
Abstract
Hydrocephalus is caused by an imbalance in cerebrospinal fluid (CSF) production and absorption, resulting in excess ventricular fluid accumulation and neurologic impairment. Current therapy for hydrocephalus involves surgical diversion of excess ventricular fluid. The water-transporting protein aquaporin-4 (AQP4) is expressed at the brain-CSF and blood-brain barriers. Here, we provide evidence for AQP4-facilitated CSF absorption in hydrocephalus by a transparenchymal pathway into the cerebral vasculature. A mouse model of obstructive hydrocephalus was created by injecting kaolin (2.5 mg/mouse) into the cisterna magna. Intracranial pressure (ICP) was ~5mm Hg and ventricular size < 0.3 mm3 in control mice. Lateral ventricle volume increased to 3.7 ± 0.5 and 5.1 ±0.5 mm3 in AQP4 null mice at 3 and 5 days after injection, respectively, significantly greater than 2.6 ± 0.3 and 3.5 ± 0.5 mm3 in wildtype mice (P < 0.005). The corresponding ICP was 22 ± 2 mm Hg at 3 days in AQP4 null mice, significantly greater than 14 ± 1 mm Hg in wildtype mice (P < 0.005). Brain parenchymal water content increased by 2% to 3% by 3 days, corresponding to ~50 μL of fluid, indicating backflow of CSF from the ventricle into the parenchymal extracellular space. A multi-compartment model of hydrocephalus based on experimental data from wildtype mice accurately reproduced the greater severity of hydrocephalus in AQP4 null mice, and predicted a much reduced severity if AQP4 expression/function were increased. Our results indicate a significant role for AQP4-mediated transparenchymal CSF absorption in hydrocephalus and provide a rational basis for evaluation of AQP4 induction as a nonsurgical therapy for hydrocephalus.
Introduction
Hydrocephalus occurs as the result of an imbalance in the production and absorption of cerebrospinal fluid (CSF), leading to accumulation of extraparenchymal fluid and expansion of the ventricular system (Fishman 1992; Johnston and Teo 2000). As the volume of intracranial contents increases, intracranial pressure (ICP) rises, decreasing cerebral blood flow, and often producing severe neurologic impairment or death. Hydrocephalus is typically divided into noncommunicating or communicating subtypes (Fishman 1992). Noncommunicating hydrocephalus is caused by an obstruction within the ventricular system, such as a tumor, which prevents CSF proximal to the obstruction from draining into the subarachnoid space, where it is reabsorbed into the venous sinuses. Communicating hydrocephalus, such as occurs after subarachnoid hemorrhage, results from impaired absorption of CSF despite patent CSF pathways. It is postulated that communicating hydrocephalus is caused by impairment in CSF absorption at the level of the arachnoid granulations as well as possible impairment of transparenchymal CSF flow (Cherian et al, 2004; Crews et al, 2004).
Both communicating and noncommunicating (obstructive) hydrocephalus may occur congenitally or might be acquired secondary to trauma, tumor, hemorrhage, or infection (Golden and Bonnemann 2003; Guyot and Michael 2000; Yoshioka et al, 2000). The primary treatment for hydrocephalus involves surgical drainage and diversion of excess CSF (Golden and Bonnemann 2003). Congenital hydrocephalus often requires the placement of a permanent ventriculoperitoneal shunt, which is frequently complicated by repeated blockage and infection, requiring multiple surgical revisions over a patient's lifetime (Li 2001). At present, there is no effective long-term medical treatment for hydrocephalus.
The astroglial water channel aquaporin-4 (AQP4) has been found to play an important role in regulating brain water balance (Amiry-Moghaddam et al, 2004; Manley et al, 2000, 2004). Aquaporin-4 is expressed at the ependymal and glial limiting borders of the brain parenchyma, which form the brain-CSF barrier, as well as in the perivascular astrocyte foot processes at the blood-brain barrier, suggesting a role in water movement between brain fluid compartments (Badaut et al, 2002; Nielsen et al, 1997). Recent studies of AQP4 in models of vasogenic edema, including brain tumor (Papadopoulos et al, 2004) and focal brain abscess (Bloch et al, 2005), suggest that AQP4 also facilitates the clearance of excess brain extracellular fluid. As intraventricular pressure rises in hydrocephalus, CSF is thought to move from the ventricular space to the brain extracellular space, causing an interstitial edema (Hiratsuka et al, 1982; Nakagawa et al, 1984). We hypothesized that AQP4 may facilitate the transparenchymal absorption of ventricular fluid into the cerebral microvasculature when the primary ventricular CSF clearance pathways are blocked.
We developed a mouse model of progressive, obstructive hydrocephalus based on the well-established kaolin injection model described in rats (Collins 1979). Kaolin-injected mice reproducibly developed an acute hydrocephalus with progressive ventriculomegaly, ICP elevation, and brain parenchymal water accumulation. We found that compared with wildtype mice, AQP4-deficient mice had an accelerated course of ventricular enlargement and ICP elevation. A multi-compartment model of CSF flow based on the experimental data predicted that AQP4-dependent fluid absorption from the parenchyma provides the primary route of CSF clearance in hydrocephalus, and that AQP4 induction would reduce ventricular enlargement and ICP elevation in hydrocephalus.
Materials and methods
Mice
Aquaporin-4 null mice were generated by targeted gene disruption as described (Ma et al, 1997). All experiments were performed on wildtype and AQP4 null mice in a CD1 genetic background matched for age/weight (6 to 8 weeks; 25 to 30 g). Protocols were approved by the University of California, San Francisco Committee on Animal Research.
