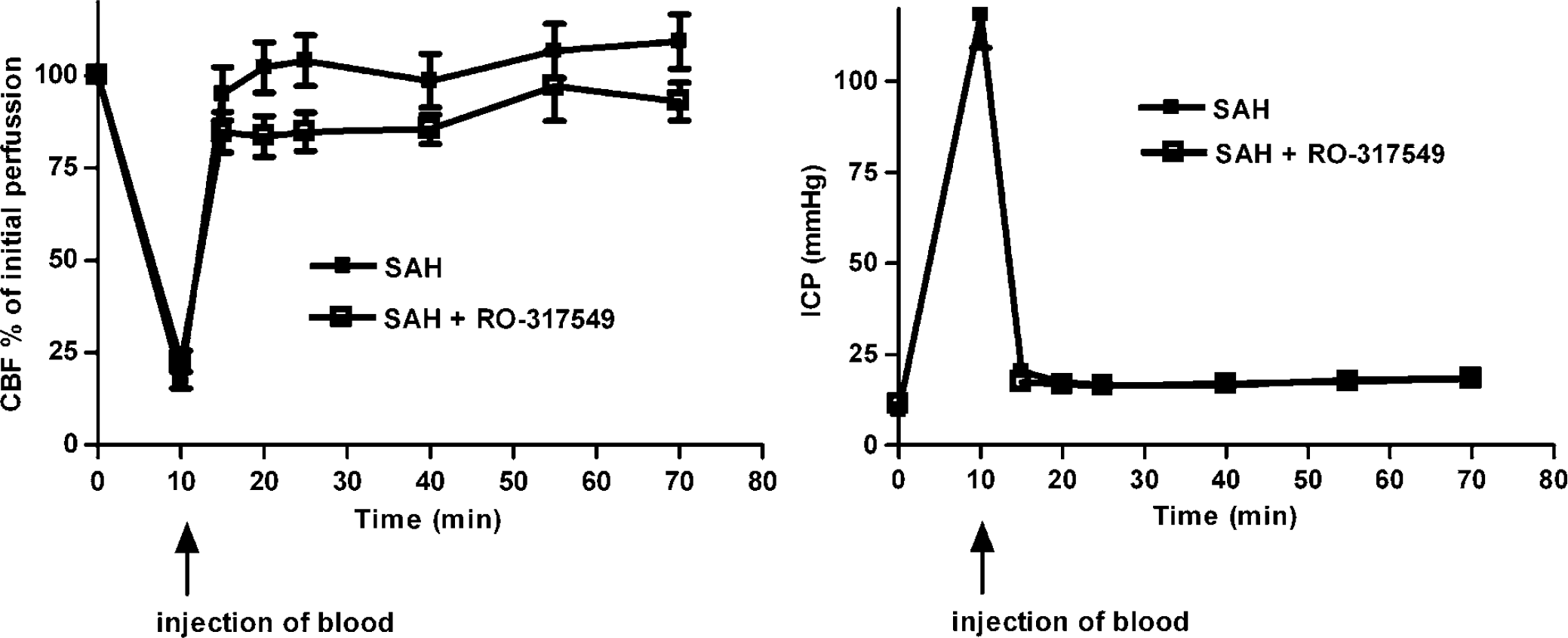
Illustration of the cortical blood flow measured with Laser Doppler flowmetry and ICP at the time of injection of blood (SAH) and 1 h after SAH.
Regional Cerebral Blood Flow
There was a significant global decrease in CBF in the SAH group (n = 5) compared with the control group (n = 5) from 60.10±3.53 to 131.30±7.04 ml/100g/min, respectively (P < 0.05). Treatment with RO-31-7549 (n = 5) prevented the marked reduction in CBF seen after SAH (Figure 2). The SAH animals showed a reduction in the rCBF in 14 of the 18 brain regions examined compared with the control operated rats (Table 1). Treatment with RO-31-7549 prevented this reduction in rCBF, and there was no difference as compared with the control group for any of the regions studied.
