
Abstract
The exact mechanism underlying regional cerebral hypoperfusion in the early phase of Alzheimer's disease (AD) is not understood. We have shown in isolated porcine cerebral arteries that stimulation of sympathetic α7-nicotinic acetylcholine receptors (α7-nAChRs) causes release of nitric oxide in parasympathetic nitrergic nerves and vasodilation. We therefore examined if β-amyloid peptides (Aβs), which play a key role in pathogenesis of AD, blocked sympathetic α7-nAChRs leading to reduced neurogenic nitrergic dilation in isolated porcine basilar arteries, using in vitro tissue bath, calcium image, and patch clamping techniques. The results indicated that Aβ1–40, but not Aβ40–1, blocked relaxation of endothelium-denuded basilar arterial rings induced by nicotine (100μ;mol/L) and choline (1 mmol/L) without affecting that induced by sodium nitroprusside or isoproterenol. In cultured superior cervical ganglion (SCG) cells, Aβ1–40, but not Aβ40–1, blocked choline- and nicotine-induced calcium influx and inward currents. The Aβ blockade of the nitrergic vasodilation and inward currents, but not that of calcium influx, was prevented by acute pretreatment with 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors mevastatin and lovastatin. These results suggest that Aβ1–40 blocks cerebral perivascular sympathetic α7-nAChRs, resulting in the attenuation of cerebral nitrergic neurogenic vasodilation. This effect of Aβ may be responsible in part for cerebral hypoperfusion occurred in the early phase of the AD, which may be prevented by statins most likely because of their effects independent of cholesterol lowering. Statins may offer an alternative strategy in the prevention and treatment of AD.
Keywords
Introduction
Alzheimer's disease (AD) is a primary neurodegenerative dementia featuring regional cerebral hypoperfusion in the early phase of disease progression (Kogure et al, 2000; Pavics et al, 1999; Johnson et al, 1998). This is consistent with the findings that reduction in regional cerebral blood flow is closely associated with the severity of dementia (Brown et al, 1996; Hirsch et al, 1997), and that severe cerebrovascular hypoperfusion can result in neuronal degeneration (Lindsberg et al, 1991; Ni et al, 1995). Accordingly, cerebrovascular hypoperfusion may initiate neuronal dysfunction and/or neuronal degeneration. This may explain that cerebrovascular disease and dementia frequently coexist in elderly patients. The exact mechanism underlying the hypoperfusion is not fully clarified.
Increased β-amyloid peptide (Aβ) production is believed to play a critical role in the pathogenesis of AD. β-Amyloid peptide has been shown to have a preferential constrictive effect on cerebral blood vessels in vivo and in vitro (Suo et al, 1998; Niwa et al, 2002; Deane et al, 2003), thereby contributing to the cerebral hypoperfusion. Thus, Aβs may act on endothelial cells and vascular smooth muscle, respectively, causing endothelial dysfunction (Chi et al, 1999; Thomas et al, 1996) and potentiation of smooth muscle constriction induced by other vasoactive substances (Niwa et al, 2001; Deane et al, 2003).
Alternatively, Aβ may block the cerebral neurogenic vasodilation. The cerebral perivascular neuron is known to play a significant role in regulating reactivity of the smooth muscle, particularly, of the large arteries at the base of the brain (Lee et al, 1975, 1976, Toda and Okamura, 2003). Possible effect of Aβ on this cerebral neurogenic vasomotor response has not been examined. We have shown the presence of α7-nicotinic acetylcholine receptors (α7-nAChRs) on cerebral perivascular postganglionic sympathetic nerve terminals originating in the superior cervical ganglion (SCG), but not on the parasympathetic nitrergic neurons (Si and Lee, 2002). This nicotinic receptor subtype has been shown to play an important role in regulating cerebral nitrergic neurogenic vasodilation (Zhang et al, 1998; Si and Lee, 2002). Activation by nicotine and choline of these sympathetic α7-nAChRs releases norepinephrine (NE), which then acts on presynaptic β2-adrenoceptors located on the neighboring parasympathetic nitrergic nerve terminals, resulting in the release of nitric oxide (NO) and vasodilation (Lee et al, 2000; Si and Lee, 2001, 2002, 2003). Since Aβs have been shown to be endogenous ligands/inhibitors for α7-nAChRs in the CNS (Dineley et al, 2002; Hashimoto and Iyo, 2002; Dougherty et al, 2003), we hypothesize that Aβs may inhibit cerebral perivascular sympathetic α7-nAChRs, leading to the blockade of cerebral neurogenic vasodilation.
Circumstantial evidence has recently indicated that patients receiving statin therapy have a reduced incidence of dementia (Atkinson, 2001; Vaughan, 2003). This may be because of the fact that statins (inhibitors of the 3-hydroxy-3-methylglutaryl coenzyme A reductase) such as mevastatin and lovastatin have been shown to inhibit Aβ synthesis (Vaughan, 2003). These lipid-lowering drugs also prevent Aβ-induced neurotoxicity in cultured neurons, and Aβ-induced vasoconstriction and production of vasoactive substances such as prostaglandin E2 and F2α in isolated rat aortic preparations (Paris et al, 2002). We, therefore, further hypothesize that statins may prevent Aβ-induced inhibition of α7-nAChR-mediated cerebral neurogenic nitrergic vasodilation. In the present study, effects of Aβ on perivascular α7-nAChR-mediated neurogenic nitrergic vasodilation in porcine basilar arteries and its prevention by statins, therefore, were examined using in vitro tissue bath, calcium imaging confocal microscopic, and patch clamping techniques. Our results indicated that Aβ1–40, but not its reverse form Aβ40–1, in a concentration-dependent manner blocked α7-nAChR-mediated sympathetic-dependent nitrergic vasodilation in isolated porcine basilar arteries, and calcium influx and inward currents in cultured porcine SCG cells. This Aβ blockade of α7-nAChR-mediated neurogenic vasodilation and inward currents was prevented by pretreatment with statins (lovastatin and mevastatin).
Materials and methods
General Procedure
Fresh heads of adult pigs (60 to 100 kg) of either sex were collected at local packing companies (Excel, Beardstown, IL, USA and Y.T, Springfield, IL, USA). The entire brain, with dura matter attached, was removed and placed in Krebs' bicarbonate solution equilibrated with 95% O2 and 5% CO2 at room temperature. The composition of the Krebs' solution was as follows (mmol/L): NaCl, 122.0; KCl, 5.16; CaCl2, 1.2; MgSO4, 1.22; NaHCO3, 25.6; ethylenediamine-tetraacetic acid, 0.03;
In Vitro Tissue Bath Studies
The ring segment (4 mm long) was cannulated with a stainless-steel rod (30-G hemispherical section) and a short piece of platinum wire and mounted horizontally in a plastic tissue bath containing 6 mL of Krebs' bicarbonate solution. The platinum wire was bent into a U shape and anchored to a gate. The stainless-steel rod was connected to a strain gauge (UC2, Gould) for isometric recording of changes in force, as described in our previous report (Lee et al, 1976). The temperature of the Krebs' solution equilibrated with 95% O2 and 5% CO2 was maintained at 37°C. Tissues were equilibrated in the Krebs' solution for an initial 30 mins and then mechanically stretched to a resting tension of 750 mg (Zhang et al, 1998).
The basilar arterial ring segments were then precontracted with U-46619 (0.3 to 3 μmol/L) to induce an active muscle tone of 0.5 to 0.75 g. Transmural nerve stimulation (TNS) at 8 Hz, nicotine (100 μmol/L), and choline (1 mmol/L) were applied to induce a relaxation. After relaxation induced by TNS, 100 μmol/L nicotine, or 1 mmol/L choline, the arteries were washed with prewarmed Krebs' solution. A similar magnitude of active muscle tone was induced with U-46619 again, and TNS was repeated (to serve as a control comparing with the relaxation elicited by TNS before the wash). Effects of different concentrations of Aβ1–40 (1 to 1000 nmol/L) were then administered, and TNS and nicotine/choline at the same concentrations before the wash were repeated. To examine possible modification of statins on Aβ effect, mevastatin (1 to 10 μmol/L) or lovastatin (1 to 10 μmol/L) was added 15 mins before Aβ application. To avoid possible development of tachyphylaxis on repeated applications of nicotinic agonists, at least 90 mins with six washes (every 15 mins) was allowed before the next application of nicotinic agonists (Zhang et al, 1998; Lee et al, 2000; Si and Lee, 2001, 2002).
