
Abstract
Pericytes are known to regulate brain capillary endothelial functions. The purpose of this study was to define the hemostatic regulatory role of human brain pericytes. We used blood–brain barrier models consisting of human pericytes grown on transwell membrane inserts and cocultured with human brain microvascular endothelial cells (HBEC), or pericytes grown in direct contact with HBEC. When grown in cocultures in which pericytes were physically separated from endothelial cells, pericytes induced significant changes in endothelial tissue plasminogen activator (tPA) messenger ribonucleic acid (mRNA) and protein: tPA mRNA level was decreased in pericyte cocultures (52% ± 25% of monocultures, P < 0.05) and tPA protein level was decreased (66% ± 23% of monocultures, P < 0.05). Pericyte effects on endothelial fibrinolysis were enhanced when the two cell types were cocultured in direct contact, with tPA protein reduced in cocultures compared with monocultures (25% ± 15% of monocultures, P < 0.05). Endotoxin (lipopolysaccharide (LPS)), used as a standardized stimulus to define brain-specific inflammation-induced change, amplified pericyteinduced enhanced release of the tPA inhibitor plasminogen activator inhibitor-1 (PAI-1); the latter was released by endothelial cells first cocultured with pericytes and then incubated with LPS in the absence of pericytes. Pericytes (in contrast to endothelial cells and astrocytes) were found to be the principal in vitro source of the serpin protease nexin-1 (PN-1), known to have primarily antithrombin effects. These in vitro findings suggest that pericytes negatively regulate brain endothelial cell fibrinolysis, while pericyte expression of PN-1 may provide endogenous anticoagulant activity.
Keywords
Introduction
Effective treatment of ischemic stroke using tissue plasminogen activator (tPA) (NINDS rt-tPA Study Group, 1995) along with extensive experimental evidence (e.g., Asahi et al, 2005; Zhang et al, 1999) have emphasized the importance of the brain's endogenous endothelial-dependent fibrinolytic system. Tissue plasminogen activator is a serine protease that processes plasminogen into proteolytically active plasmin, a key regulator in fibrinolysis. Plasminogen activator inhibitor-1 (PAI-1) inhibits the activation of plasminogen into plasmin by forming stable 1:1 complexes with tPA. Endothelial fibrinolysis is substantially influenced by the balance between tPA and PAI-1, and reduced fibrinolytic capacity predisposes to increased risk of thrombosis (Juhan-Vague et al, 2003). Prior work has shown both coordinate expression of endothelial tPA and PAI-1 (Santell and Levin, 1988) and dissociated expression of the two proteins (Shi et al, 1992). Fibrinolysis may also be modulated by the serpin (serine protease inhibitor) protease nexin-1 (PN-1), which can inhibit tPA and is a potent inhibitor of thrombin (Richard et al, 2004; Vaughan et al, 1994).
The relationship between fibrinolysis and stroke has been further emphasized by work showing that genetically determined impairment of tPA release significantly increases the risk of lacunar stroke (Jannes et al, 2004), and that an important stroke prevention drug increases the release of brain endothelial tPA (Kim et al, 2005). Moreover, there is evidence that stroke risk is transiently increased after systemic infections (Paganini-Hill et al, 2003; Smeeth et al, 2004) and that this might be explained by impairment of fibrinolysis associated with these infections (Macko et al, 1996). Experimental stroke models using tPA-deficient mice have shown both protective (Wang et al, 1998) and deleterious effects of the tPA deficiency (Tabrizi et al, 1999). The current view is that deleterious effects of tPA in ischemic stroke, for example, hemorrhagic conversion and enhanced excitotoxicity and edema formation, probably require that tPA leave the intravascular space; clot lysis effects of tPA appear to predominate as along as tPA remains intravascular (Kaur et al, 2004; Pfefferkorn and Rosenberg, 2003).
The blood–brain barrier (BBB) is a highly organized microenvironment with functions well beyond the traditional view of a ‘gatekeeper’. We have previously shown an important regulatory role for the BBB in the expression of endothelial-dependent hemostasis factors. Astrocytes regulate brain microvascular endothelial fibrinolysis as well as thrombomodulin expression via transforming growth factor-beta (TGF-β) (Kim et al, 2003; Tran et al, 1996, 1998, 1999). The consequence of brain-specific hemostasis regulation is the creation of a microenvironment at the BBB that is primarily procoagulant.