Regional cerebral blood flow 48 h postsubarachnoid haemorrhage
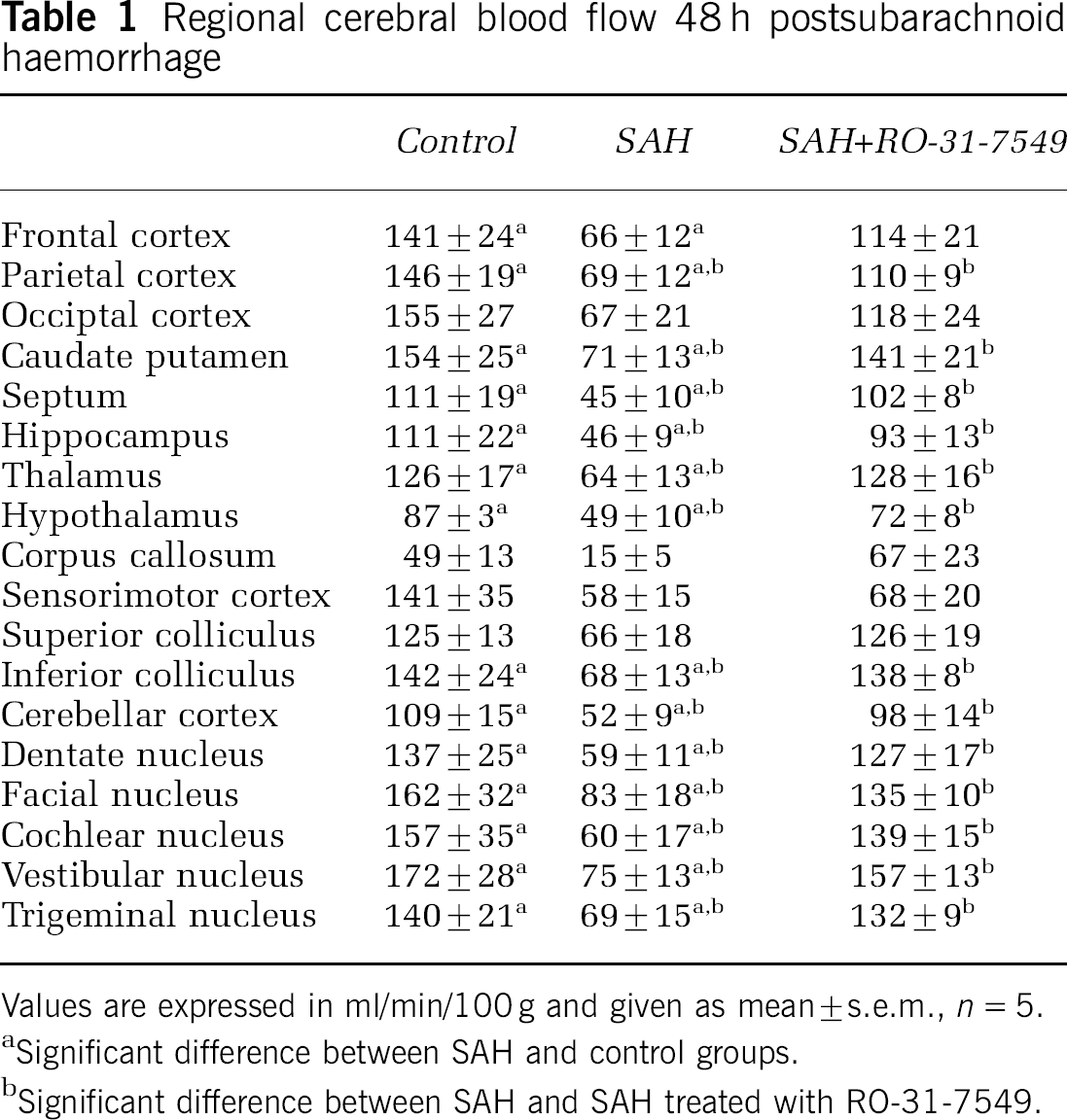
Values are expressed in ml/min/100g and given as mean±s.e.m., n = 5.
Significant difference between SAH and control groups.
Significant difference between SAH and SAH treated with RO-31-7549.
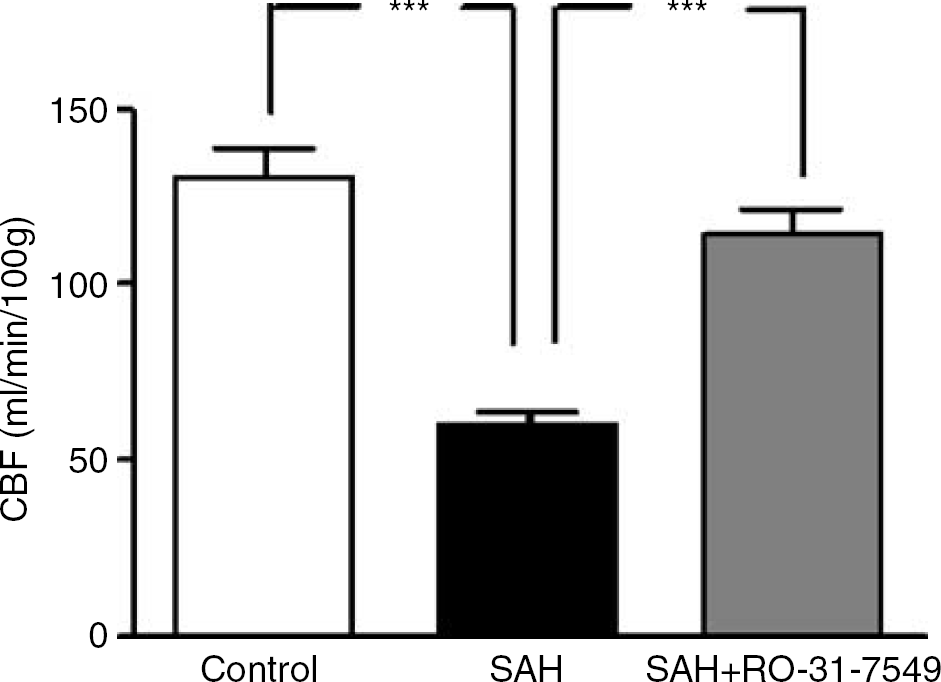
Effect of treatment with the PKC inhibitor RO-31-7549 on the global CBF after induced SAH in rats. There is a reduction in the global CBF in the SAH (n = 5) compared with the control rats (n = 5). Treatment with RO-31-7549 (n = 5) prevented this reduction in CBF. Data were obtained by an autoradiographic method and data are expressed as mean ± s.e.m., ***P < 0.001.