For TNS, tissues were electrically, transmurally stimulated with a pair of electrodes through which 100 biphasic square-wave pulses of 0.6 ms in duration and 200 mA in intensity were applied at various frequencies (Zhang et al, 1998). Stimulation parameters were continuously monitored on a Tektronix oscilloscope. The neurogenic origin of this TNS-induced response was verified by its complete blockade by tetrodotoxin (TTX) (0.3 μmol/L). At the end of each experiment, papaverine (PPV) (100 μmol/L) was added to induce a maximum relaxation. The magnitude of a vasodilator response was expressed as a percentage of the maximum response induced by PPV (Zhang et al, 1998).
For examining effects of experimental drugs on relaxation induced by isoproterenol or sodium nitroprusside (SNP), concentration–response relationships for these two vasodilators were obtained by a cumulative technique in arteries without endothelial cells in the presence of active muscle tone induced by U-46619. After the arterial rings were washed with prewarmed Krebs' solution, a similar magnitude of active muscle tone was again induced by U-46619. The experimental drugs were then added, and 15 mins later, concentration–response relations for isoproterenol or sodium nitroprusside were repeated. EC50 values (the concentration that produces 50% of the maximum relaxation) were determined for each arterial ring. From these values, the geometric means EC50 with 95% confidence intervals (Fleming et al, 1972) were calculated.
The endothelial cells of all arterial ring segments were mechanically removed by a standard brief gentle rubbing of the intimal surface with a stainless-steel rod having a diameter (25 to 30 G) equivalent to the lumen of the arteries (Zhang et al, 1998; Lee et al, 2000). A complete removal of endothelial cells was verified by lack of effect of N-nitro-
Superior Cervical Ganglion Cell Culture
Freshly dissected SCG from animals were placed in cold Hibernate A (Life Technologies, Carlsbad, CA, USA) solution (Si and Lee, 2001). After being cut into smaller pieces, the ganglia were transferred to Mg2+/Ca2+-free Hank's balanced salt solution (HBSS) containing papain (2 U/mL; Sigma, St Louis, MO, USA), collagenase D (1.2 mg/mL; Boehringer Mannheim, Germany) and dispase (4.8 mg/mL, Gibco, Carlsbad, CA, USA), and were incubated for 50 mins at 37°C. Cells were released by gentle trituration at the end of the incubation. The cell suspension was centrifuged at 300g for 5 mins. The pellet was gently resuspended in Neurobasal culture medium (Life Technologies), containing B27 (1:50 dilution, Life Technology),
Intracellular Calcium Imaging
Between 3 and 7 days in culture, the SCG cells were used to examine effects of nicotine and choline on calcium influx in these cells by confocal microscopy. The cells were washed with physiologic buffer (130 mmol/L NaCl, 5 mmol/L KCl, 10 mmol/L HEPES, 5 mmol/L glucose, 2 mmol/L CaCl2, 2 mmol/L MgCl2, pH 7.3), and were loaded with 3 μmol/L fluo-4, AM in physiologic buffer and incubated at room temperature for 30 mins. The cells were washed with calcium indicator-free buffer to remove any dye that is nonspecifically associated with the cell surface, and then incubated for additional 30 mins to allow complete de-esterification of intracellular AM esters. Nicotine (100 μmol/L) or choline (1 mmol/L) was then applied and the calcium influx measured. Aβ1–40 at 0.1 to 1 μmol/L was added 15 mins before application of nicotine, choline, or KCl (50 mmol/L). Calcium fluorescence images were examined with a Fluoview Olympus confocal microscope (Melville, NY, USA). Fluo-4 was excited at 488 nm, and emitted fluorescence was filtered with a 535 ± 25 nm bandpass filter and read into a computer running Fluoview software and quantified (Si and Lee, 2002).
Electrophysiologic Study
Electrophysiologic study using patch-clamp recording was performed as described previously (Liu et al, 2000). In brief, a glass coverslip containing cultured neurons was transferred from the growth medium to a recording chamber containing the extracellular recording solution (in mmol/L: 140 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, 10 HEPES, pH 7.35, and 320 mOsm) on a phase-contrast microscope (Olympus IMT2, Japan). The choline-induced current was recorded with the whole-cell configuration of the patch-clamp technique (Hamill et al, 1981; Liu et al, 2000) at room temperature. Recording electrodes were manufactured from capillary glass (PG52151-4, World Precision Instruments, Sarasota, FL, USA). After filling with intracellular solution (in mmol/L: 145 K-gluconate, 10 KCl, 1 EGTA, 10 HEPES, 5 K-ATP, and 0.25 GTP (pH 7.35)), electrode impedance in the extracellular recording solution was about 5 M. Standard extracellular solution was constantly perfused at 2 ml/min. All recordings were conducted at room temperature.