Kaolin-Induced Hydrocephalus
Mice were anesthetized with 2.5% Avertin (2,2,2-tribro-moethanol, 125 mg/kg intraperitoneally, Sigma-Aldrich, St Louis, MO, USA). The head was secured in a stereotactic frame (Benchmark, Neurolab, St Louis, MO, USA) and a cm midline vertical incision was made over the back of the neck from the top of the occiput to C1. The muscular layers were divided and the dural membrane overlying the cisterna magna was exposed. In all mice, 10 μL of a kaolin suspension (250 mg/ml in PBS, Sigma-Aldrich, St Louis, MO, USA) was injected into the cisterna magna through a 30-gauge needle attached to a 50 μL gas-tight glass syringe (Hamilton, Reno, NV, USA). The muscle layers and skin were sutured closed and mice were allowed to recover. Control wildtype and AQP4 null mice received 10 μL cisternal injections of sterile PBS. In total, > 80% of mice survived the injection procedure. After recovery from anesthesia, mice were kept at room temperature and provided with standard chow and water ad libitum. They were examined daily for clinical signs of illness. Daily weights and rectal temperature were recorded, as well as an assessment of global neurologic function (see below).
Neurologic Score
Mice were scored for global neurologic function using a modified neurologic scale as follows: 5 = normal, 4 = decreased scavenging activity, decreased scatter reflex, 3 = no spontaneous scavenging, loss of scatter reflex, ataxia, = nonpurposeful movements, 1 = loss of righting reflex, 0 = dead (Matkowskyj et al, 1999). Mice were assessed before kaolin injection and every 24 hours thereafter.
Intracranial Pressure Measurement
Anesthetized mice were immobilized in a stereotactic frame and a 1 cm midline incision was made over the vertex of the skull. A 1 mm burr hole was drilled 3 mm posterior and 3 mm lateral to the bregma using a micromotor drill (Foredom, Bethel, CT, USA). A parenchymal pressure catheter (SPR-320, Millar Instruments, Houston, TX, USA) with 660 μm tip diameter connected to a pressure control unit (TC-510, Millar Instruments) was inserted to a depth of 2 mm. Intracranial pressure was recorded at 50 Hz using a TC-100 recording system (Biopac, Santa Barbara, CA, USA). Rectal temperature was maintained at 37°Cto 38°C by a heating lamp during ICP measurements. Intracranial pressure values were averaged over 2 to 5 mins.
Ventricular Volume
Anesthetized mice were perfused with 10 mL of a neutralbuffered 10% formalin solution (Sigma-Aldrich, St Louis, MO, USA) by puncture of the left cardiac ventricle, after which brains were removed and immersed in a formalin solution for 72 h. Fixed brains were sectioned using a vibratome (Vibraslice NVSLM1, World Precision Instruments, Sarasota, FL, USA) to obtain serial 300-mm-thick sections from the anterior horn of the lateral ventricles to the cerebral aqueduct. Slices were mounted on glass slides and imaged at × 12 magnification under a surgical microscope (SMZ1500, Nikon, Melville, NY, USA) equipped with a cooled CCD camera (CoolSnapHQ, Photometrics, Tucson, AZ, USA). The lateral ventricle area in each section was measured using image analysis software (ImageJ, NIH) for computation of total lateral ventricular volume.
Brain Water Content
Brain water content was determined by gravimetric analysis as described previously (Marmarou et al, 1978). A bromobenzene/kerosene gradient column was constructed and calibrated with potassium sulfate standards. Anesthetized mice were decapitated, and freshly excised brains were immediately cooled in a sealed container on ice. Coronal sections of 2 mm thickness were cut on a brain block using the optic chiasm as the anterior landmark. Using a punch biopsy needle, 2 mm3 tissue sections were obtained from periventricular brain and placed in the gravimetric column. Tissue height within the column was recorded after 1 min, and specific gravity was calculated from the standard calibration. Data from two to four tissue fragments from both hemispheres were averaged for each mouse. Specific gravities were converted to % brain water as described by Nelson et al (1971).
Immunohistochemistry
Anesthetized mice were perfused by cardiac puncture with 10 mL of a neutral-buffered 10% formalin solution, after which brains were removed and immersed in formalin for 24 h. The fixed tissue was processed through graded concentrations of ethanol, followed by Citrisolv (Fisher Scientific, Los Angeles, CA, USA), and embedded in paraffin. Sagittal sections of 10 μm thickness were cut by microtome, deparaffinized in Citrisolv, and rehydrated in graded ethanols. After blocking with goat serum, sections were incubated with a rabbit anti-AQP4 polyclonal antibody (1:200, Chemicon, Temecula, CA, USA), followed by a Cy3-conjugated sheep anti-rabbit IgG secondary antibody (1:200, Sigma-Aldrich, St Louis, MO, USA) for immunofluorescence.
Aquaporin-4 Immunoblot
Brains of anesthetized mice were freshly excised and homogenized in 250 mmol/L sucrose, 10 mmol/L Tris-HCl, 2 μg/mL aprotinin, 2 μg/mL pepstatin A, 2 μg/mL leupeptin, and 100 μmol/L Pefabloc at pH 7.4. The homogenate was centrifuged at 2700g for 10 mins at 4°C to remove debris and the supernatant containing crude membranes was loaded on a 4% to 12% SDS polyacrylamide gel (10 μg/lane). The protein was then transferred to a polyvinylidene difluoride membrane and incubated with rabbit anti-AQP4 antibody (1:1000) followed by anti-rabbit IgG horseradish peroxidase-linked antibody (1:10,000, Amersham Biosciences, Piscataway, NJ, USA), and visualized using enhanced chemiluminescence (Boehringer Mannheim, Indianapolis, IN, USA).