Western Blot
Since PKC isotypes can act differentially depending on their phosphorylation state, we investigated the activation of the eight PKC isotypes. The study revealed that only phosphorylation of PKCδ (148% ± 25%) and PKCα (164%±26%) were significantly increased after SAH as compared with sham (100%). Treatment with the PKC inhibitor RO-31-7549 significantly prevented the upregulation of pPKCδ (49% ± 8%) and pPKCα (54% ± 8%) protein levels in the cerebral arteries as compared with the SAH (P < 0.05) (Figure 3A–3D).
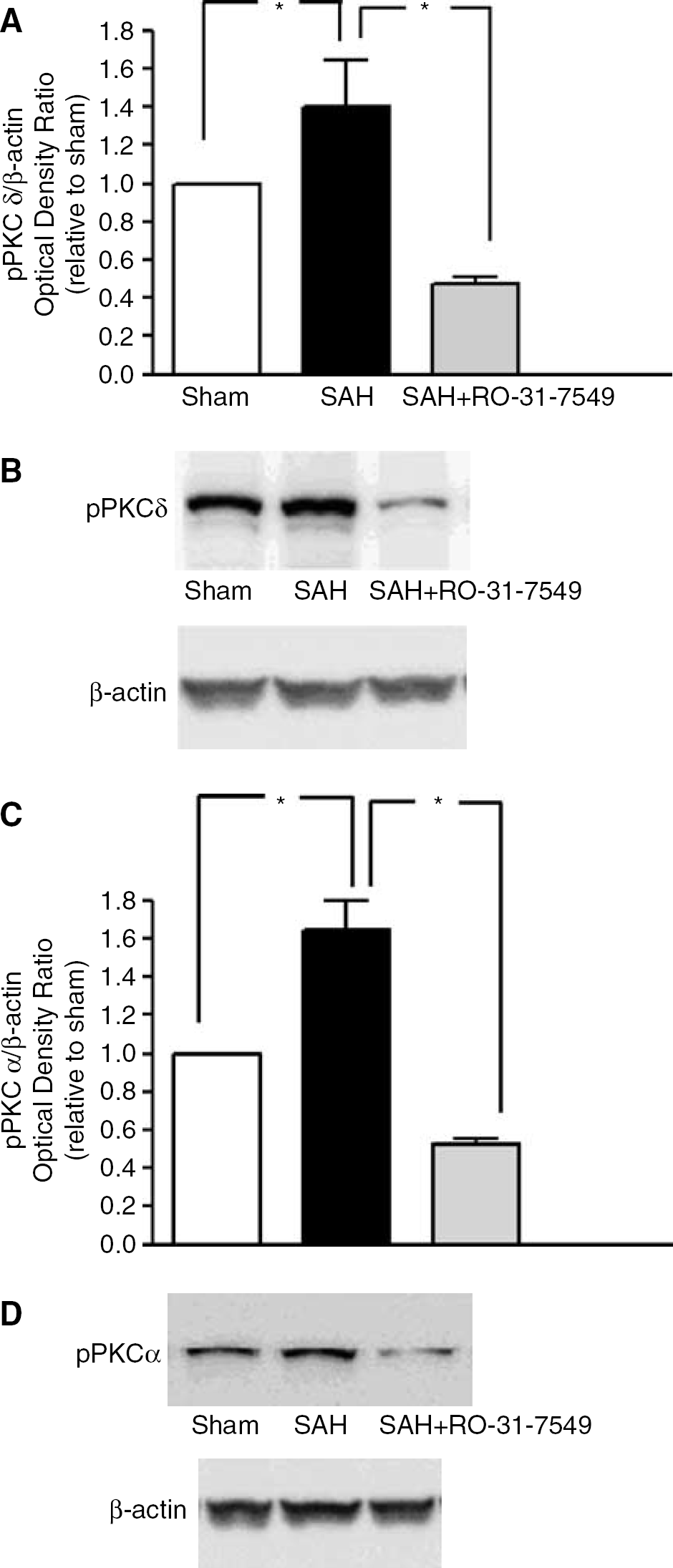
Effect of treatment with the PKC inhibitor RO-31-7549 in cerebral arteries on the protein levels of pPKCδ and pPKCα after induced SAH in rats. Data are presented as the pPKC/β-actin mean optical density ratio relative to sham; (
In vitro Pharmacology
K+ -induced contractions did not differ significantly between the vessels from the three groups sham, SAH and SAH + RO-31-7549 (Table 2). Emax and pEC50 values for respective group are presented in Table 2. In addition, there was no difference in relaxant response to carbachol between the three groups studied (data not shown).