A Master-8 (AMPI, Jerusalem, Israel) controlled SF-77B Perfusion Fast-Step (Warner Instrument Corp., Hamden, CT, USA) solution exchange system was used for rapid application of drugs onto SCG neurons. This system used a three-barreled glass perfusion head (<20 ms exchange at the cell). The distance from the perfusion head to the cell was ~200 μm with flow controlled manually using a micromanipulator. External drug application was delivered by gravity flow from a linear array of quartz tubes. The recording chamber was continuously perfused and cells were exposed to a constant flow of bath solution between drug applications.
Drugs Used and Statistical Analysis
The following drugs were used: (−)-nicotine, choline chloride,
Results were expressed as means ± s.d. Statistical analysis was evaluated by analysis of variance (ANOVA) and Student's t-test for paired or unpaired samples as appropriate. The P <0.05 level of probability was accepted as significant.
Results
Choline-, Nicotine-, and Transmural Nerve Stimulation-Induced Neurogenic Vasodilation in Porcine Basilar Arteries
Consistent with our previous reports (Si and Lee, 2002, 2003), the porcine basilar arteries without endothelial cells, in the presence of active muscle tone induced by U-46619 (0.3 μmol/L), were relaxed exclusively on TNS (8 Hz), and applications of nicotine (100 μmol/L) or choline (1 mmol/L) (Figures 1 and 2). The relaxation induced by nicotine and choline was significantly blocked by TTX (0.3 μmol/L, n = 7, data not shown), and was abolished by
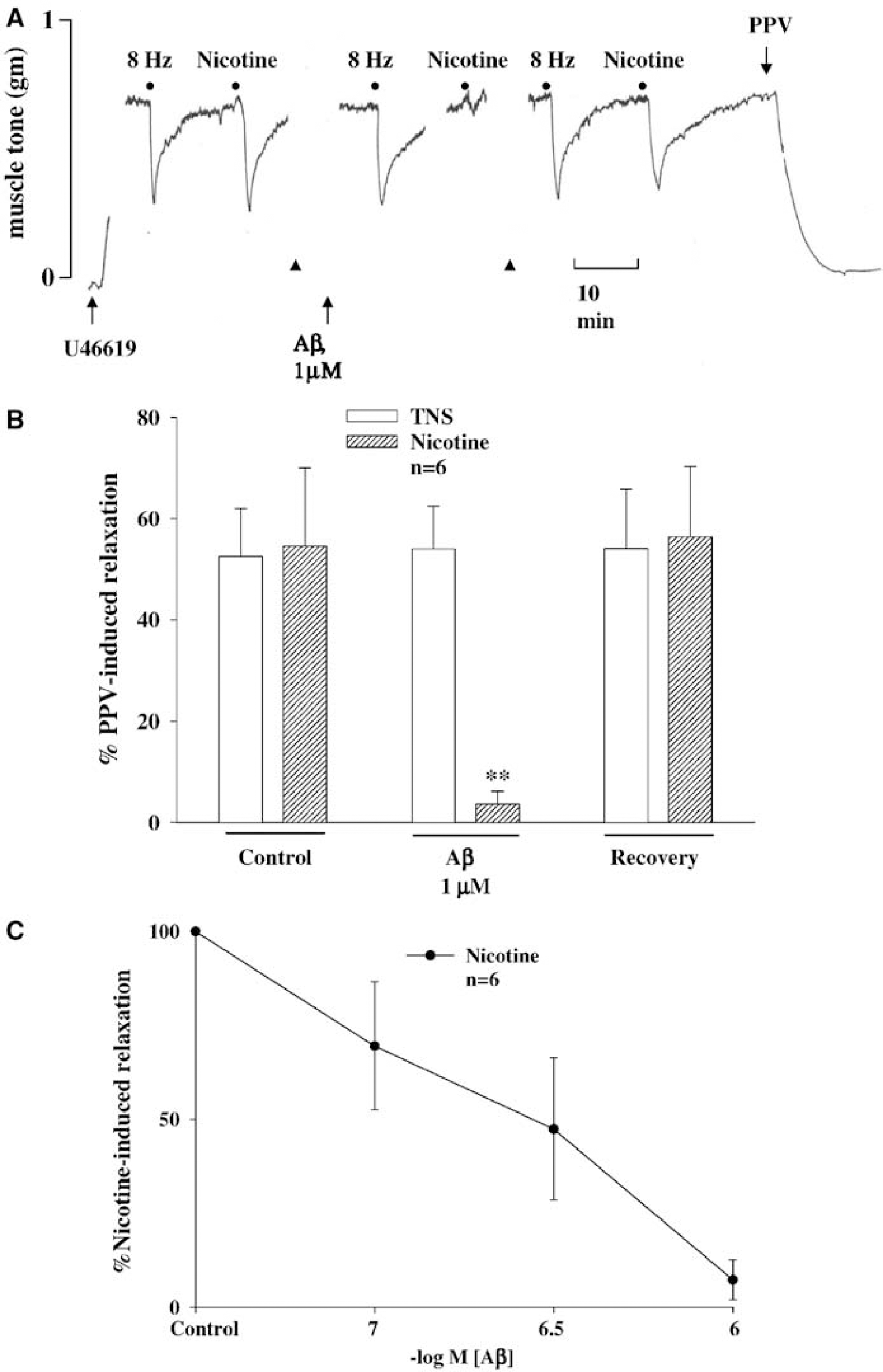
Effects of Aβ1–40 on nicotine (Nic) and transmural nerve stimulation (TNS)-induced relaxation of porcine basilar arteries without endothelial cells. (
Aβ1–40 Inhibited Nicotine- and Choline-Induced Neurogenic Vasodilation
Since TNS at 8 Hz, nicotine at 100 μmol/L, and choline at 1 mmol/L induced maximum relaxation, these parameters, which have previously been used by us and others (Toda and Okamura, 2003; Zhang et al, 1998; Lee et al, 2000; Si and Lee, 2001, 2002, 2003), were used in the subsequent studies. As reported previously by many investigators, neurogenic vasodilation induced by nicotinic agonists diminished on repeated applications of this agonist with short time intervals (Zhang et al, 1998; Si and Lee, 2002). Accordingly, in the present study, a 90 mins interval with six washes was allowed before repeating each application of nicotine and choline. Three consecutive, reproducible relaxations induced by nicotine (100 μmol/L) or choline (1 mmol/L) were obtained, which were not significantly different (Zhang et al, 1998; Si and Lee, 2002). Furthermore, the relaxation elicited by repeated TNS at 8 Hz, like other reports in the porcine basilar arteries (Zhang et al, 1998; Lee et al, 2000), was reproducible and was not different.
In basilar arteries (without endothelial cells) in the presence of active muscle tone induced by U-46619 (0.3 μmol/L), relaxation induced by nicotine (100 μmol/L) and choline (1 mmol/L) was blocked by Aβ1–40 in a concentration-dependent manner (Figures 1A–1C and 2A and 2B, n = 6). These concentrations of Aβ, however, did not affect the basal or active vascular tone or TNS-elicited relaxation (Figures 1A, 1B, and 2A). The IC50 values for Aβ1–40 against nicotine- and choline-induced relaxation were 2.12 (0.85 to 7.64) × 10−7 mol/L and 1.89 (0.74 to 6.03) × 10−7 mol/L, respectively, which were not significantly different (P>0.05). The Aβ blockade of relaxation induced by nicotine and choline was reversible after washing off Aβ (Figures 1A, 1B, and 2A). As a negative control, Aβ40–1 at similar concentrations never affected relaxation induced by TNS, nicotine, or choline (n = 5, data not shown).
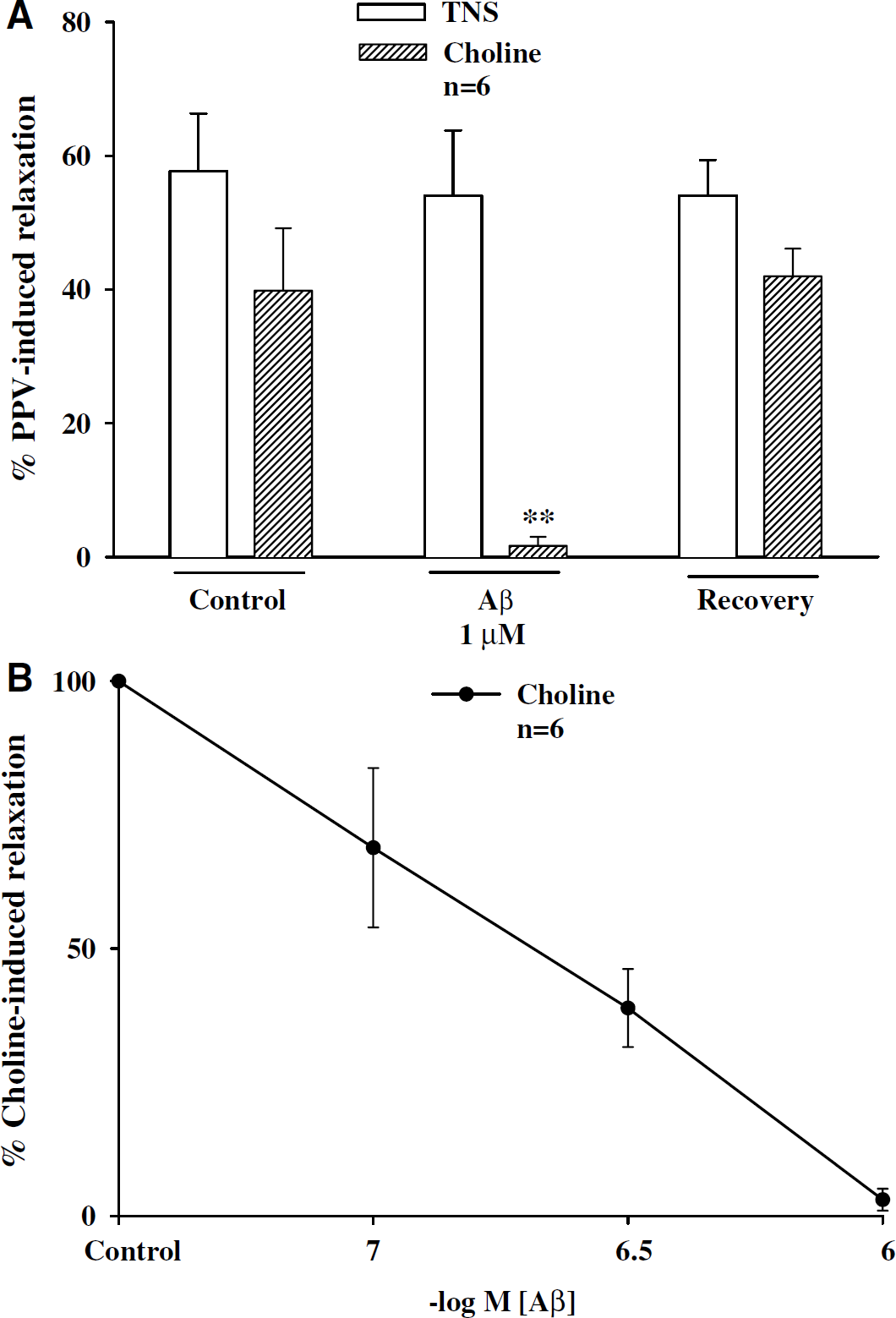
Effects of Aβ1–40 on choline and transmural nerve stimulation (TNS)-induced relaxation of porcine basilar arteries without endothelial cells. In the presence of active muscle tone induced by U-46619 (0.3 μmol/L), β-amyloid peptide (Aβ) at 1 μmol/L almost abolished choline (1 mmol/L)-induced relaxation in porcine basilar arterial ring (estimated as the percent of papaverine (PPV)-elicited maximum relaxation) without affecting the relaxation elicited by TNS at 8 Hz. The results were summarized in (
Aβ1–40 did not Affect Sodium Nitroprusside- or Isoproterenol-Induced Relaxation in Basilar Arteries
With active muscle tone induced by U-46619 (0.3 μmol/L), porcine basilar arteries denuded of endothelial cells relaxed on application of SNP and isoproterenol in a concentration-dependent manner (Figure 3), a result similar to that reported previously (Lee et al, 2000). Aβ1–40 (1 μmol/L) did not affect relaxation induced by SNP (10 nmol/L to 0.1 mmol/L) or isoproterenol (1 nmol/L to 3 μmol/L) in arteries denuded of endothelial cells (Figure 3). The EC50 values for SNP in control and in the presence of Aβ were 5.16 (2.15 to 14.78) × 10−6 mol/L and 5.83 (2.54 to 13.65) × 10−6 mol/L, respectively (n = 7, P>0.05), and those for isoproterenol were 2.54 (0.75 to 7.82) × 10−7 mol/L and 2.93 (0.88 to 9.53) × 10−7mol/L, respectively (n = 7, P>0.05).