While there is substantial evidence showing the role of astrocytes in brain-specific hemostasis regulation, little is known of the role of pericytes. Brain pericytes cover 22% to 32% of the outer aspect of rat capillaries (Sims, 1991), and are present in isolated preparations in the ratio of one pericyte per three brain capillary endothelial cells (Pardridge, 1999). While pericytes are separated from endothelial cells by basement membrane, gap junctions and ‘peg-and-socket’ contacts between pericytes and endothelial cells have been reported (Cuevas et al, 1984; Wakui et al, 1989). Brain pericytes have functional properties consisting of contraction, mediation of inflammation, and regulation of endothelial cell activity (Nag, 2003). Pericyte-endothelial coculture studies have shown the capacity of pericytes to induce and rapidly associate with capillary-like structures, as well as activating endothelial cells and upregulating integrin (Balabanov and Dore-Duffy, 1998; Minakawa et al, 1991; Robinson et al, 1990). We hypothesized that brain pericytes have an important role in regulating brain endothelial fibrinolysis factors.
Materials and methods
Cell Preparation
Human brain microvascular endothelial cells (HBEC) were harvested from normal human brain cortical tissue (provided by Applied Cell Biology Research Institute, Kirkland, WA, USA at passage 2). Human brain microvascular endothelial cells (Figure 1A) were cultured in CSC medium (Applied Cell Biology Research Institute, Kirkland, WA, USA) containing 10% fetal bovine serum (FBS). Identification of endothelial cells was confirmed by immunoreactivity (more than 95%) for von Willebrand factor antibody (Dako, Carpinteria, CA, USA) and uptake of acetylated LDL labeled with 1-1′-dioctacecyl-l-l-3-3-3′-3′-tetamethyl-indocarbocyanine perchlorate (Biomedical Technologies, Stoughton, MA, USA). Human brain microvascular endothelial cells at passages 7 to 11 were used for experiments. Human pericytes were obtained from normal human brain cortical tissue (Applied Cell Biology Research Institute, Kirkland, WA, USA, provided at passage 2). Pericytes (Figure 1B) were identified by positive immunoreactivity (more than 90%) for α smooth muscle actin, positive immunoreactivity for vimentin intermediate filaments, negative immunoreactivity for von Willebrand factor and glial fibrillary acidic protein (Dore-Duffy, 2003; Fujimoto and Singer, 1987), and absence of hill and valley morphology. Pericytes at passage 4 to 7 were used for experiments. Human astrocytes were prepared by Cambrex (Walkersville, MD, USA, provided at passage 2), and obtained from 17.5- to 23-week gestation fetus brain. Astrocytes were maintained on Dulbecco's modified Eagle's medium (DME) containing 10% FBS. Astrocytes were characterized by > 95% immunoreactivity for glial fibrillary acidic protein. Astrocytes used for these experiments were taken from passage 3 to 7.
Human brain capillary endothelial cells (
Pericyte–Endothelial Cocultures
We studied pericyte–endothelial interactions using transwell cocultures (Corning Costar, Cambridge, MA, USA) and direct-contact mixed cocultures. Endothelial cells and pericytes were grown on surfaces coated with 0.1% gelatin (Sigma, St Louis, MO, USA). For the transwell cocultures, when confluent HBEC monolayers formed, culture medium was changed with fresh medium. Cocultures were established by transferring confluent pericyte transwell inserts over endothelial cells. After 48 h incubation, endothelial cells and conditioned media were collected for assays. For direct-contact mixed cocultures, pericytes were plated over endothelial cells. Endothelial cell counts were performed on both mono- and coculture preparations after 48 h incubation; endothelial cell counts were similar for monoculture versus coculture transwell preparations, or for direct contact preparations (data not shown). For endotoxin (lipopolysaccharide (LPS)) effects on fibrinolysis, 1 μg/mL of LPS (Sigma, St Louis, MO, USA) was incubated with HBEC for 6 h at the end of 48 h coculture period. For neutralizing antibody studies, 20 μg/mL of anti-TGF-β monoclonal antibody (R&D Systems Inc, Minneapolis, MN, USA) was incubated with HBEC. For levels of PN-1 protein and messenger ribonucleic acid (mRNA), conditioned media and cell pellets of HBEC, human astrocytes (Cambrex, Walkersville, MD, USA), and pericytes were collected.