Contractile effects of ET-1 and 5-CT in MCA and basilar arteries
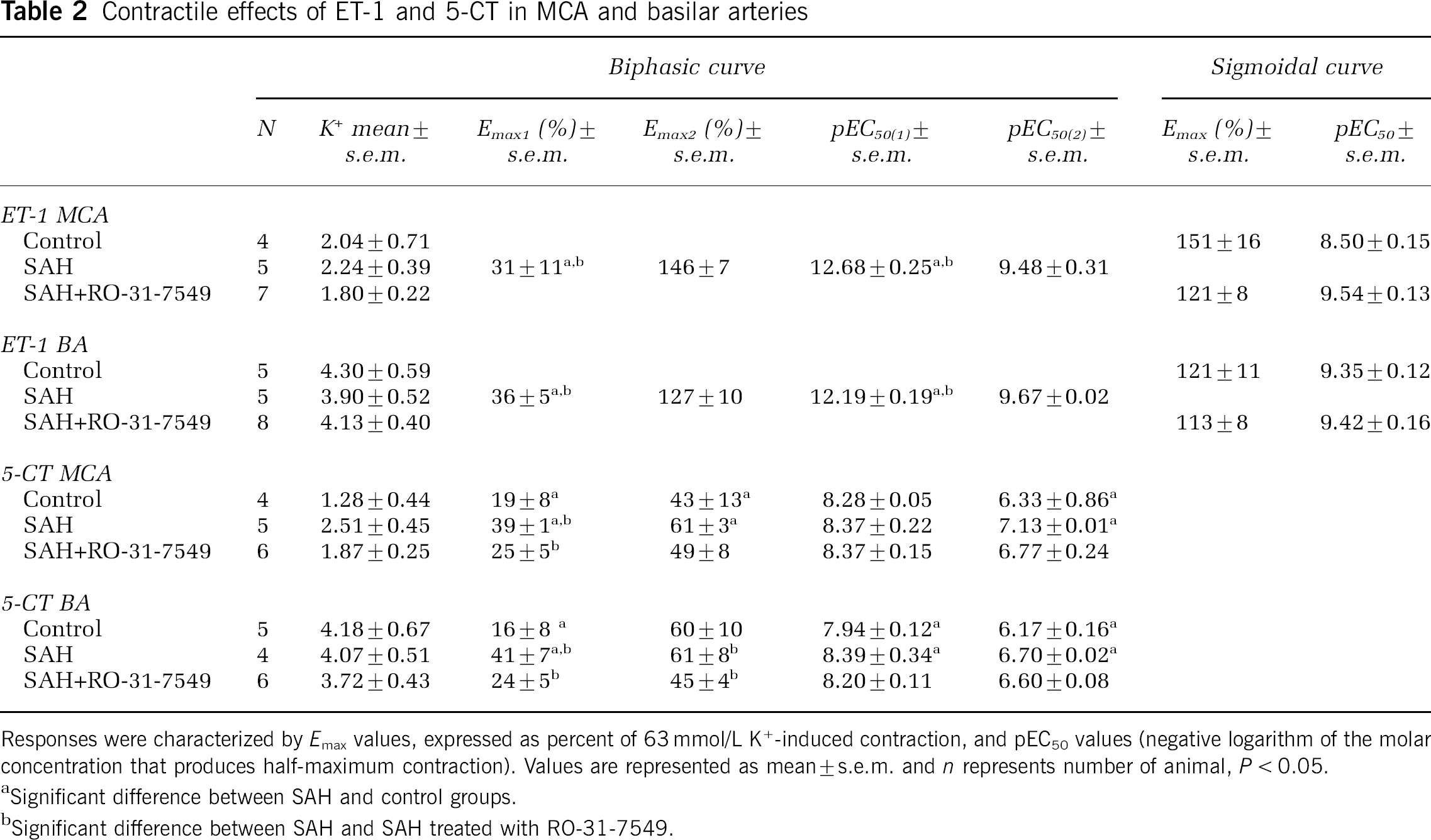
Responses were characterized by Emax values, expressed as percent of 63 mmol/L K+-induced contraction, and pEC50 values (negative logarithm of the molar concentration that produces half-maximum contraction). Values are represented as mean±s.e.m. and n represents number of animal, P < 0.05.
Significant difference between SAH and control groups.
Significant difference between SAH and SAH treated with RO-31-7549.
Contractile Response to Endothelin-1
In the MCA and BA from SAH rats (n = 5) ET-1 showed a leftward shift of the curve, which indicates an enhanced contractile response to ET-1 as compared with the sham-operated rats (n = 4 to 5) where a normal sigmoidal curve was obtained (Figure 4A and 4B; Table 2). Treatment with RO-31-7549 (n = 7 to 8) produced a significantly attenuated ET-1 induced response, compared with the rats with induced SAH (P < 0.05). Interestingly, there was no significant difference in the contractile response between sham and RO-31-7549 treated rats (Figure 4A and 4B; Table 2).