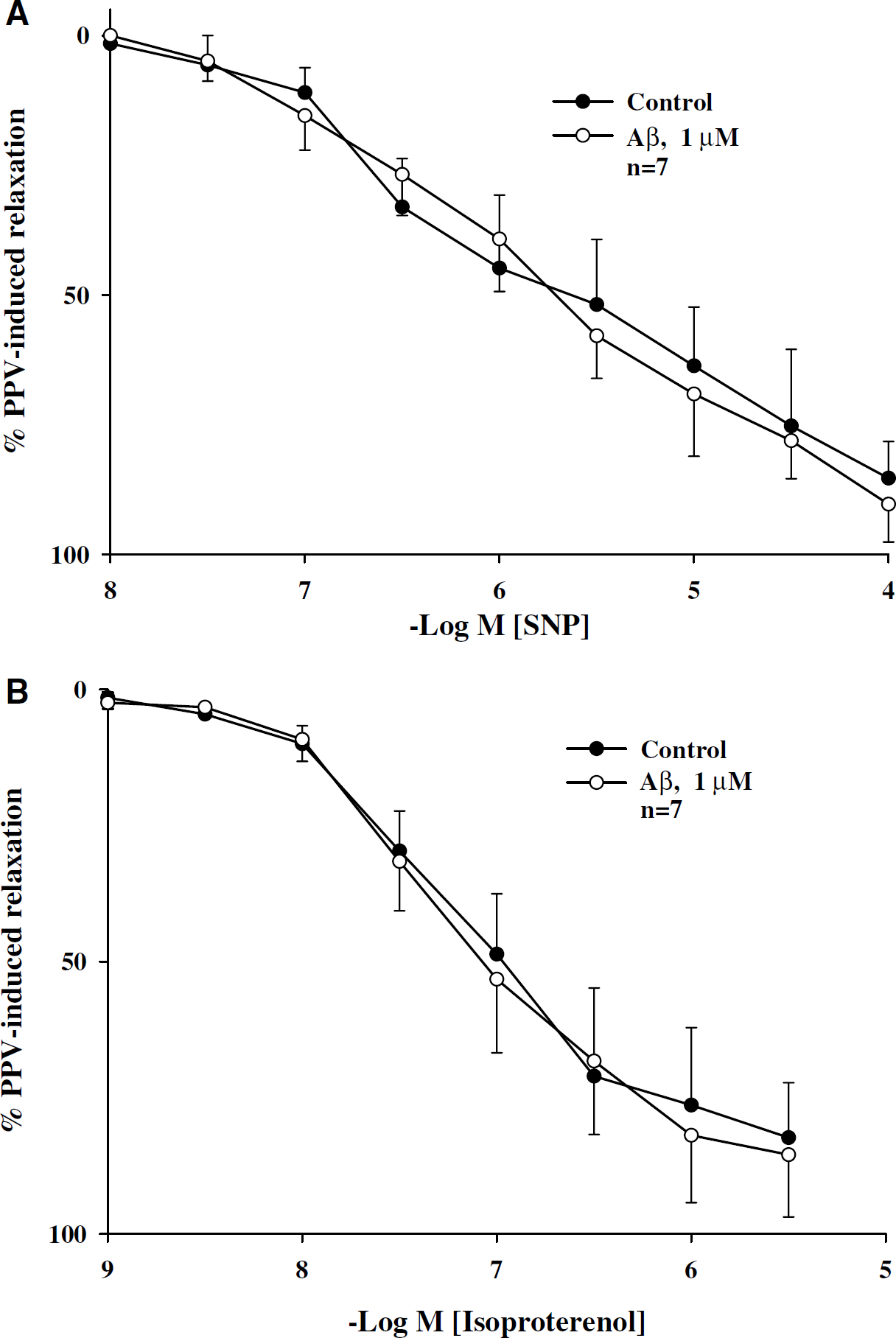
Effects of Aβ1–40 on sodium nitroprusside (SNP)- and isoproterenol-induced relaxation in porcine basilar arteries without endothelial cells. In the presence of active muscle tone induced by U-46619 (0.3 μmol/L), porcine basilar arterial rings relaxed on application of SNP (
Statins Prevented β-Amyloid Peptide Inhibition of Nicotine- and Choline-Induced Relaxation in Porcine Basilar Arteries
In basilar arteries (without endothelial cells) in the presence of active muscle tone induced by U-46619 (0.3 μmol/L), Aβ1–40 inhibition of relaxation induced by nicotine (100 μmol/L) and choline (1 mmol/L) was prevented by pretreatment with mevastatin (10 μmol/L, n = 5, Figure 4), and lovastatin (10 μmol/L, n = 6, Figure 5). Mevastatin or lovastatin alone (at 10 μmol/L) had no effect on basilar arterial tone or relaxation induced by TNS (Figures 4 and 5), nicotine, or choline (data not shown). Mevastatin or lovastatin at lower concentration (1 μmol/L) slightly but insignificantly reversed Aβ1–40 inhibition of relaxation induced by nicotine (100 μmol/L) or choline (1 mmol/L) (n = 4, data not shown). A higher concentration (10 μmol/L) of mevastatin and lovastatin, therefore, was used in the subsequent studies for calcium influx and patch-clamp experiment.