Mathematical Model of Hydrocephalus
A multi-compartment, lumped parameter model was developed to simulate the dynamics of obstructive hydrocephalus and altered AQP4 expression. As diagrammed in Figure 4A, the model included compartments for the cerebral vasculature, brain parenchyma, and CSF space, the latter comprised of ventricular and extra-ventricular subarachnoid spaces, but commonly referred to as the ‘ventricular’ compartment. Each compartment was defined by a volume (V) and pressure (P). The system was driven by fixed rates of CSF production comprised of a choroidal component (JCP) in the ventricular compartment, and a parenchymal component (JBP) representing capillary filtration and metabolic water production in the brain parenchyma. Water movement between compartments was driven by intercompartmental hydrostatic pressure gradients. The flows (J) between adjacent compartments at any given time (t) were computed as:
Parenchyma to Capillary Flow:
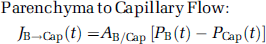
Parenchyma to Ventricle Flow:
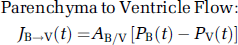
Ventricle to Venous Sinus Flow:
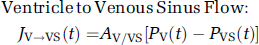
where A is the apparent permeability coefficient, incorporating the AQP4-dependent barrier water permeability and the surface area of water exchange (see Table 2 for parameter values). The term AB/Cap represents the apparent blood-brain barrier permeability and the term AB/V represents the apparent blood-CSF barrier permeability. Alteration of these values allows for changes in AQP4-dependent barrier permeability to simulate the effects of modifying AQP4 expression levels. Accumulation of water in the parenchymal (B) and ventricular (V) compartments was related to net pressure-driven flow. The change in compartmental volume (dV) was determined at each time step by summing the flows into and out of each compartment:
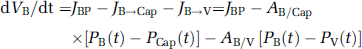
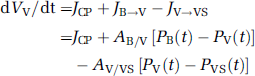
Subsequent compartmental pressures at each time step were then determined from volumes according to a compliance relationship, C(v):
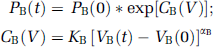
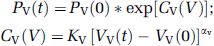
where K and α are compliance coefficients determined empirically from the wildtype data fit. Obstruction of CSF outflow causing hydrocephalus was modeled by reducing the permeability of ventricular outflow into the venous sinus (Av/vs) to 1% of its original value over 24h. A logistic function was used to model the time course of decreasing CSF outflow permeability after kaolin injection:
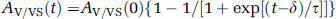
where δ and τ are empirically determined time constants. The model was allowed to run for 24 h of simulated time with normal physiologic parameters to ensure a stable steady state before simulated obstruction of CSF outflow. CSF outflow permeability was then reduced according to the logistic function above over the next 24 h, and the model was performed to 7 days after obstruction. All simulations were run using 1-min time steps. Hydrocephalus in wildtype versus AQP4 null mice was modeled by varying the permeability at the AQP4-containing interfaces (AB/Cap and AB/v). Simulations were written in Objective-C using Xcode (Apple, Redwood City, CA, USA) and run on a PowerBook G4.
Statistics
Data are expressed as mean ± standard error (s.e.). Significance between experimental groups was determined by a two-tailed Student's t-test assuming a 95% confidence interval.
Results
Hydrocephalus Model
Injection of kaolin into the cisterna magna resulted in CSF accumulation and ventricular enlargement. By 5 days after injection there was significant expansion of the lateral, third, and fourth ventricles, as seen in Figure 1A, as well as fluid accumulation within the central canal of the spinal cord (data not shown). Control mice injected with sterile saline did not develop ventricular enlargement. To examine the communication between the intraventricular and extraventricular CSF spaces, Evans blue dye was injected into the lateral ventricles of mice 3 days after kaolin injection. Blue dye was seen in the cisterna magna within 1 min of injection, and flowed freely into the subarachnoid space surrounding the spinal cord (Figure 1B). Dye was not seen in the subarachnoid space of the cranial convexity after craniotomy with intact dura.
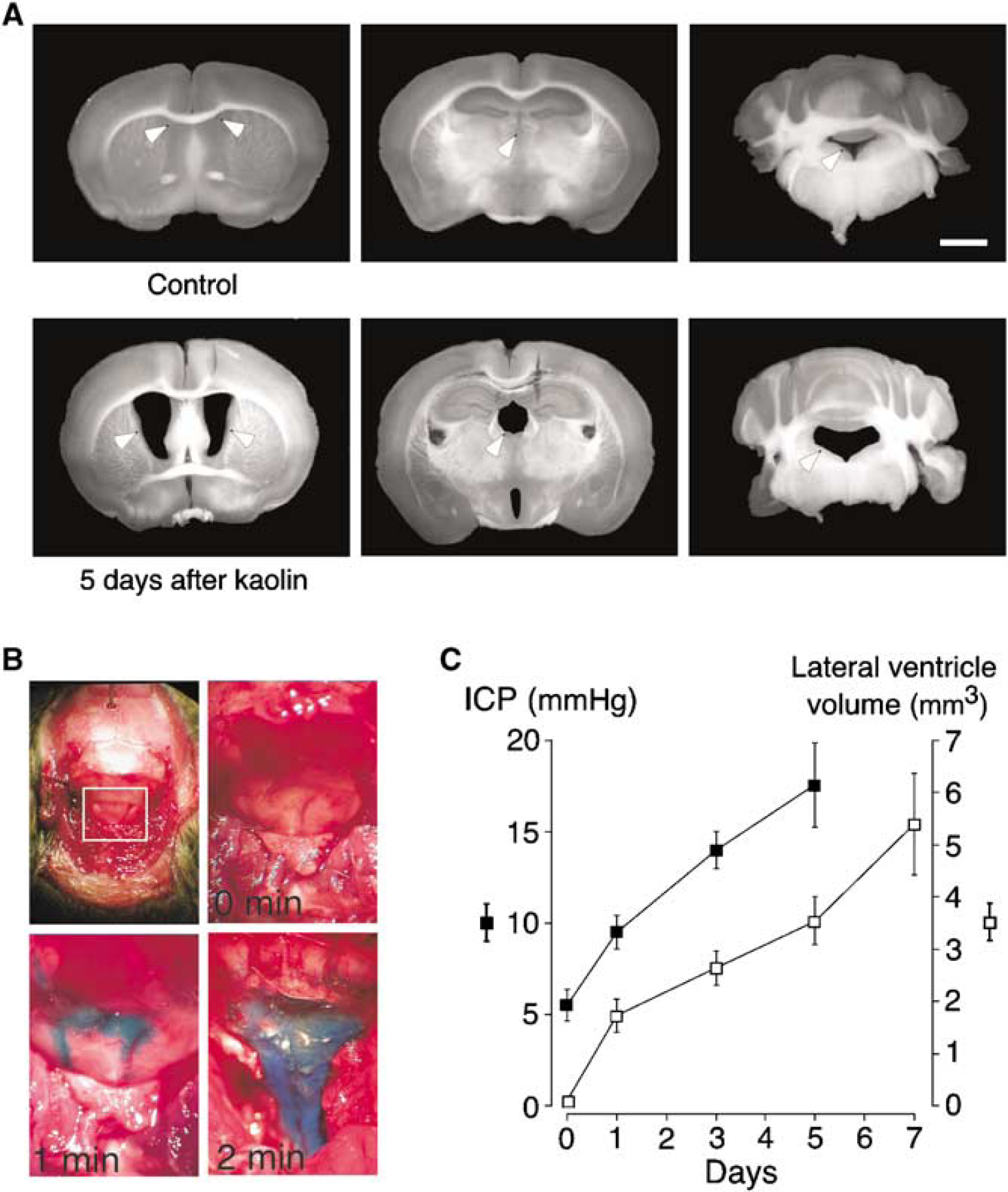
Mouse model of hydrocephalus produced by kaolin injection into the CSF. (
The magnitude of ventricular enlargement associated with hydrocephalus was quantified by summing the lateral ventricle areas from 12 to 14 serial coronal sections of formalin-fixed mouse brains. Lateral ventricle volume in wildtype mice increased from < 0.3 mm3 before kaolin administration to 5.7 ± 1.0 mm3 at 7 days after injection (Figure 1C). The associated elevation in ICP was measured using a micropressure transducer introduced into the brain parenchyma through a small cranial burr hole. At 5 days after kaolin injection, ICP increased by ~12mm Hg above baseline (Figure 1C). Intracranial pressure could not be measured accurately at 7 days after kaolin injection because introduction of the pressure transducer resulted in significant CSF leakage from the burr hole site, resulting in a rapid decrease in ICP.