Enzyme Immunoassay for Tissue Plasminogen Activator, Plasminogen Activator Inhibitor-1 and TGF-β
Enzyme immunoassay was performed using microtiter plates coated with goat anti-tPA IgG or mouse anti-PAI-1 IgG (American Diagnostica Inc., Greenwich, CT, USA). In all, 20 μL of tPA and PAI-1 standards and 20 μL of test samples were added to the wells. After 1 h incubation on an orbital plate shaker at 600 r.p.m., the conjugate (50 μL) was added and incubated for an additional 30 mins. The plate was washed four times with buffer (phosphate, NaCl, EDTA, and Tween 20) before the addition of 100 μL OPD/H2O2 substrate. After 15 mins, the reaction was terminated by the addition of 100 μL, 1.5 mol/L H2SO4, and absorbance was read at 492 nm. Levels of tPA and PAI-1 in media conditioned for 48 h by endothelial cells were typically in the range of 55 ± 17 and 130 ± 29 ng/mL, respectively. Levels of tPA and PAI-1 in media conditioned for 48 h by pericytes alone were typically in the range of 15 ± 13 and 125 ± 13 ng/mL, respectively. Isoforms of TGF-β from conditioned media were also measured by enzyme-linked immunosorbent assay (Promega Inc., Madison, WI, USA).
Polymerase Chain Reaction Studies
Total ribonucleic acid (RNA) was isolated with RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Complementary deoxyribonucleic acid (cDNA) was synthesized from 2 μg of total RNA in a total volume of 20 μL. Ribonucleic acid was incubated in 11 μL of DEPC-treated water at 65°C for 5 mins and quickly on ice. The RNA was then added to the transcription solution containing: oligo dT primers (1.5 μmol/L), Tris-HCl (50 mmol/L, pH 8.3), KCl (75 mmol/L), MgCl2 (3 mmol/L), dNTP (0.5 mmol/L), RNase inhibitor (1 U/μl), and Avian Myeloblastosis Virus reverse transcriptase (13.3 U/μL). The reaction was performed at 42°C for 50 mins and terminated at 52°C for 15 mins. Complementary deoxyribonucleic acid was stored at −20°C until used.
TaqMan real-time quantitative polymerase chain reaction (PCR) was performed in a Gene Amp 5700 Sequence Detector (Applied Biosystems, Foster City, CA, USA) using 4 μL cDNA, 1 × TaqMan buffer A, MgCl2 (5.5 mmol/L), deoxyadenosine triphosphate (dATP) (200 μmol/L), deoxycytidine triphosphate (dCTP) (200 μmol/L), deoxyguanosine triphosphate (dGTP) (200 μmol/L), deoxyuridine triphosphate (dUTP) (400 μmol/L), forward and reverse primers (200 nmol/L, PAI-1 forward primer, 5′GCCCCACT TCTTCAAGCTGTT3′, reverse primer, 5′TGGCTCTCTCCA CCTCTGAAA3′; tPA forward primer, 5′GGCCTTGTCTCC TTTCTATTCG3′, reverse primer, 5′GCGGCTGGATGGGTA CAGT3′; PN-1 forward primer, 5′TTCCAACCCGAGAACA CAAAG3′, reverse primer, 5′AGCATTGGACTTGATAGGA TTTC3′; β-actin forward primer, 5′CAAGTACTCCGTGT GGATCGG3′, reverse primer, 5′GCTGATCCACATCTGC TGGA3′), TaqMan probe (100 nmol/L, PAI-1, FAM-TCCA CTTGCTTGACCGTGCTCCG-TAMRA; tPA, FAM-TGACAT GAGCCTCCTTCAGCCGCT-TAMRA; PN-1, FAM-AACGC ACTTTCGTGGCAGCCGAC-TAMRA; β-actin, FAM-TCCA TCCTGGCCTCGCTGTCCA-TAMRA), AmpErase (0.01 U/μL), and AmpliTaq Gold DNA Polymerase (0.025 U/μL) in a 50 μL volume (Li and Wang, 2000; Medhurst et al, 2000). Polymerase chain reaction was initiated at 50°C for 2 mins to allow AmpErase UNG incubation to remove uracil-contaminated cDNA, followed by AmpliTaq Gold activation at 95°C for 10 mins, and 40 cycles consisting of 95°C for 15 secs and 60°C for 1 min. During TaqMan real-time PCR amplification, the probe with attached reporter dye and quencher dye hybridized to an internal sequence on the gene region of interest. Throughout the amplification process, AmpliTaq Gold polymerase cleaved the dyes from the probe, generating a fluorescent signal from the released reporter dye. The fluorescent signal was measured in real time and proportional to the amount of PCR product present. Concentrations were derived from the threshold cycle number (CT), which is the number of amplification cycles required for the detected fluorescent signal to reach a designated threshold value. A standard curve, generated from three-fold serial dilutions of HBEC cDNA, was used to determine the target gene and β-actin concentrations. β-Actin mRNA is a housekeeping gene control. Relative quantities of tPA and PAI-1 were adjusted to β-actin levels (i.e., dividing by β-actin quantity).