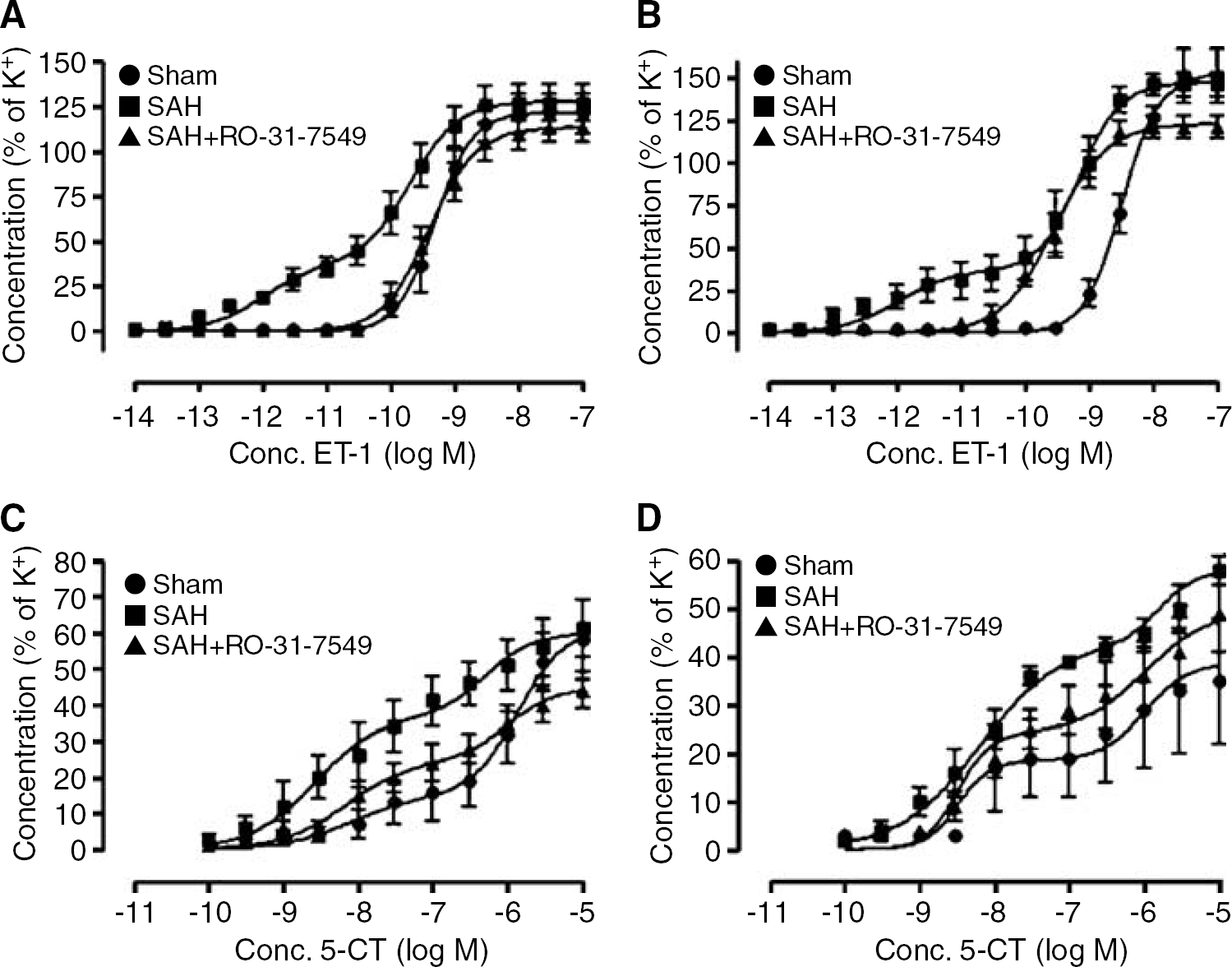
Concentration–response curves elicited by cumulative application of ET-1 and 5-CT in rat cerebral arteries. (
In the present study, application of the specific ETB receptor agonist S6c alone did not give rise to any contraction in rats with induced SAH, in rats with induced SAH and treated with PKC inhibition, or in sham operated rats in the MCA or the BA (data not shown). Desensitization of the ETB receptor with S6c induced an attenuated contractile response to ET-1 in the rats with induced SAH (data not shown). This implicates that the enhanced contractile effect after SAH is ETB dependent. This is in support of studies previously published with selective endothelin receptor antagonists (Hansen-Schwartz and Edvinsson 2000; Hansen-Schwartz et al, 2003b)
Contractile Response to 5-carboxamidotryptamine
5-CT gave rise to a biphasic concentration-dependent contraction, indicating the presence of two receptors 5-HT1B and 5-HT2A as verified by previous detailed antagonist studies (Hoel et al, 2001). In both MCA and BA from rats with induced SAH (n = 4 to 5) 5-CT gave rise to an elevated Emax(1), Emax(2) and pEC50(2) as compared with the sham-operated rats (n = 4 to 5) (P < 0.05, Figure 4C and 4D; Table 2). In BA treatment in vivo with RO-31-7549 (n = 6) down-regulated, both the first 5-HT1B and the second 5-HT2A phases as compared with the rats with induced SAH (Figure 4C). In the MCA treatment with RO-31-7549 (n = 6) significantly reduced the Emax(1) (P < 0.05) and had a tendency to decrease the Emax(2), pEC50(1) and pEC50(2) compared with the SAH induced rats (Figure 4D, Table 2).
Real-time Polymerase Chain Reaction
The standard curves for each primer pair had almost similar slopes, demonstrating that EF-1, ETA, ETB, and 5-HT1B cDNA were amplified with the same efficiency (data not shown). In each PCR experiment, a no template control was included, and there were no signs of contaminating nucleic acids in the samples. Electrophoresis verified only one product for each primer pair at the expected size (data not shown). Since the results from the brain arteries MCA and BA (n =7 to 10) were identical, they were added together in the statistical analysis. The results showed elevated levels of ETB receptor mRNA relative to the amount of EF-1 mRNA in the cerebral arteries from the rats with induced SAH compared with the sham operated rats (P < 0.05). Treatment with RO-31-7549 inhibited this ETB receptor mRNA upregulation (Figure 5A). There was no difference in the expression of ETA receptor mRNA levels between the three groups sham, SAH and SAH treated with RO-31-7549 (data not shown). There was a significant upregulation of 5-HT1B receptor mRNA levels in SAH compared with sham operated rats. Treatment in vivo with RO-31-7549 prevented this 5-HT1B receptor mRNA upregulation as compared with SAH (P < 0.05) (Figure 5B).