The clinical course of hydrocephalus was followed by general assessment of illness, including body weight, temperature, and neurologic score, as summarized in Table 1. Despite a significant increase in ventricular volume and ICP, the mice showed only a modest decline in global neurologic function. No focal neurologic deficits were observed; however, all mice developed a global tremor present at rest and exacerbated by action, which began on day 1 after kaolin injection and progressed until death. In a small pilot experiment, 4 out of 5 of wildtype mice died by 10 days after kaolin injection, with the surviving mice returning to a normal, pre-hydrocephalus condition as determined by non-invasive clinical measures.
Clinical characteristics of wild-type and AQP4 null mice after kaolin injection
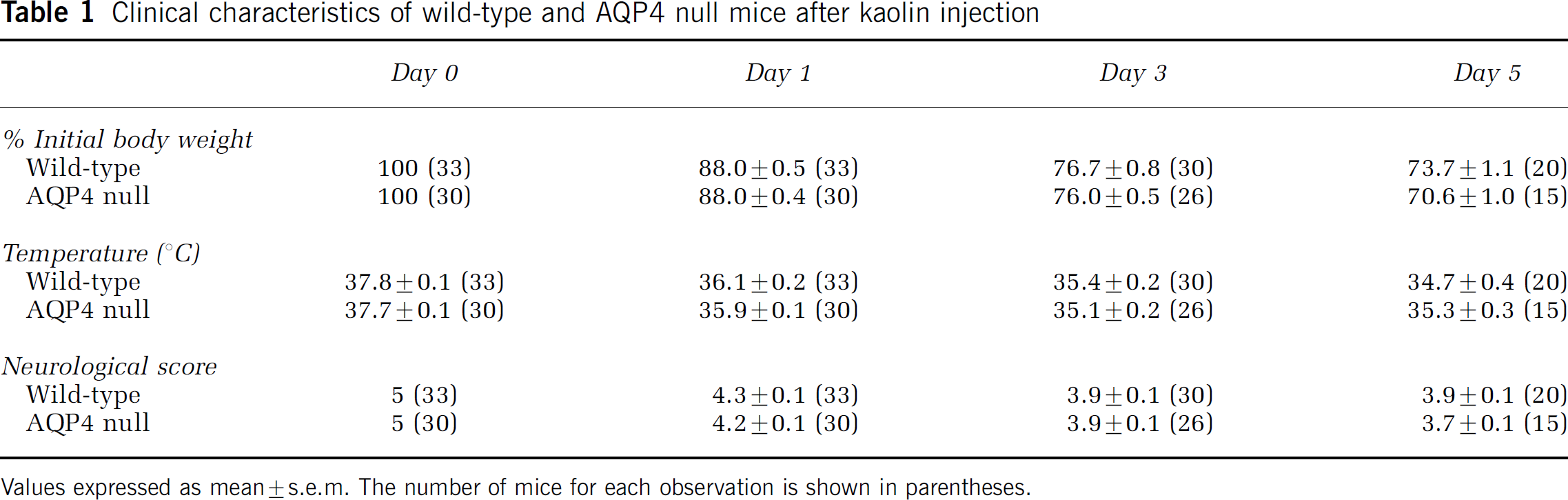
Values expressed as mean 7 s.e.m. The number of mice for each observation is shown in parentheses.
Definition of parameters for hydrocephalus model

Accelerated Progression of Hydrocephalus in Aquaporin-4 Null Mice
The photographs in Figure 2A show increased ventricular expansion after kaolin injection in the AQP4 null mice compared with wildtype mice. Lateral ventricle volume measured in formalin-fixed brains showed no significant differences in volume at baseline or at 1 day after kaolin injection. However, by 3 days after injection, lateral ventricle volume was 3.7 ± 0.5 mm3 in AQP4 null mice versus 2.6 ± 0.3mm3 in wildtype mice (P < 0.05), and increased to 5.1 < 0.5 mm3 in AQP4 null mice versus 3.5 ± 0.5 mm3 in wildtype mice (P < 0.005) by 5 days (Figure 2B). Intracranial pressure was 22 ± 2 mm Hg in AQP4 null mice at 3 days, significantly greater than that of 14 ± 1 mm Hg in wildtype mice (P < 0.005, Figure 2C). Intracranial pressure could not be measured in AQP4 null mice later than 3 days after kaolin injection because of CSF leakage from the burr hole, as mentioned above for wildtype mice at 7 days.