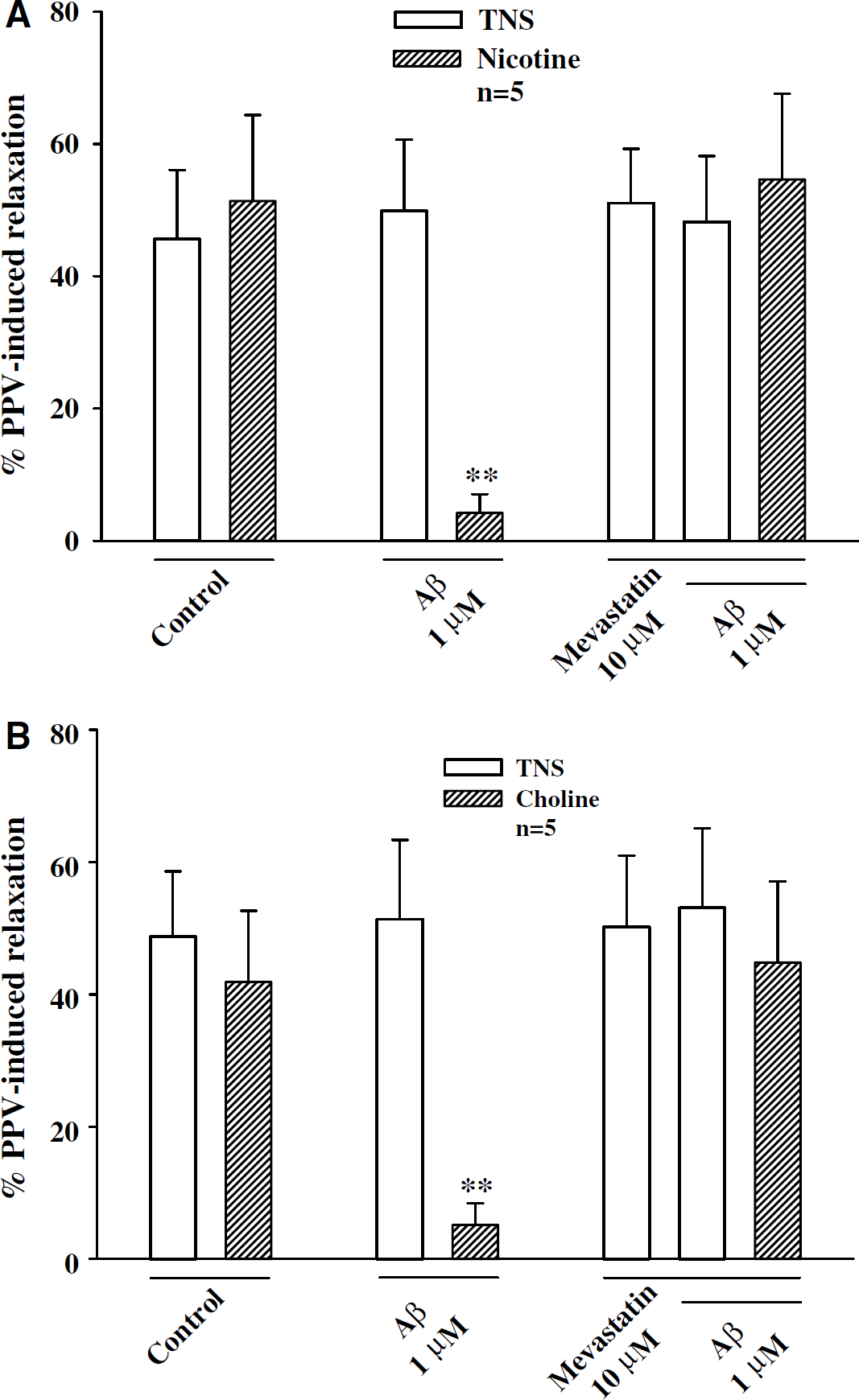
Mevastatin prevented Aβ1–40 inhibition of nicotine-and choline-induced relaxation of porcine basilar arteries without endothelial cells. Basilar arterial rings were preincubated with mevastatin (10 μmol/L) for 15 mins before application of β-amyloid peptide (Aβ), which inhibited nicotine- (
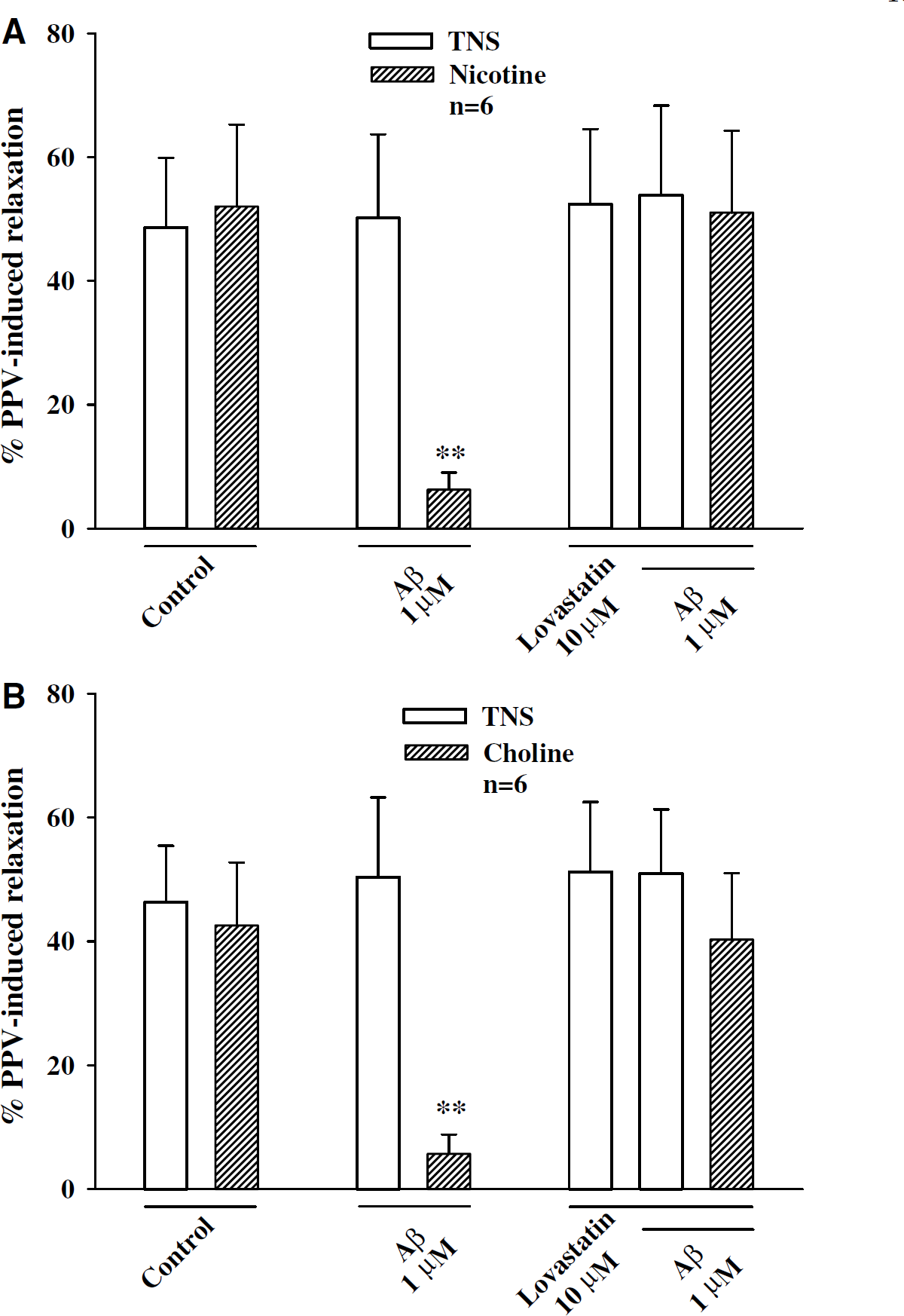
Lovastatin prevented Aβ1–40 inhibition of nicotine- and choline-induced relaxation of porcine basilar arteries without endothelial cells. Basilar arterial rings were preincubated with lovastatin (10 μmol/L) for 15 mins before application of β-amyloid peptide (Aβ), which inhibited nicotine- (
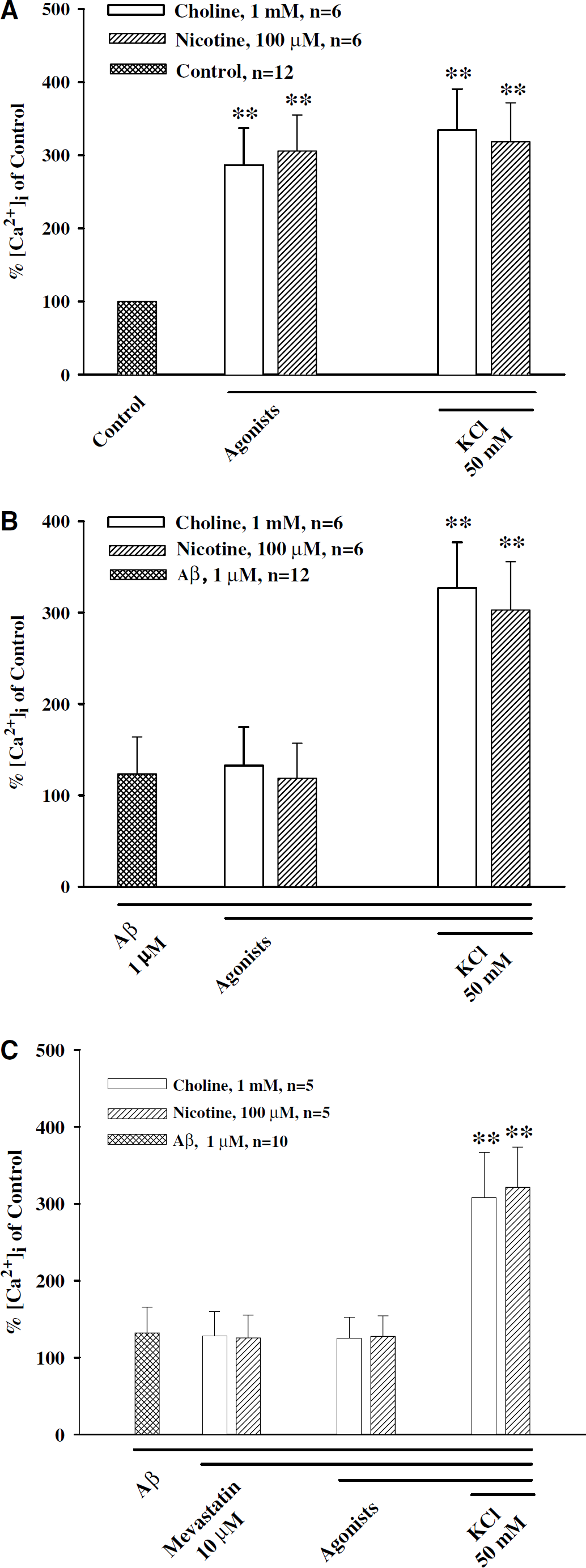
Aβ1–40 Blocked Nicotine- and Choline-Induced Calcium Influx in the Porcine Superior Cervical Ganglion Neurons
Cultured SCG cells, like cerebral perivascular sympathetic neurons in whole-mount arterial preparations, have been shown to contain dense α7-nAChRs (Si and Lee, 2001, 2002), which form membrane cation channels possessing high Ca2+ permeability (Sargent, 1993). Using the intracellular calcium imaging indicator fluo-4, AM to examine calcium influx, both nicotine (100 μmol/L) and choline (1 mmol/L) induced a significant increase in calcium image in the SCG cells (Figures 6B and 7A), a result similar to our previous reports (Si and Lee, 2002, 2003). Addition of KCl (50 mmol/L) did not further increase intracellular calcium. KCl (50 mmol/L) on its own induced similar level of calcium influx as after nicotine or choline application (Si and Lee, 2002, 2003) did not further increase intracellular calcium. The nicotine- and choline-induced calcium influx in the SCG cells was blocked in cells pretreated with Aβ1–40 (1 μmol/L), which alone did not significantly affect basal intracellular calcium level (Figures 6D, 6E, and 7B, n = 12). As a negative control, Aβ40–1 did not affect choline- and nicotine-induced calcium influx (n = 6, data not shown).