Western Blot Assay
For PN-1 protein, cell pellets were lysed in lysis buffer (Cell Signaling Technology Inc., Beverly, MA, USA) and conditioned medium was precipitated with 10% trichloroacetic acid and prepared for Western blotting. Sodium dodecyl sulfate (SDS)-polyacrylamide gels and buffers were prepared as described by Laemmli (Laemmli, 1970). Samples were applied to a 10% Polyacrylamide gel and then subjected to electrophoresis. After transferring to PVDF membrane (Millipore, Bedford, MA, USA), the membrane was incubated with tris-buffered saline (TBS) containing 5% milk for 1 h, then incubated with antibodies (1:500) for 1 h. Primary polyclonal antibody directed against human PN-1 sequence was raised in rabbit (Abgent, San Diego, CA, USA). The membrane was washed with 0.1% Tween 20 in TBS for 30 mins. HRP-labeled secondary antibody (1:2000) was then incubated for 1 h. The washed membranes were processed with the chemiluminescent detection system (Pierce, Rockford, IL, USA). Densitometrie analysis was performed to quantitate Western blotting protein bands.
Statistical Analysis
Data are presented as mean ± standard deviation for each group. Statistical comparisons between groups were performed using unpaired Student's t-tests. Differences were considered significant for P < 0.05.
Results
We examined human pericyte effects on endothelial fibrinolysis factors tPA and PAI-1 in our coculture systems. Transwell pericyte-endothelial cocultures (endothelial cells and pericytes physically separated) showed significant reductions in tPA mRNA levels (Figure 2; 52% ± 25% of monocultures, P < 0.05). Pericyte–endothelial cocultures also produced reductions in tPA protein (66% ± 23% of monocultures, P < 0.05). Direct-contact mixed cocultures showed further reduction of tPA protein compared with monocultures (Figure 3; 25% ± 15% of monocultures, P < 0.05). Taken together, these findings suggest pericyte-dependent transcriptional regulation of endothelial tPA expression.
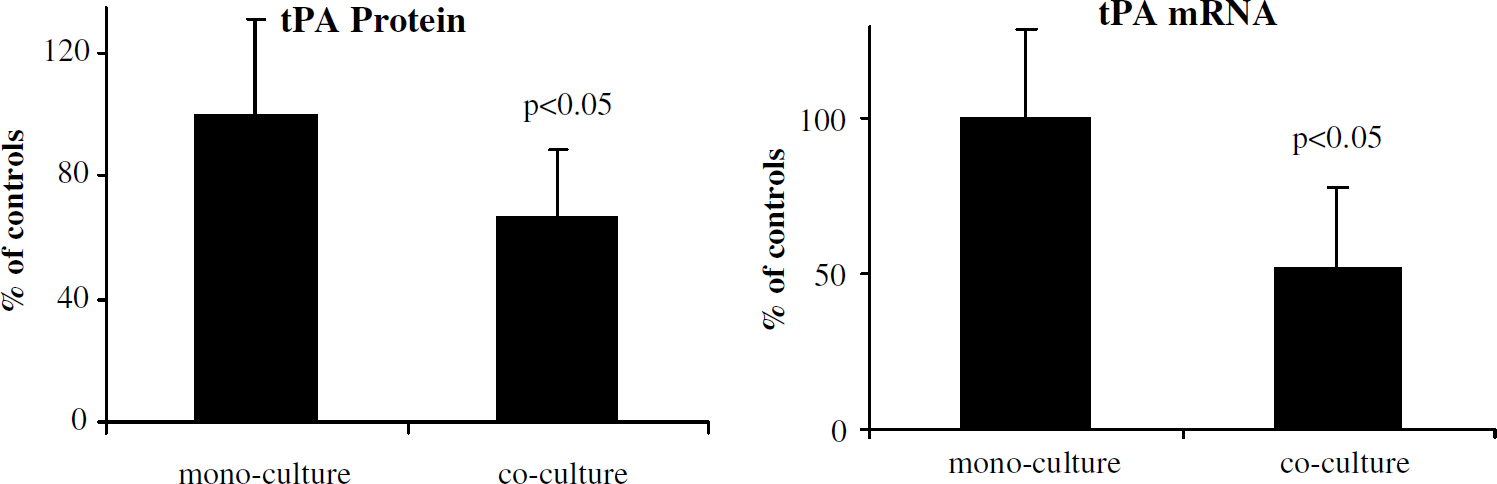
Pericyte regulation of tPA expression by endothelial cells. Assay of tPA protein in culture media from mono- and coculture preparations showed a significant reduction in tPA in 48-h pericyte–endothelial cocultures; Taqman reverse transcriptase (RT)-PCR analysis showed significant decrease in tPA mRNA in pericyte–endothelial cocultures compared with monocultures. Data are pooled from nine independent experiments.