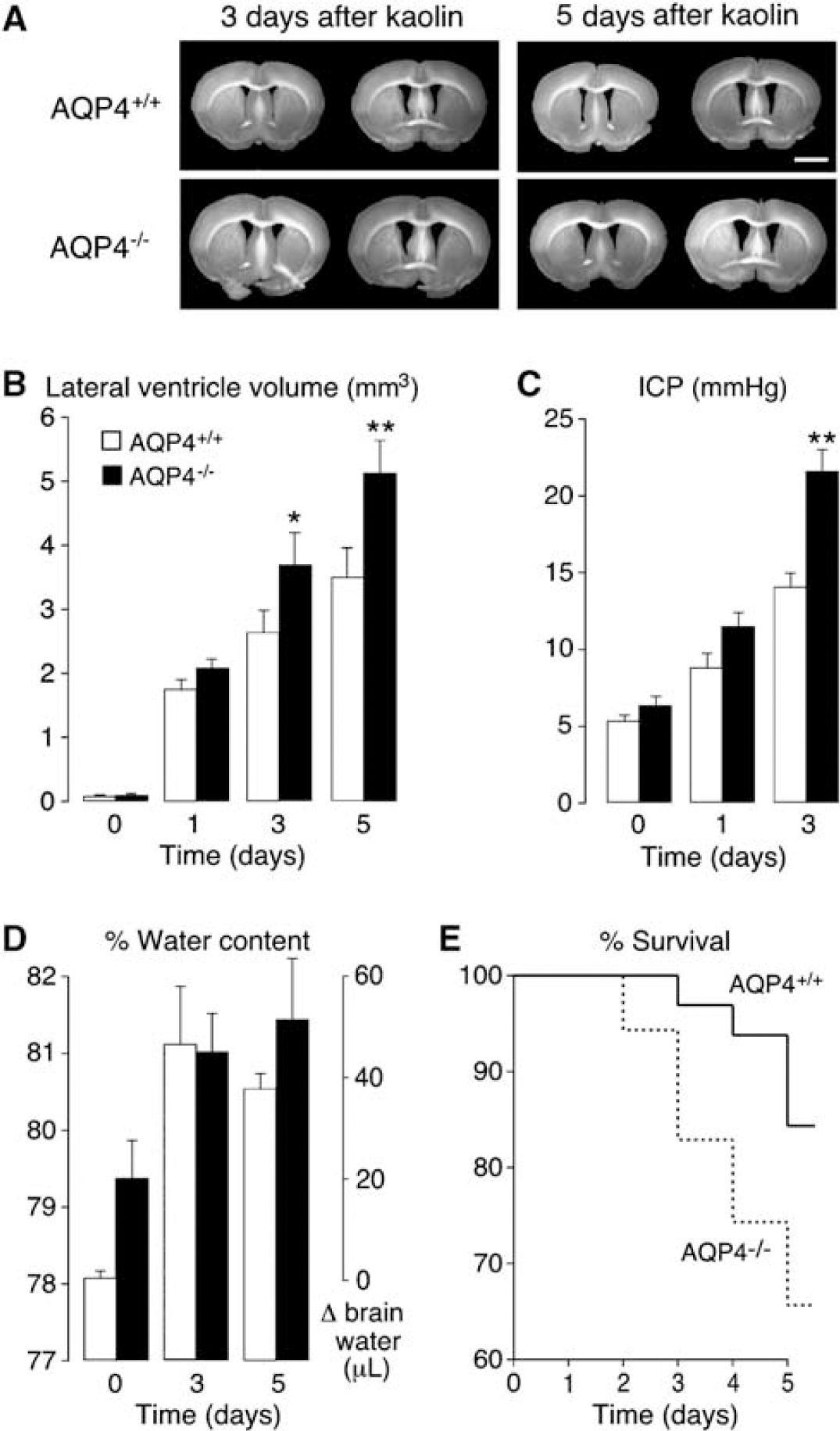
Accelerated progression of hydrocephalus in AQP4 null mice. (
Increased CSF accumulation and elevated ventricular pressure can produce interstitial edema with excess fluid accumulation in brain parenchyma, a well-documented complication of hydrocephalus (Braun et al, 1997; Fishman 1992; Hiratsuka et al, 1982). The water content of periventricular brain parenchyma was measured by column gravimetry over the course of hydrocephalus to evaluate the magnitude of CSF backflow from the ventricular to the parenchymal compartments. At baseline, AQP4 null mice had slightly greater brain water than wildtype mice (79.5% ± ± 0.5% versus 78.1% ±7 0.8%, P 0.01) as reported previously (Bloch et al, 2005). Figure 2D indicates that by 3 days after injection brain water content increased significantly to ~81% in wildtype and AQP4 null mice, and remained elevated at 5 days. However, the parenchymal water content did not differ significantly between wildtype and AQP4 null mice at 3 or 5 days.
A small survival study showed that 66% of AQP4 null mice survived to 5 days after injection as compared with 84% of wildtype mice (Figure 2E). Most mice showed only mild-to-moderate decline in neurologic function before death, which occurred rapidly, probably as a result of acute brain herniation secondary to elevated ICP.
Aquaporin-4 Expression in Hydrocephalus
Increased expression of AQP4 is a common feature in many neuropathologies, including stroke, tumor, trauma, and infection (Badaut et al, 2003; Saadoun et al, 2002, 2003; Vizuete et al, 1999). Although the mechanism of AQP4 upregulation remains unknown, increased AQP4 protein expression in wildtype mice can exacerbate the phenotypic differences between wildtype and AQP4 null mice, significantly affecting ICP and survival (Bloch et al, 2005; Papadopoulos and Verkman 2005). Surprisingly, histologic sections from control and kaolin-injected wildtype mice stained for AQP4 showed similar ependymal and perivascular staining, without obvious differences in expression (Figure 3). Aquaporin-4 expression by immunoblot analysis showed similar AQP4 protein expression before versus 5 days after kaolin injection (Figure 3D). Densitometric analysis of an immunoblot from 5 mice before kaolin injection and 5 mice at 5 days after kaolin injection showed a trend towards a minor (< 2-fold) increase in AQP4 expression, which was not statistically significant (not shown).