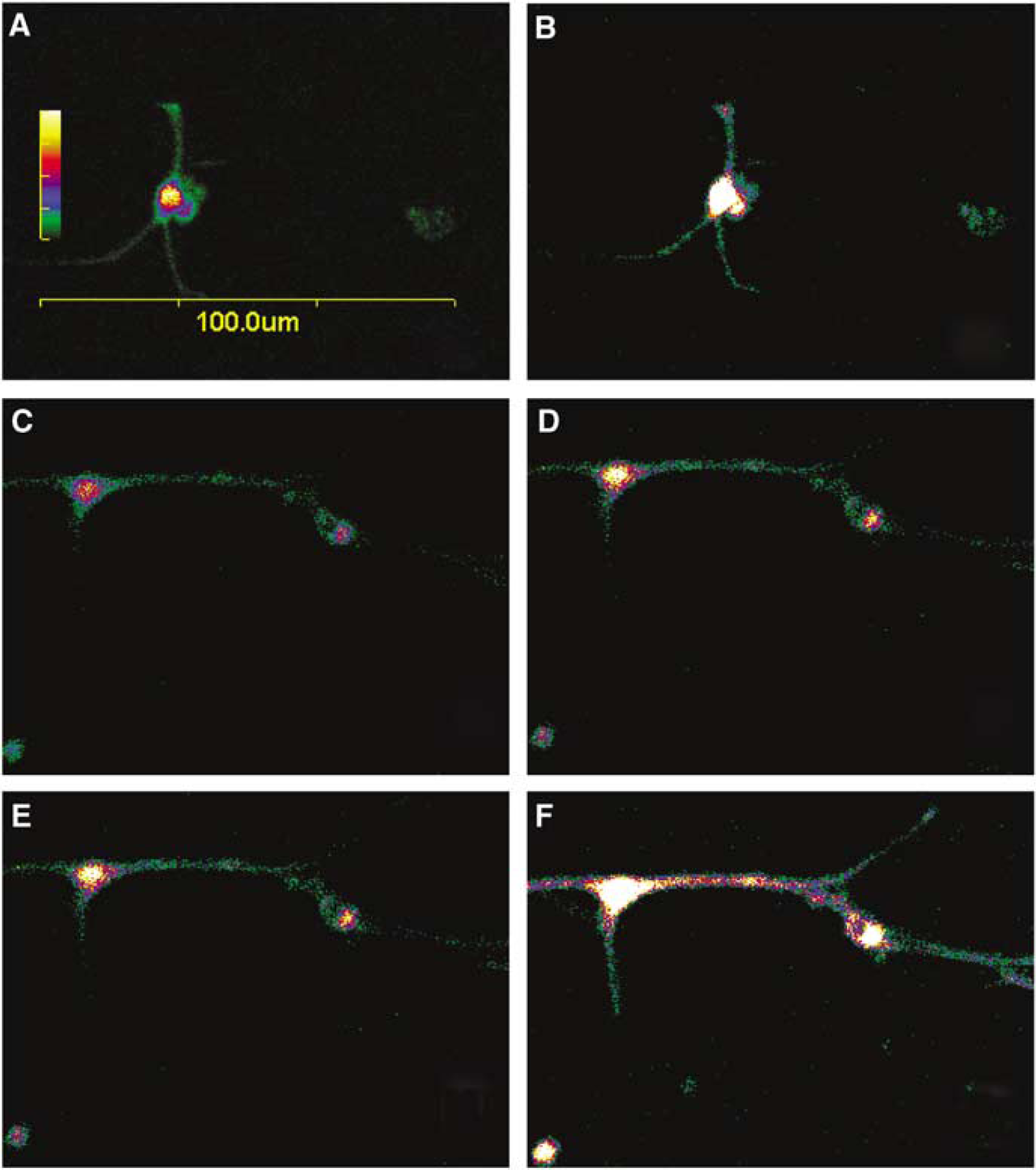
Effects of β-amyloid peptide (Aβ) on choline-induced calcium influx in cultured porcine superior cervical ganglion (SCG) cells. The neuronal cells were loaded with fluo-4, AM (3 μmol/L) in physiologic buffer and incubated at room temperature for 30 mins, and the basal calcium image in each cell served as own control (
Aβ1–40 blockade of nicotine- and choline-induced calcium influx in the porcine superior cervical ganglion (SCG) cells. Cultured SCG cells were loaded with fluo-4, AM (3 μmol/L) in physiologic buffer and incubated at room temperature for 30 mins. Nicotine (100 μmol/L) or choline (1 mmol/L) was applied to the medium and changes in the calcium image in the neuronal cells were determined. Both choline and nicotine induced a significant calcium influx (
Despite of the presence of Aβ1–40 blockade of calcium influx induced by nicotine and choline, KCl (50 mmol/L) still induced a significant calcium influx in the same cells (Figures 6F and 7B), which was comparable to that seen in preparations in the absence of Aβ (Figure 7A). Mevastatin and lovastatin (10 μmol/L), which alone did not affect basal calcium, did not prevent Aβ blockade of choline- or nicotine-induced calcium influx in the SCG cells (Figure 7C, n = 5 for each group).
Statins Prevented β-Amyloid Peptide-Blocking Effect of Choline-Induced Inward Currents in the Porcine Superior Cervical Ganglion Neurons
The whole-cell mode of the patch-clamp technique was used to record responses evoked by choline, an α7-nAChR-selective agonist (Si and Lee, 2002), in porcine SCG neurons. The inward currents were evoked by 10 mmol/L choline (Figure 8A, n = 10). The choline-induced currents were blocked after 5 mins perfusion of the neurons with 1 μmol/L of Aβ1–40 (Figures 8A and 8B, n = 10), which Per se did not evoke a current (data not shown). The blockade by Aβ1–40 was prevented by 5 mins perfusion of 10 μmol/L of lovastatin and mevastatin (Figures 8A and 8B, n = 5 each). Application of statins alone did not elicit any current (n = 10, data not shown). The choline-induced currents were fully recovered after washing off Aβ (Figure 8B, n = 10). Aβ40–1 did not affect choline- or nicotine-induced inward current (n = 4, data not shown).