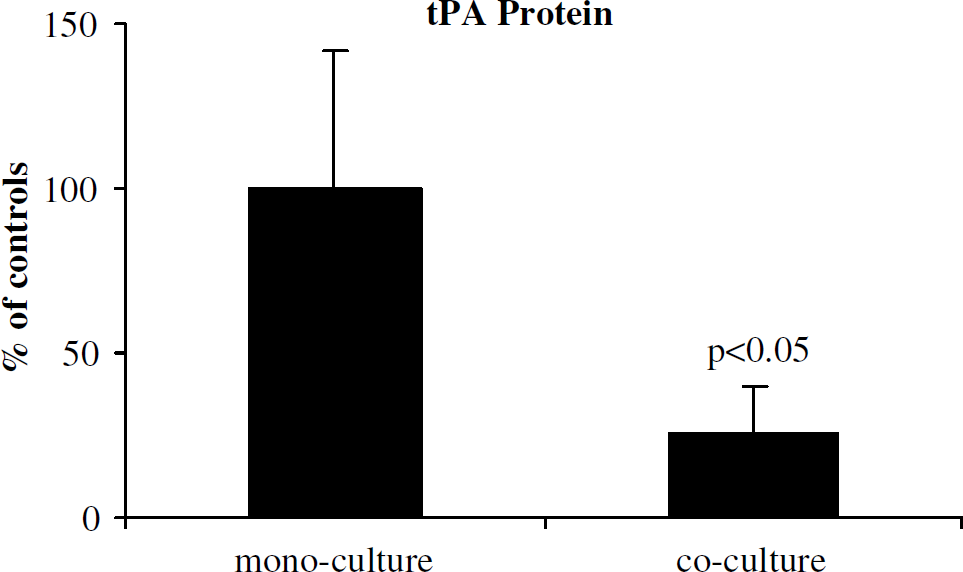
Mixed coculture effect on tPA regulation. Assay of tPA protein in culture media from mono- and coculture preparations showed a significant reduction in tPA. Data are pooled from three independent experiments.
Previous studies showed increased expression of active TGF-β after astrocyte-endothelial cultures, as well as TGF-β downregulation of tPA, while enhancing PAI-1 in vitro (Kim et al, 2003; Tran et al, 1999). We assayed conditioned media for total and active TGF-β after 48 h incubation to evaluate potential TGF-β contribution to the observed changes in tPA expression. Pericyte–endothelial transwell cocultures (versus monocultures) showed small and statistically insignificant increases in levels of total TGF-β1 (412 ± 68 versus 357 ± 45 pg/mL) or active TGF-β1 (91 ± 26 versus 83 ± 12 pg/mL). Furthermore, addition of TGF-β neutralizing antibody did not block pericyte-mediated change of tPA (coculture 44% ± 7% of monocultures, coculture + Ab 36% ± 2% of monocultures). In direct-contact mixed pericyte–endothelial cocultures, cocultures (compared with monocultures) showed nonsignificant increases in total (1126 ± 184 versus 884 ± 136 pg/mL) and active (248 ± 42 versus 185 ± 2 pg/mL) TGF-β1.
We next examined the effects of endotoxin (LPS) on brain microvascular endothelial cells in the presence and absence of pericytes. After incubation with LPS for 6 h, there was more reduction of tPA and enhancement of PAI-1 compared with cocultures without LPS treatment. Cocultures with LPS showed approximately 50% reduction in tPA protein (Figure 4; 49% ± 8% of monocultures, P < 0.05), increased PAI-1 protein (144% ± 27% of monocultures, P = 0.06), and 100% increase in PAI-1 mRNA (200% ± 82% of monocultures, P < 0.05) compared with controls.