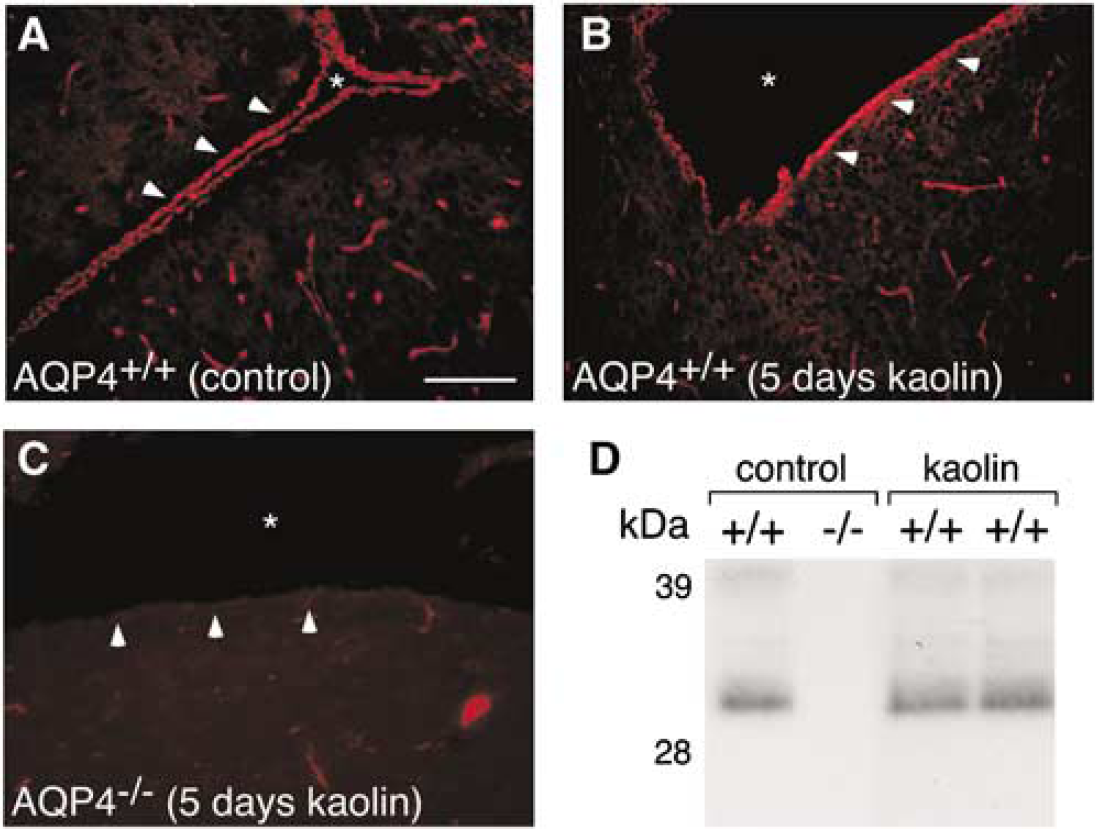
Aquaporin-4 protein expression in mouse model of hydrocephalus. Aquaporin-4 immunostaining of AQP4+/ +control mouse (A), AQP4+/+ (B), and AQP4−/-- (
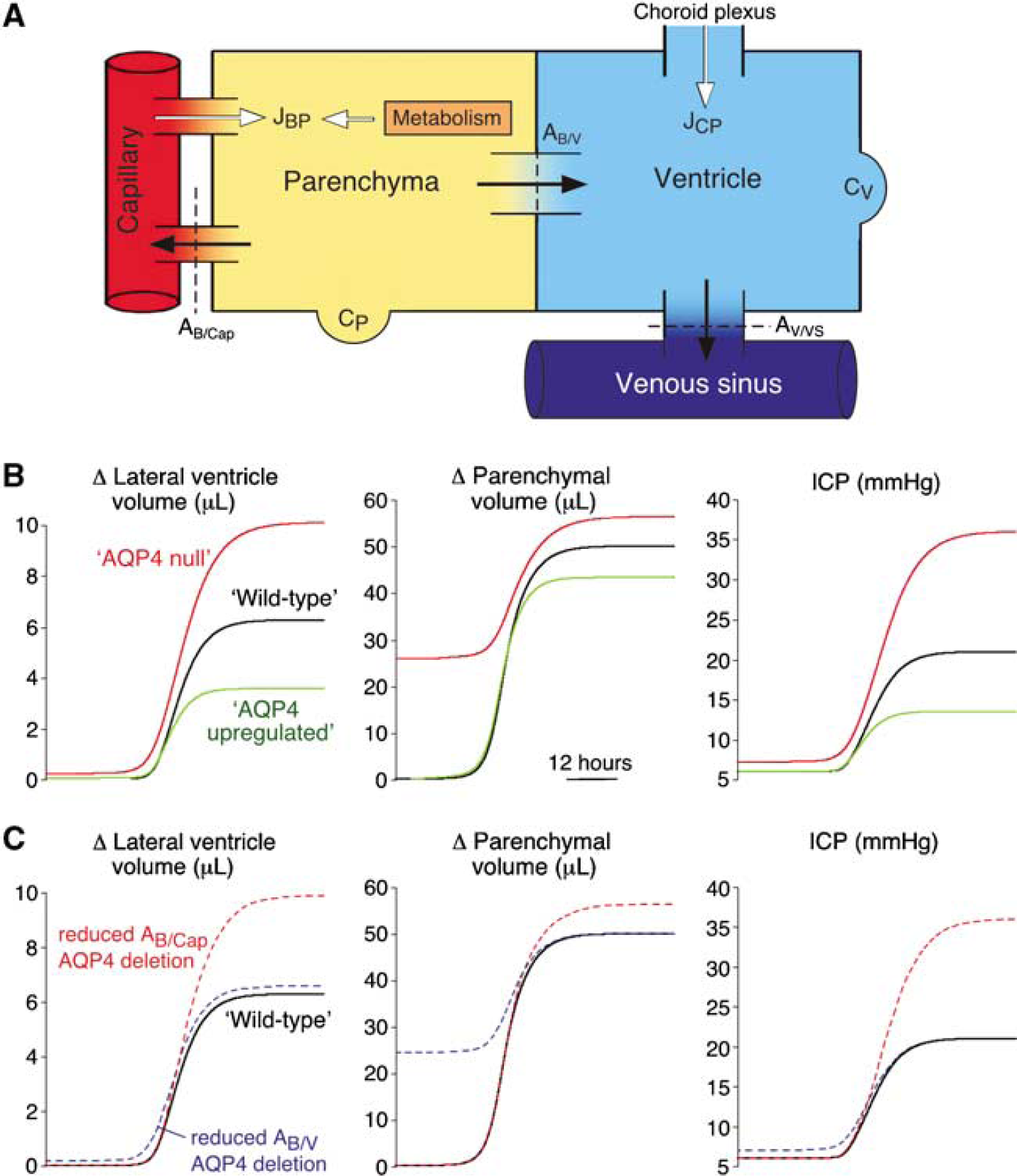
Multi-compartment model of hydrocephalus. (
Computational Modeling of Hydrocephalus
The dynamics of hydrocephalus was modeled as a multi-compartment system as diagrammed in Figure 4A. As described in Materials and methods, the system was driven by fixed rates of CSF production in the ventricular and parenchymal compartments (JCP and JBP). Total CSF production in the mouse has been reported as ~0.3 μL/min (Oshio et al, 2005), with previous estimates (from several species) of choroidal fluid secretion representing 40% to 100% of total CSF production (Cserr 1971; Curl and Pollay 1968; Milhorat et al, 1971). Here, the CSF production was set at 0.3 μL/min with 2/3 of the production from ventricular (choroid plexus) and 1/3 from parenchymal sources, which include metabolic water production and capillary filtration. Baseline pressure for the parenchymal compartment was set at 6 mm Hg based on the experimental data. As summarized in Table 2, the pressure gradient from parenchyma to ventricle was taken as 0.5 mm Hg, and mean cerebral capillary pressure was set equal to parenchymal pressure to eliminate net hydrostatically induced fluid flow at baseline. Permeabilities at each interface (A
Figure 4B shows model predictions for changes in lateral ventricle volume, parenchymal water volume, and parenchymal pressure (within 5% of ventricular pressure at all times) for kaolin-induced CSF outflow obstruction in wildtype mice (black curves). The magnitudes of the maximal volume and pressure changes were comparable to those measured at 5 days, though the model predicted a somewhat more rapid progression than was observed experimentally. Several factors may account for the difference in detailed kinetics between the model and experiment. Kaolin-induced hydrocephalus is reported in other animals to produce marked histologic changes in choroid plexus and b 50% decrease in CSF production (Hochwald et al, 1969; Marlin et al, 1978; Nakamura and Hochwald 1983). Also, ICP elevation during the progression of hydrocephalus is likely to reduce cerebral blood flow and cause capillary collapse at higher pressures, effectively decreasing parenchymal-capillary permeability. These factors, as well as possible alterations in parenchymal and ventricular compliances over the several-day course of disease progression, would slow the rate of ventricular expansion and the increase in ICP. However, since quantitative data on the kinetics of these processes are not available, we could not include these phenomena in the computations.