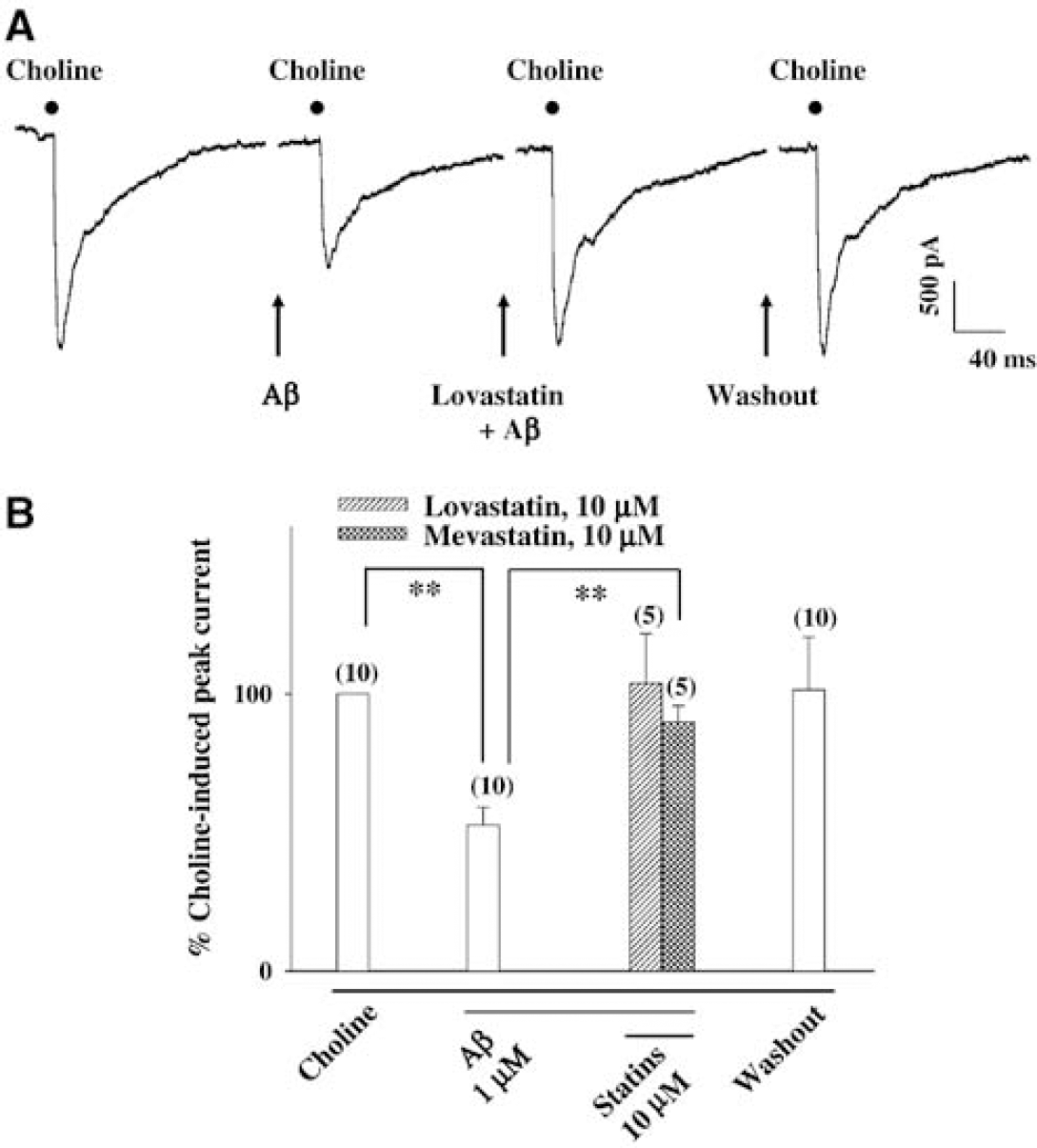
Aβ1–40 inhibition and its prevention by statins of choline-induced inward currents in the porcine superior cervical ganglion (SCG) cells. Choline induced a fast decaying currents rectify inwardly in cultured SCG neurons (
Discussion
The major findings of the present study are that Aβ1–40 inhibits the presynaptic α7-nAChRs located on the cerebral perivascular sympathetic neurons resulting in a decreased nitrergic neurogenic dilation in isolated porcine basilar arteries, and that this Aβ1–40 inhibition of α7-nAChRs is prevented by statins. The inhibition of cerebral neurogenic vasodilation by Aβ1–40 may in part underlie cerebral hypoperfusion, an early feature of AD (Kogure et al, 2000; Pavics et al, 1999; Johnson et al, 1998). β-Amyloid peptide may have early pathogenic consequences by blocking cerebral nitrergic vasodilation in the AD process before its deposition in the brain parenchyma and cerebral blood vessels, and dementia onset. Statins, which reversed Aβ effects as shown in the present study, may be useful in the prevention and treatment of AD.
Ample evidence has been presented to indicate that Aβ, a peptide produced by processing the amyloid precursor protein, is a major component of the senile plagues present in the brain and cerebral blood vessels of patients with AD (Dickson, 1997; Atkinson, 2001). Evidence also suggests that Aβs play crucial roles in the brain dysfunction associated with AD (Dickson, 1997; Atkinson, 2001). Mechanisms by which Aβs exert their pathogenic effects, however, have not been fully clarified. Regional cerebral hypoperfusion is one of the earlier clinical manifestations in patients with AD (Kogure et al, 2000; Pavics et al, 1999; Johnson et al, 1998), and that Aβs have been shown to impair the function of the cerebral circulation (Iadecola, 2003). It has been shown that Aβs induce vasoconstriction in vivo and in vitro, and enhance constriction of vascular smooth muscle induced by other vasoactive substances such as endothelin and thromboxane analog U-46619 (Thomas et al, 1996; Suo et al, 1998; Niwa et al, 2002; Deane et al, 2003). β-Amyloid peptides also are known to attenuate endothelium-dependent dilation in isolated peripheral and cerebral arteries (Thomas et al, 1996, 1997; Crawford et al, 1998; Iadecola et al, 1999; Price et al, 2001). These findings provide a logical explanation for cerebral vasoconstrictive effect of Aβ. Our present results provide an additional mechanism underlying the Aβ-induced vasoconstriction in large cerebral arteries by inhibiting cerebral perivascular neurogenic nitrergic vasodilation.
Both Aβ1–40 and Aβ1–42 have been shown to be vasoactive in causing cerebral and peripheral vasoconstriction and enhancing that induced by other vasoactive substances (Thomas et al, 1996, 1997; Crawford et al, 1998; Price et al, 2001). Cerebral amyloid angiopathy (CAA) is an integral part of the AD process with frequent amyloid deposition in cerebral blood vessels (the media and the adventitia of the medium and small arteries), which destroys wall integrity (Atkinson, 2001). It is known that Aβ1–40 is the predominant isoform found in the walls of cerebral blood vessels, although Aβ1–42 is the major isoform deposited in senile plaques. It also has been shown that in the CAA-affected cerebral blood vessels, an increase in Aβ1–40, but not Aβ1–42, is found in cortical and meningeal microvessels (Alonzo et al, 1998). Furthermore, some reports indicate that Aβ1–40 is significantly more vasoactive than Aβ1–42 both in vitro (Crawford et al, 1998) and in vivo (Niwa et al, 2001). For these reasons, we focused on the possible effect of Aβ1–40 on the cerebral neurogenic vasodilation in the present study.