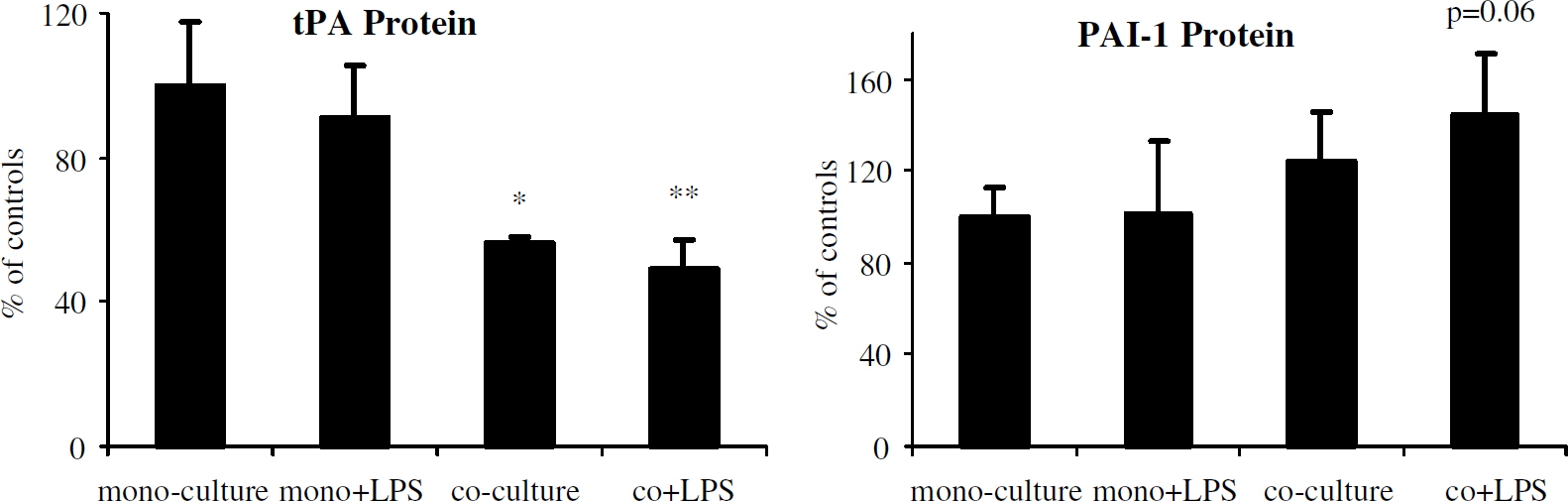
Endotoxin effects on tPA and PAI-1. Assay of tPA and PAI-1 proteins in culture media from mono- and pericyte coculture preparations showed enhanced reduction in tPA protein and increase in PAI-1 in 48-h pericyte–endothelial cocultures with endotoxin (LPS) for the last 6 h incubation. Pooled data from three independent experiments are presented as mean ± s.d. (*P < 0.05 compared with monocultures; **P < 0.01 compared with monocultures, P < 0.05 compared with monocultures with LPS).
To determine whether endotoxin-induced increased PAI-1 originated from HBEC or pericytes, we analyzed the response to LPS by endothelial cells after pericyte conditioning. Human brain microvascular endothelial cells were incubated as monocultures or pericyte cocultures for 48 h. After removing pericytes, the media were replaced with fresh media and HBEC were incubated with or without LPS for 6 h. Endothelial cells, previously cocultured with pericytes and then treated with LPS, showed significant increase in PAI-1 protein (Figure 5). Thus, the PAI-1 increase originated from endothelial cells, which responded to pericyte conditioning.
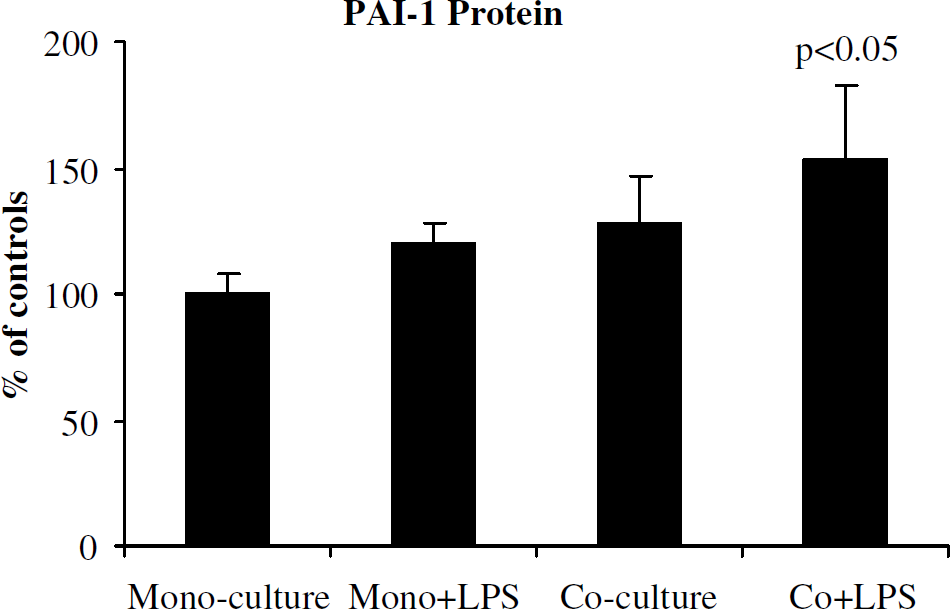
Endothelial response to endotoxin with pericyte conditioning. Human brain microvascular endothelial cells were incubated as monocultures or pericyte cocultures for 48 h. After removing pericytes, media were replaced with fresh media and HBEC incubated with or without LPS for 6 h. After 6 h, conditioned media were collected and assayed for PAI-1 protein. Endothelial cells (previously cocultured with pericytes) treated with LPS showed significant increase in PAI-1, showing that PAI-1 increase originated from endothelial cells, which responded to pericyte conditioning. Data from three independent experiments are presented as mean ± s.d. (P < 0.05 compared with monocultures).