Having selected model parameters from data on wildtype mice, AQP4 deletion was simulated by reducing twofold the permeabilities of the parenchyma-capillary (AB/Cap) and parenchyma-ventricle (AB/V) interfaces (Figure 4B, red curves). Before reducing the CSF outflow, the system was allowed to reach a new baseline steady state with a mild increase in parenchymal volume (equivalent to a 1.5% increase in brain water content in AQP4 null mice), as was found experimentally. Following outflow obstruction, maximal ventricular volume and parenchymal pressure increased by 61% and 71% more in AQP4 null mice as compared with that simulated for wildtype mice, in agreement with our experimental results. Also, as found experimentally, parenchymal water was predicted to increase to a similar absolute volume in the wildtype and AQP4 null mice.
Given the experimental evidence for AQP4 in the transparanchymal clearance of accumulated ventricular fluid, AQP4 induction was also simulated to predict the potential therapeutic benefit of AQP4 upregulation. In Figure 4B (green curves), a twofold increase in permeabilities of the parenchyma-capillary and parenchyma-ventricle interfaces produced a 43% decrease in maximal ventricular volume and a 36% decrease in ICP, as compared with the wildtype mice.
To examine which of the two AQP4-containing interfaces might be rate-limiting for transparenchymal fluid clearance, the permeabilities of the parenchyma-capillary and parenchyma-ventricle interfaces were separately reduced by twofold. As seen in Figure 4C (blue curve), reduction in permeability of the parenchyma-ventricle barrier (AB/V) produced only a mild increase in parenchymal volume and had little effect on the progression of hydrocephalus. In contrast, reduction in permeability of the parenchyma-capillary barrier (AB/Cap) recapitulated closely the effects of the AQP4 deletion simulation, suggesting that flow across the blood-brain barrier is rate-limiting in transependymal reabsorption of CSF into the cerebral vasculature (Figure 4C, red curve).
Discussion
Cisternal kaolin injection is a well-characterized model of acute, obstructive hydrocephalus described for over 50 years in rats and larger animals (Collins 1979; Hochwald et al, 1969; Nakagawa et al, 1984). Kaolin is believed to induce an inflammatory response in the posterior fossa, causing meningeal fibrosis and obstruction of CSF flow into the subarachnoid space of the cranial cavity (Azzi et al, 1999; DeFeo et al, 1979). In this study, we report the first model of kaolin-induced hydrocephalus in mice. After kaolin injection, mice reliably developed acute hydrocephalus with enlargement of the lateral, third, and fourth ventricles. The cerebral aqueduct was also enlarged and patent throughout its length (data not shown), excluding an inflammatory aqueductal stenosis as the cause of obstruction. As found for kaolin-induced hydrocephalus in larger animals, administration of Evans blue dye into the lateral ventricles of mice showed communication between ventricular and extra-ventricular CSF compartments, with restricted flow into the cranial subarachnoid space.