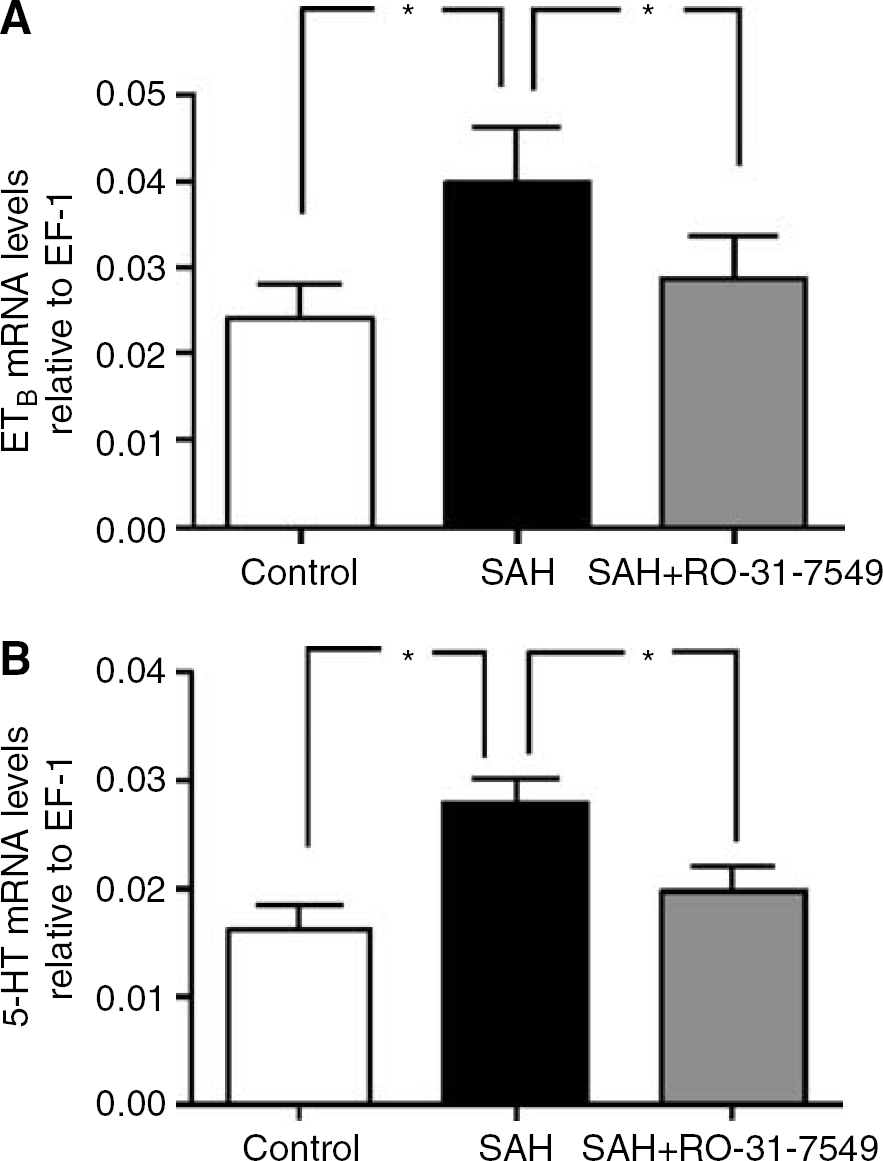
Effect of treatment with PKC inhibitor RO-31-7549 in cerebral arteries on the mRNA levels of ETB and 5-HT1B receptors after experimental induced SAH in rats. (
Immunohistochemistry
Selective antibodies towards the ETB and 5-HT1B receptors visualized the smooth muscle cell localization of these receptors using confocal microscopy (n = 3 in each of the groups). Double immunohistochemistry staining versus smooth muscle actin, expressed in the smooth muscle cells, and CD31, expressed in the endothelial cells, were performed to verify their localization (data not shown). The ETB receptor protein was expressed on the smooth muscle cells and this signal was increased in SAH (163±17 %) as compared with sham (100±10%) (P < 0.05). As can be seen in Figure 6, similar results were observed for the 5-HT1B receptor protein expression in SAH; it was increased from 1367 19% compared with sham 100±10% (P < 0.05). Treatment with the PKC inhibitor RO-31-7549 prevented the upregulation of ETB (123± 39%) and 5-HT1B (81±18%) receptors protein level in the smooth muscle cell layer as compared with the SAH (Figure 6).