Finally, we analyzed the expression of PN-1. We measured PN-1 protein and mRNA levels in endothelial cells, astrocytes, and pericytes. Pericytes showed markedly increased protein levels in cell extracts and conditioned media as well as mRNA levels compared with endothelial cells (Figure 6).

Protease nexin-1 expression. (Left) Western blot analysis shows PN-1 protein in conditioned media (Top) and cells extracts (Bottom). (Right) Taqman RT-PCR analysis shows significant increase in PN-1 mRNA in pericyte monocultures compared with endothelial monocultures or astrocyte monocultures (P < 0.05 compared with endothelial cells and astrocytes).
Discussion
We have analyzed the hemostatic regulatory role of brain pericytes by coculturing pericytes with HBEC. We showed that pericytes induced decreased endothelial expression of tPA mRNA and protein. These pericyte effects were not blocked by TGF-β neutralizing antibodies, indicating that effects were not mediated by TGF-β. Pericyte effects on HBEC were present when the two cell types were physically separate, suggesting the presence of a soluble pericyte-derived factor; however, pericyte effects were enhanced with direct pericyte–endothelial contact. Pericytes also amplified the enhanced release of PAI-1 induced by endotoxin, the latter acting as a standardized inflammatory stimulus. Finally, pericytes (but not endothelial cells or astrocytes) expressed robust amounts of the antithrombin and antifibrinolytic molecule PN-1. Taken together, these findings suggest an important and complex role for pericytes at the BBB, coordinating hemostasis within the microvasculature with net antifibrinolytic effects.
We have shown pericyte regulation of endothelial fibrinolysis when pericytes are physically separate from HBEC (transwell cocultures); pericytes in direct contact with HBEC had more profound antifibrinolytic effects. Pericytes have previously been shown to affect a variety of endothelial functions (Antonelli-Orlidge et al, 1989; Darland and D'Amore, 2001; Dente et al, 2001; Hellstrom et al, 2001; Hori et al, 2004; Sato and Rifkin, 1989). Pericytes cultured with microvascular EC produce increased endothelial electrical resistance and decreased permeability (Dente et al, 2001). A soluble pericyte-derived factor, thought to be angiopoietin-1, has been shown to increase the endothelial expression of occludin, a key BBB protein (Hori et al, 2004). Our transwell coculture results indicate pericyte regulation of endothelial fibrinolysis at least in part via a soluble factor. However, pericyte-endothelial paracrine interactions regulating fibrinolysis are enhanced with direct cell-cell contact.
Previously, we reported that astrocytes, another cellular constituent of the BBB, have antifibrinolytic effects that are mediated by a soluble factor, TGF-β. Active TGF-β was increased during astrocyte-endothelial coculture and produced a dose-dependent response (Tran et al, 1999). Our pericyte–endothelial cultures showed only small and insignificant increases in TGF-β after coculture. Importantly, TGF-β neutralizing antibody did not attenuate the pericyte-mediated alterations of tPA. Therefore, the mechanism of pericyte regulation of endothelial fibrinolysis has yet to be determined.
We have shown that pericytes amplify LPS-induced enhancement of endothelial PAI-1. This finding has important implications, given emerging studies showing a transient increased risk of ischemic stroke after incidental systemic infection (Paganini-Hill et al, 2003; Smeeth et al, 2004), a phenomenon that might be mediated by impaired fibrinolysis (Macko et al, 1996). Our findings of LPS-induced reduction in tPA release are consistent with some (Bevilacqua et al, 1986; Muth et al, 2004), but not all (Hanss and Collen, 1987), prior work; our data do not address the relative contributions of endothelial- and pericyte-derived tPA after coculture incubation with LPS.