Mice lacking AQP4 had accelerated progression of kaolin-induced hydrocephalus compared with wildtype mice. By 3 days after kaolin injection, AQP4 null mice had significantly greater ventriculomegaly and ICP than wildtype mice, with the differences increasing further by 5 days. Interestingly, parenchymal water content was increased to a similar level in the wildtype and AQP4 null mice, which may reflect the compliance properties of the brain parenchyma limiting maximal volume expansion.
Normally, extracellular fluid in the brain parenchyma is believed to flow into the cerebral ventricles or subarachnoid space and exit the central nervous system through venous sinuses via the arachnoid granulations. Milhorat et al (1971) showed that CSF production in monkeys was reduced by only 40% after complete choroid plexotomy, indicating a significant parenchymal fluid component of total CSF production. In mice, deletion of aquaporin-1, a water channel expressed in the apical membrane of choroid plexus epithelial cells and believed to facilitate CSF fluid secretion, reduced CSF production by only 25% (Oshio et al, 2005). Several studies have shown that fluorescently or radioactively labeled tracers injected into brain ECS move into the CSF by bulk flow (Marmarou et al, 1994; Reulen et al, 1978; Wrba et al, 1998). Wrba et al (1998) showed that tracer clearance into the CSF in rabbits was dependent on ICP, with reduced clearance into the ventricular space and no transcortical clearance into the subarachnoid space when ICP was greater than 14 mm Hg. In hydrocephalus, ventricular fluid accumulation and increased intraventricular pressure drive fluid flow from the CSF space into the parenchymal extracellular space. MRI studies of human hydrocephalus and histologic evidence from animal models show increased water accumulation within the brain parenchyma, predominantly in subventricular white matter, which has been termed interstitial edema (Braun et al, 1997; Hiratsuka et al, 1982; Ulug et al, 2003). Tracers injected into the lateral ventricles of cats and rats rendered hydrocephalic by cisternal kaolin injection were found to accumulate within the perivascular space of subventricular and subcortical white matter, indicating reversal of flow from the ventricles into the parenchyma (Nakagawa et al, 1985; Sahar et al, 1969). In this study, we also found significant elevation of brain water content in the periventricular tissue of wildtype and AQP4 null mice, indicating considerable interstitial edema. We propose, therefore, that with obstruction of normal CSF flow in hydrocephalus, the transparenchymal route for CSF clearance into the cerebral microvasculature becomes the main route for CSF exit. Unfortunately, this alternative pathway is not sufficient to halt the progression of hydrocephalus, and to date no therapeutic targets have been identified to enhance transparenchymal CSF absorption.
The enhanced progression of hydrocephalus in AQP4 null mice was likely due to reduced transparenchymal CSF absorption. Since AQP4 is expressed in ependymal cells and the subependymal glia limitans, as well as in astrocyte foot processes, deletion of AQP4 could reduce CSF flow across the brain-CSF barrier into the parenchyma and/or reduce the clearance of extracellular parenchymal fluid across the blood-brain barrier into the vasculature. Given the relatively low resistance of the ventricular ependyma compared with the tight blood-brain barrier, AQP4-facilitated flow across the brain-CSF barrier is likely to be minimal. This is supported by the simulations in Figure 4C, which show that a 2-fold reduction in permeability at the ependymal barrier alone would have little effect on the progression of hydrocephalus. In addition, histologic data from animal models of hydrocephalus indicate that the ependymal cells become flattened and develop paracellular rifts with progressive ventricular dilation, further reducing the resistance to paracellular flow (Azzi et al, 1999). We therefore postulate that AQP4 in perivascular astrocyte foot processes is the main molecular route for parenchymal extracellular fluid clearance across the blood-brain barrier. As seen in Figure 4C, a twofold reduction in permeability of the blood-brain barrier alone closely reproduced the simulation of ‘AQP4 deletion’ in Figure 4B. Nakagawa et al (1984) examined the endothelial tight junctions of cerebral capillaries by electron microscopy in a rat model of kaolin-induced hydrocephalus. They found inter-endothelial microfluid pockets between tight junctions with progressive hydrocephalus, suggesting the development of a significant hydrostatic gradient across the capillary wall. This may provide a sufficient driving force to greatly slow the progression of hydrocephalus if AQP4-dependent permeability at the blood-brain barrier could be enhanced.
In most intracranial pathologies, including stroke, hemorrhage, trauma, tumor, and infection, AQP4 has been found to be upregulated (Badaut et al, 2003; Saadoun et al, 2002, 2003; Vizuete et al, 1999), perhaps limiting the possibility of therapeutic induction, and in some cases representing a maladaptive response worsening cerebral edema. Although the mechanism for AQP4 upregulation in these disease states has yet to be determined, evidence from astrocyte cell culture experiments suggest that AQP4 may be upregulated by changes in extracellular osmolality (Arima et al, 2003) and hormonal factors (Gu et al, 2003). In addition, reactive astrocytosis induced by inflammatory cytokines is often associated with increased AQP4 (Badaut et al, 2003; Bloch et al, 2005). Unexpectedly, we did not find significantly altered AQP4 protein expression in the hydrocephalus model here, despite marked changes in ICP and brain water. The reason for the absence of AQP4 upregulation may be that hydrocephalus is not associated with large changes in ECS osmolality and cytokines. In hydrocephalus, the interstitial edema is composed of normal CSF and maintains an isotonic ECS. These findings suggest that therapeutic AQP4 upregulation in hydrocephalus may be possible to increase extracellular fluid clearance at the blood-brain barrier. The clinical benefit of AQP4 induction is predicted by the multi-compartment model, in which only a twofold increase in AQP4-dependent permeability would reduce ventricular enlargement and associated ICP by >40%, and could have a significant impact on clinical outcome.
In summary, we found that AQP4 deletion in mice accelerates the progression of kaolin-induced hydrocephalus by decreasing parenchymal extracellular fluid clearance at the blood-brain barrier. A multi-compartment model of CSF flow dynamics based on the experimental data suggested that, with obstruction of normal CSF outflow, AQP4-dependent, transparenchymal clearance of CSF into the cerebral vasculature is the predominant route of CSF exit. Our results thus identify a new molecular target to enhance CSF absorption and provide a rational basis for evaluation of AQP4 induction as a nonsurgical therapy for hydrocephalus.