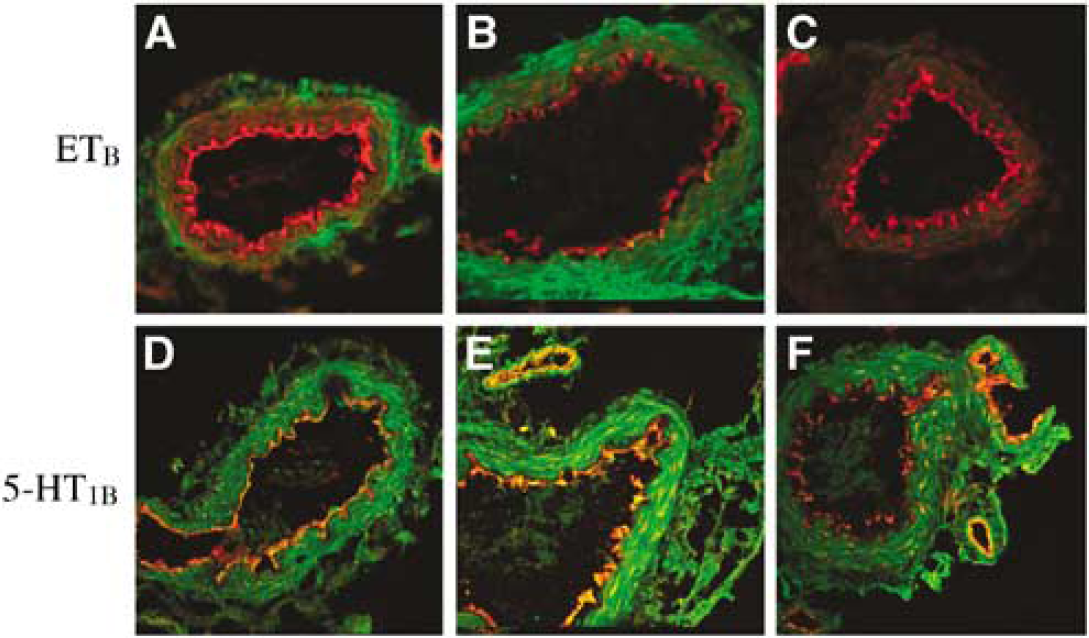
Sections from the basilar artery showing ETB and 5-HT1B immunoreactivity in the smooth muscle cell layer (green layer). The red layer is illustrating the CD31. (
Discussion
This is the first study to show that intracisternal administration of a specific PKC inhibitor totally prevents the SAH induced reduction in total global and rCBF as compared with control. At the same time, the associated upregulation of cerebrovascular ETB and 5-HT1B receptors was prevented and the specific functional responses, mRNA and protein expressions were not different from sham animals.
We hypothesized that there is a correlation between the changes in CBF and cerebrovascular effects induced by SAH.
The CBF reduction was a global phenomenon and was present in all the brain regions studied. This agrees well with reports using the same SAH model in which flow and metabolism were reduced and in addition there were signs of neuronal loss, indicative of cerebral ischaemia (Prunell et al, 2004). The degree of reduction in CBF correlates with worsening of the clinical grade after a cerebrovascular insult such as SAH (Heilbrun et al, 1972). Treatment with the PKC inhibitor RO-31-7549 prevented the reduction in both global and rCBF as examined at 48 h, but it had no acute effect per se or altered the initial rise in ICP or the drop in cortical blood flow. This agrees with preliminary data that showed that RO-31-75-49 had no effect on the contractility when it was given just before induction of SAH (present data) or directly applied on the isolated arteries (unpublished data). Furthermore, it was observed that there was no difference in the local cortical blood flow response between the two groups SAH and SAH treated with RO-31-7549 during the acute phase. This suggests that the positive effect that we observed following treatment with the PKC inhibitor RO-31-7549 arise when the blocker is administrated after the induction of SAH, which is of particular clinical relevance.
Clearly, an increased sensitivity of the cerebral arteries toward endogenous agonists would lead to an increased vascular tone given an unchanged concentration of the agonist. Endothelin and 5-HT are produced locally in endothelial cells (Ralevic and Burnstock, 1996). The detailed analysis of isolated artery segments in vitro has shown that RO-31-7549 potently inhibits the upregulation of ETB and 5-HT1B receptors during organ culture (Hansen-Schwartz et al, 2002). With this in mind we carefully examined the receptor upregulation in cerebral arteries that occur after SAH (Hansen-Schwartz et al, 2003a, b). The pharmacological analysis of cerebral arteries after SAH revealed contraction at much lower concentrations of ET-1 and 5-CT than in arteries from control animals. In animals that are treated with the PKC inhibitor RO-31-7549 showed contractions identical to those seen in sham. ET-1 induced a contraction of control arteries that is blocked by selective ETA receptor antagonist, and thus represents a rather pure population of ETA receptors (Hansen-Schwartz et al, 2003b). There is no direct functional response to the selective ETB stimulation with S6c in sham or SAH arteries. However, the available antagonist data show that blocking the ETB receptor through desensitization with S6c attenuates the first upregulated phase after SAH, while it does not affect the second phase (ETA receptor), indicating the presence of a functional ETB receptor. Previous data have revealed that blocking the ETB receptor with the selective antagonist IRL1038 results in similar findings (Hansen-Schwartz et al, 2003a). A possible explanation for this phenomenon is a dimerization mechanism between the two receptors to form an ETA-ETB receptor heterodimer in the recognition of ET-1, a typical bivalent ligand (Harada et al, 2002). In the present study, we have shown with confocal immunohistochemistry that SAH induced an increased expression of ETB receptors in the smooth muscle cell layer in cerebral arteries. This increase in expression of ETB receptors was normalized by treatment with PKC inhibitor.