The functional significance of cerebral nitrergic vasodilator innervation in controlling cerebral circulation is well recognized (Lee, 2000; Toda and Okamura, 2003), although other factors such as functional hyperemia may play a more important role in regulating regional CBF (Niwa et al, 2000; Zonta et al, 2003; Iadecola, 2003). Recently, α7-nAChRs located on the sympathetic nerves have been shown to play an important role in mediating cerebral nitrergic neurogenic vasodilation (Zhang et al, 1998; Si and Lee, 2001, 2002). Activation of the sympathetic α7-nAChRs by nicotinic agonists has been shown to release NE, which then diffuses to act on β2-adrenoceptors located on the neighboring nitrergic nerves, causing the release of NO from these nerves with resultant vasodilation (Zhang et al, 1998; Lee et al, 2000; Si and Lee, 2001, 2002, 2003). In the present study, this α7-nAChR-mediated neurogenic vasodilation was attenuated by Aβ1–40 but not by reverse peptide Aβ40–1. Similar blockade was observed for Aβ1–42 (our preliminary data). The Aβ1–40 attenuation of the nicotine- and choline-induced neurogenic vasodilation appears to be because of its blockade of the sympathetic α7-nAChRs. This is consistent with the findings by others that Aβ is an antagonist for neuronal α7-nAChRs (Svensson and Nordberg, 1999; Wang et al, 2000; Liu et al, 2001; Petit et al, 2001; Dineley et al, 2002; Dougherty et al, 2003), and that Aβ1–40 acts directly on the α7-nAChRs expressed in Xenopus oocytes (Dineley et al, 2002). This is supported further by the present finding that Aβ1–40 blocks nicotine- and choline-induced calcium influx in the SCG neurons, which is known to be mediated by α7-nAChRs (Si and Lee, 2002). Similar results also were found for Aβ1–42 (our preliminary data). This is also consistent with the findings by others that Aβ1–42 inhibits nicotine-induced α7-nAChR-mediated presynaptic calcium influx in the rat hippocampus and neocortex (Dougherty et al, 2003).
It should be noted that the Aβ concentration used in the present study is higher than that reported in endothelial cells (Price et al, 2001). It is possible that the cerebral perivascular nerves may be less sensitive to Aβ than are the endothelial cells. In addition, variations in effective Aβ concentrations may also result from different experimental paradigms to be used. In cultured cells, exogenously administered Aβ can reach its receptors almost directly. Thus, it is conceivable that lower Aβ concentrations are enough to exert its biologic actions. However, in whole-mount tissues like those used in the present study, perivascular nerve endings are imbedded in the adventitia where physical barriers exist to preclude direct nerve contact with Aβ, thereby necessitating higher concentrations of Aβ to observe an effect. Additional reason may be that relatively high concentrations of agonists, namely nicotine and choline, were applied such that a correspondingly higher concentration of ‘antagonists’, or Aβ, is needed to counteract their actions.
It has been shown that aggregated Aβs may alter membrane fluidity and its response, suggesting that aggregated Aβs are pathogenic (Ross and Poirier, 2004). In our present study, Aβ was freshly prepared several hours before the experiment. While we cannot completely rule out the possibility that Aβ aggregation may occur during such a short period of time, numerous reports have indicated that it typically takes 1 to 3 days for Aβ to fully aggregate (Ross and Poirier, 2004). At this moment, whether Aβ aggregation plays an important role in the observed vasoconstrictive effects remains unknown. Since soluble Aβ is known to inhibit endothelial NO release and vasodilation (Price et al, 2001; Gentile et al, 2004), we speculate that both soluble and aggregated Aβ may be pathogenic.
The exact mechanism of Aβ blockade of α7-nAChR remains unclear. The intracellular events after Aβ binding on the SCG are not exactly clear (Verdier and Penke, 2004). Without solid knowledge of the existence of Aβ-specific membrane receptors, it is premature to speculate on the interplay between nicotine- and Aβ-induced intracellular signal transduction. This important issue requires further investigation.
As already been stated, α7-nAChR-mediated cerebral neurogenic vasodilation involves activation by NE of β2-adrenoceptors on the nitrergic neurons leading to release of NO and vasodilation (Si and Lee, 2002). The possibility that Aβ1–40 may attenuate neurogenic dilation by blocking either β2-adrenoceptors on the nitrergic neurons and/or NO–cyclic 3‘, 5’-guanosine monophosphate coupling in the smooth muscle cells, therefore, was examined. These two possibilities, however, are unlikely, since Aβ1–40 did not affect relaxation induced by either isoproterenol (a nonspecific β-adrenoceptor agonist) or sodium nitroprusside (an NO ‘releaser’). Since cerebral nitrergic neurogenic vasodilation is predominant in the neurogenic control of the cerebral circulation (Lee, 2000; Toda and Okamura, 2003), Aβ blockade of cerebral nitrergic neurogenic vasodilation, therefore, may suggest a likely reduction of the cerebral blood flow leading to cerebral hypoperfusion.
Statins, which are inhibitors of the 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis), have been shown to inhibit Aβ-induced neurotoxicity in cultured neuronal cells, and vasoconstriction and production of vasoactive substances such as prostaglandin E2 and prostaglandin F2α in the rat aorta (Paris et al, 2002). Observational studies in human have recently suggested that patients receiving statin therapy have a reduced incidence of dementia (Vaughan, 2003). Our present study showed that mevastatin and lovastatin (two most commonly used anticholesterol drugs) prevented the Aβ blockade of nicotine- and choline-induced relaxation (Figures 4 and 5). Results from the present patch clamping study also indicated that choline-induced inward currents, which are mediated by α7-nAChRs (Long et al, 2003), in cultured porcine SCG neurons were attenuated by Aβ1–40, and the attenuation was prevented by statin pretreatment. These results suggest the possible prevention by acutely administered statins of Aβ-induced inhibition of α7-nAChR-mediated transmitter release and cerebral nitrergic neurogenic vasodilation. It has been reported by others that chronic statin treatment may increase CBF via endothelial nitric oxide synthase (eNOS) activation (Budzyn et al, 2004; Yamada et al, 2000). Thus, both acute and chronic effects of the statin on neurogenic and endothelial components may contribute to its beneficial effects on the cerebral circulation.
Aβ1–40 attenuation of choline-induced calcium influx, unlike that of choline-induced neurogenic vasodilation or inward currents, however, was not prevented by statin pretreatment. This may be because of already desensitized α7-nAChRs to the initial calcium influx on Aβ preincubation (Dougherty et al, 2003). It is possible that statins prevent Aβ inhibition of sympathetic α7-nAChR-mediated nitrergic vasodilation of cerebral arteries by restoring the noncalcium component of the inward currents via α7-nAChRs. The exact mechanism for the statins in this specific effect remains unknown. Interestingly, a recent report has indicated that simvastatin (another anticholesterol drug) may reduce NMDA-induced whole-cell currents and protect cortical culture against NMDA neurotoxicity but does not affect the NMDA-induced increase in intraneuronal calcium concentrations (Zacco et al, 2003), suggesting possible existence of calcium-independent pathways that may be acted on by statins.
Since no cholesterol synthesis inhibitors other than statin was used in the present study, it is difficult to conclude whether the observed statin effect on Aβ-mediated attenuation of nitrergic neurogenic dilation depends on its inhibitory action on cholesterol synthesis. However, the statin prevention/protection effect is most likely because of its noncholesterol-lowering effect because of their acute effects in ‘reversing’ vasorelaxation and inward currents induced by choline and nicotine. In addition to lowering cholesterol synthesis, statins may possess pleiotropic beneficial actions including antioxidant, anti-inflammation, and improvement of endothelial functions with cholesterol-independent components (Kwak et al, 2003). Results from clinical studies also have found that subjects taking statins have a lowered risk of developing dementia than those who did not exhibited hyperlipidemia or those who were not receiving lipid-lowering agents, suggesting that the effect of statins was independent of lipid-lowering effect (Atkinson, 2001). It is not likely because of binding of statins to Aβ resulting in a neutralization of Aβ either (Paris et al, 2002). The intriguing possibility of statin itself being capable of binding to α7-nAChR and molecular mechanisms underlying statins-mediated prevention of Aβ effects require further investigation.
In summary, we have shown that Aβ1–40 blocks nicotine- and choline-induced sympathetic α7-nAChR-mediated cerebral nitrergic dilation in isolated porcine basilar arteries, providing a possible explanation that vascular Aβ can cause a decreased release of neurogenic NO and cerebral neurogenic vasodilation, that is, ‘vasoconstriction‘. A significant constriction of large arteries is expected to reduce the blood flow to the whole brain. This together with the reported Aβ effect on regional functional hyperemia (Niwa et al, 2000) may in part underlie cerebral hypoperfusion, an early feature of AD. These data further suggest an early pathogenic role for Aβs in the AD process by blocking α7-nAChR-mediated vasomotor response that precedes Aβ deposition and dementia onset. This potentially detrimental effect of Aβ may be prevented by statins, suggesting that statins may offer an alternative strategy in the prevention and/or treatment of AD. Furthermore, the in vitro tissue bath technique used in the present study appears to be a useful platform for searching for new anti-AD drugs acting on the α7-nAChR.