Our findings are consistent with earlier studies showing LPS-induced increased PAI-1 expression in endothelial cultures (Emeis and Kooistra, 1986; Hanss and Collen, 1987; Ruan et al, 2001). Prior work has shown endothelial cell activation in experimental endotoxemia (Kneidinger et al, 1996), and our in vitro observation of LPS-induced increased PAI-1 release is likely duplicated in vivo (Emeis et al, 1995; Paloma et al, 1995). Moreover, we show that LPS-enhanced PAI-1 release was augmented by pericytes. These pericyte-dependent effects occurred even after pericytes were removed from endothelial cells incubated with LPS. These findings indicate a conditioning effect of pericytes on endothelial cells by a soluble factor, and suggest a unique, brain-specific hemostasis response to an inflammatory stimulus. Such a response may contribute to a thrombotic predilection within the brain's vasculature in the setting of an infectious or inflammatory challenge. The soluble factor mediating this response has yet to be determined.
We show that pericytes, but not astrocytes or endothelial cells, are a robust source of PN-1 in vitro: pericytes synthesize PN-1 mRNA and secrete PN-1 protein into conditioned media. Protease nexin-1 is a 43–50 kDa member of the serpin superfamily and has powerful inhibiting activity on serine proteases despite its near absence from plasma (Richard et al, 2004). At the cell surface, PN-1 forms a stable complex with the protease's catalytic site serine residue; the complex is internalized and degraded (Richard et al, 2004; Vaughan et al, 1994). Free PN-1 is bound to proteins of the extracellular matrix, and its binding to heparin-like glycosaminoglycans increases by a 1000-fold its interaction with thrombin. Under physiological conditions, PN-1 is primarily an inhibitor of thrombin (Richard et al, 2004; Vaughan et al, 1994). The importance of PN-1 expression in brain may relate to the paucity of brain thrombomodulin. The thrombomodulin–thrombin complex activates protein C, thus reversing the procoagulant effects of thrombin. Thrombomodulin is expressed extensively on systemic, but not brain, endothelial cells (Tran et al, 1996; Wong et al, 1991). Given the limited expression of brain thrombomodulin, as well as the limited presence of brain endothelial tPA, abundance of any molecule with substantial anticoagulant activity is notable. Thus, pericyte-dependent PN-1 might have a role compensatory for underexpression of other endogenous anticoagulants in the brain.
Regulation of hemostasis is a dynamic process, with constitutive expression of hemostasis factors providing a baseline state that is modifiable by a number of inductive influences. For example, localization studies of tissue factor have shown restricted expression of tissue factor in nonendothelial elements of the cortical microvasculature (del Zoppo et al, 1992). Tissue factor originates in astrocytes (Eddleston et al, 1993) and pericytes (Bouchard et al, 1997), and astrocyte tissue factor expression is enhanced by endotoxin (Erlich et al, 1999). Similarly, astrocyte expression of PN-1 in vivo has been shown (Choi et al, 1990), and this appears to be modified under pathologic conditions. In Alzheimer's disease, for example, there is reduction of perivascular PN-1 expression (Vaughan et al, 1994), and thrombin–PN-1 interactions are thought to contribute to neuronal degeneration (Choi et al, 1995). It is not known how endotoxin as well as other inductive influences may modify the expression of PN–1.
Our study is limited largely by the fact that it is an in vitro investigation, and any extrapolation to the in vivo setting must be performed cautiously. Moreover, we have used relatively mature pericytes, rather than primary cells, and cannot rule out the possibility that a different response might be obtained using earlier passage cells. Multiple configurations of seeding pericytes and endothelial cells are possible using transwell inserts, and it is possible that different culture conditions might have yielded different results. Our study does not address the relative contributions of PN-1 by pericytes and astrocytes in vivo. We have, however, focused on using human cells, with the expectation that our findings would ultimately be more applicable to the clinical setting.
In conclusion, we show a substantial role for pericytes negatively regulating endothelial fibrinolysis in vitro. These findings are consistent with prior work showing reduced brain endothelial tPA both in vitro and in vivo (Grau et al, 1997; Levin and del Zoppo, 1994). However, hemostasis regulation by pericytes has additional important dimensions: Pericytes amplify enhanced release of PAI-1 induced by endotoxin, and also appear to contribute PN-1 to the microvascular milieu. Protease nexin-1, in turn, may contribute anticoagulant activity to the brain's microcirculation, already characterized as having relative deficiencies of important systemic anticoagulants. Thus, pericytes appear to have an important role in mediating brain microvascular hemostasis, with effects that are largely, but not exclusively, antifibrinolytic.