Previously data have shown that rat cerebral arteries contain a pure population of 5-HT2A receptors (Hoel et al, 2001). These data were based on functional agonist and antagonist experiments in combination with PCR experiments. As a model to mimic phenotypic alterations that occur during cerebrovascular disease organ culture of isolated arteries segments were used. In such a condition, the 5-HT1B receptor was seen to be expressed at significant levels (Hoel et al, 2001) and similar to that of SAH (Hansen-Schwartz et al, 2003a). The 5-HT1B receptor localization was studied with confocal microscopy. We observed that SAH induced an increased expression of 5-HT1B receptors in the smooth muscle cells of the medial layer in cerebral arteries. The PKC inhibitor prevented this increased expression of 5-HT1B receptors on the smooth muscle cells.
Previous studies have shown that upregulation of ETB and 5-HT1B receptors in the cerebrovascular smooth muscle cells depends on enhanced transcription (Hansen-Schwartz et al, 2003a, b). This event is critically dependent on PKC and can at least in vitro be inhibited by specific PKC inhibitors (Hansen-Schwartz et al, 2002; Henriksson et al, 2003). Here, we show for the first time that this can be performed in vivo.
The fact that we have shown involvement of at least two cerebrovascular receptor systems in experimental SAH alludes to the possibility of the involvement of several receptor systems making it attractive to search for a key factor involved in this upregulation. In experimental models mainly in dogs of SAH studying PKC, increased levels of activated PKC have been observed (Fujikawa et al, 1999; Laher and Zhang 2001; Nishizawa et al, 2003, 2000). This implicates that PKC is involved in the development of cerebral ischaemia. However, these studies focused on a direct effect of PKC in the contraction-excitation mechanism which is an acute effect. The question they asked was if PKC inhibition could reverse SAH induced contraction via inhibition of elements in the contraction-excitation process. Our PKC inhibitor, however, did not have such a direct antivasoconstrictor effect. The PKC family comprises at least 12 cloned isozymes of which the specific isoforms PKCα and PKCδ may be involved in vasospasm after SAH (Nishizawa et al, 2000). This is in agreement with our study. We examined eight PKC isoforms, which are expressed in cerebral arteries. Only PKCδ and PKCα were increasingly activated after SAH suggesting that these particular PKC-subunits are the primary subunits involved in ETB and 5-HT1B receptor upregulation. Accordingly, specific PKC inhibition decreased the amount of activated PKCδ and PKCα. This agrees well with other studies performed on stroke. Treatment with more specific PKC inhibitors such as rottlerin or chelerytine may in part inhibit the progression of vasospasm after SAH (Nishizawa et al, 2003; Obara et al, 2005). Recent evidence supports a role for brain PKCδ in reperfusion injury and its inhibition improves histopathologic and behavioural outcome after stroke in vivo (Bright et al, 2004; Chou and Messing, 2005). In the MCAO model, it has been observed that mice lacking PKCγ develop smaller infarcts than wild-type mice (Chou and Messing, 2005). It has been shown that PKCε and PKCα are involved in sustained vasoconstriction induced by oxyhemoglobin in cerebral arteries (Wickman et al, 2003). Although activation of PKCα, δ and ε have been detected, the role of PKC after SAH still remains unclear. The three principal PKC isoforms that are expressed in the cerebral arteries are PKCα, δ and ε, that is, the classic and novel PKC's (Way et al, 2000). None of the available PKC inhibitors are specific for a single subunit. We therefore decided on using an inhibitor that blocks specifically the classic and novel PKC's, which is the property of RO-31-7549.
Our results revealed that the PKC δ and α, signal transduction pathway is involved in the upregulation of ETb and 5-HT1B receptors after SAH as verified in vivo functionally, with protein determination and mRNA levels. However, the exact mechanisms causing the receptor upregulation remain elusive. There are however at least two possible ways: Our hypothesis suggests that a sudden change in ICP induces a switch-on of a transcriptional mechanism that responds to changes in vessel wall tone. Another possibility could be that the extravasated blood exerts a local effect on the cerebral arteries. When extravasated the red blood cells lysate and give rise to free oxyhemoglobin, which in turn generates superoxide radicals, activate cytokines, which may activate stress pathways. This could in turn activate intracellular signal transduction pathways that act via protein kinases to enhance transcription of proteins and subsequently cause the receptor upregulation. Since both the change in ICP and the extravasated blood could be possible causes of the cerebral ischaemia and cerebral vasoconstriction observed following SAH, we decided to avoid the injection of saline in our sham-operated rats. Preliminary data from our laboratory exist to support our rationale.
In conclusion, the present study suggests that PKC d and a play an important role in the pathogenesis of cerebral ischaemia after SAH. Since vasoconstriction and reduction in CBF are results of the SAH our study is of high clinical relevance. This is the first study to show that specific inhibition of the PKC subunits prevents the reduction in global and rCBF, the receptor upregulation and attenuates the vasoconstriction mediated by ETB and 5-HT1B receptors in cerebral arteries after SAH.
Footnotes
Acknowledgements
We are grateful for the scientific advice from Professor NA Svendgaard and N Diemer and the technical assistance of M Nielsen and K Eskesen.