
Abstract
The present study combined molecular and neuroimaging techniques to examine if free radical-mediated damage to barrier function in hypoxia would result in extracellular edema, raise intracranial pressure (ICP) and account for the neurological symptoms typical of high-altitude headache (HAH) also known as acute mountain sickness (AMS). Twenty-two subjects were randomly exposed for 18 h to 12% (hypoxia) and 21% oxygen (O2 (normoxia)) for collection of venous blood (0 h, 8 h, 15 h, 18 h) and CSF (18 h) after lumbar puncture (LP). Electron paramagnetic resonance (EPR) spectroscopy identified a clear increase in the blood and CSF concentration of O2 and carbon-centered free radicals (P > 0.05 versus normoxia) subsequently identified as lipid-derived alkoxyl (LO•) and alkyl (LC•) species. Magnetic resonance imaging (MRI) demonstrated a mild increase in brain volume (7.0 ± 4.8mL or 0.6% ± 0.4%, P > 0.05 versus normoxia) that resolved within 6 h of normoxic recovery. However, there was no detectable evidence for gross barrier dysfunction, elevated lumbar pressures, T2 prolongation or associated neuronal and astroglial damage. Clinical AMS was diagnosed in 50% of subjects during the hypoxic trial and corresponding headache scores were markedly elevated (P > 0.05 versus non-AMS). A greater increase in brain volume was observed, though this was slight, independent of oxidative stress, barrier dysfunction, raised lumbar pressure, vascular damage and measurable evidence of cerebral edema and only apparent in the most severe of cases. These findings suggest that free-radical-mediated vasogenic edema is not an important pathophysiological event that contributes to the mild brain swelling observed in HAH.
Introduction
Headache is the hallmark phenotypical characteristic of acute mountain sickness (AMS) and is associated with, if not the trigger for, other vegetative symptoms including nausea, emesis, dizziness, anorexia, lassitude and insomnia (Hackett and Roach, 2001). Symptoms typically present in otherwise healthy individuals within 6 to 12 h of arrival at high-altitude depending on individual susceptibility, rate of ascent and prior exposure (Schneider et al, 2002). Though debilitating, AMS is generally benign but may progress in severe cases and with continued ascent above 4000m to high-altitude cerebral edema (HACE), that if left untreated may result in death due to brain herniation (Bärtsch and Roach, 2001). Current opinion suggests that AMS is a subclinical form of HACE and that both syndromes share a common pathophysiology (Hackett, 1999b).
Some neuroimaging studies have tentatively indicated an association between extracellular vasogenic edema and neurological sequelae present in severe AMS and HACE (Hackett et al, 1998; Matsuzawa et al, 1992; Roach and Hackett, 2001) subsequent to intracranial hypertension and mechanical compression or biochemical activation of pain-sensitive structures (Sanchez del Rio and Moskowitz, 1999). However, the precise mechanisms responsible for disruption of the BBB in hypoxia await investigation despite widespread speculation for a mechanical vascular leak due to raised capillary hydrostatic pressure and impaired cerebral autoregulation. Though altered hemodynamics may prove a contributory factor, animal studies have clearly identified that an additional and as of yet unidentified stimulus is prerequisite to induce changes in barrier function and associated symptoms (Krasney, 1997).
Emerging evidence suggests a potential pathophysiological role for free radicals, which is not unreasonable since the cerebral endothelium and associated vasculature is particularly susceptible to damaging redox reactions (Bailey, 2003). Our laboratory-based studies have identified that inspiratory normobaric hypoxia activates oxidative stress and is compounded by physical exercise (Bailey et al, 2001a), an established risk factor for AMS (Roach et al, 2000). We have since identified a selective increase in putative biomarkers of free radical-mediated lipid peroxidation and sarcolemmal membrane damage in AMS (Bailey et al, 2001b, Bailey 2004b), with effective neuroprotection conferred during antioxidant prophylaxis at terrestrial high altitude (Bailey and Davies, 2001).
Thus, to provide a more comprehensive examination of the potential pathophysiological role of free radicals, we exposed healthy volunteers to normobaric hypoxia for 18 h, resulting in an inspired partial pressure of oxygen (PIO2) that corresponded to an altitude of 4600 m. Field studies incorporating hypobaric hypoxia (Schneider et al, 2002) have established a comparable prevalence and severity of AMS with that induced in the laboratory using normobaric hypoxia (Swenson et al, 1997). Blood and CSF samples were taken before and during hypoxia and the combined application of electron paramagnetic resonance (EPR) spectroscopy and magnetic resonance imaging (MRI) permitted, for the first time, a direct examination of the temporal relationship between altered redox state, brain morphology and neurological symptoms during the evolution of clinical AMS. We hypothesized that normobaric hypoxia would cause free radical-mediated damage to barrier function, resulting in a vasogenic edema. This, in turn, would cause mild brain swelling and raise intracranial pressure (ICP), causing compression of pain-sensitive structures, thus activating neurovascular headache. However, our findings suggest that free radical-mediated damage to barrier function is not an important pathophysiological event responsible for the mild brain swelling that was observed in AMS.
Materials and methods
Sample
Twenty two apparently healthy subjects (14 men, 8 women) aged 24 ± 2 years were recruited for the present study after written informed consent was obtained according to the ethical requirements of the University of Heidelberg. Specific exclusion criteria included any history of chronic headache disorder or habitual antioxidant vitamin supplementation.
Experimental Design
Subjects were randomly assigned to perform two trials in a thermoneutral environmental chamber, each separated by a 6-week recovery period. Each trial involved an 18 h passive exposure incorporating an overnight stay to a normobaric hypoxic (12% inspiratory fraction of oxygen (FIO2)) and control normobaric normoxic (21% FIO2) environment. The hypoxic trial incorporated an ambient partial pressure of oxygen (PO2 = ≈ 90 Torr), which equated to a terrestrial altitude of ≈ 4600 m (Schneider et al, 2002). Blood samples were coded specifically to blind the investigating scientist who completed the metabolic analyses.
Experimental Procedures
A summary of the timing and experimental procedures performed during the study is outlined in Figure 1. Magnetic resonance imaging was performed during the hypoxic trial only due to limited access. On the morning of the study and 7 h before venous sampling, a baseline MRI scan was performed in normoxia (−7 h). Venous sampling was initiated in normoxic room air before subjects entered the chamber at the predetermined FIO2 (0 h). Further samples were obtained before bed rest (8 h) and within 30mins of waking (15 h) during each chamber exposure. After a further 1 h, subjects were fitted with a neoprene mask connected to a two-way nonrebreathing valve (Hans Rudolph, 2400 series, USA), which they wore until venous sampling was completed (18 h). The inspiratory port was connected by a 2 m length of Falconia tubing to a 250 L Douglas bag that contained medical grade quality normoxic (21% O2, balanced nitrogen) or hypoxic (12% O2, balanced nitrogen) gas mixtures that were delivered by pressurized gas cylinders. Subjects exited the chamber (still breathing the designated FIO2) and were transported to the neuroimaging unit for a second MRI (16 h exposure). After a comprehensive neurological examination, 10 mL of lumbar cerebrospinal fluid (CSF) was obtained (18 h) via lumbar puncture (LP) obtained in the lateral decubitus position according to standard clinical procedures. A final venous sample (18 h) was taken and subjects were disconnected from the respective inspirates and allowed to recover for 6 h in the prone position under medical supervision. A third and final MRI in normoxia (24 h) was obtained before medical discharge.
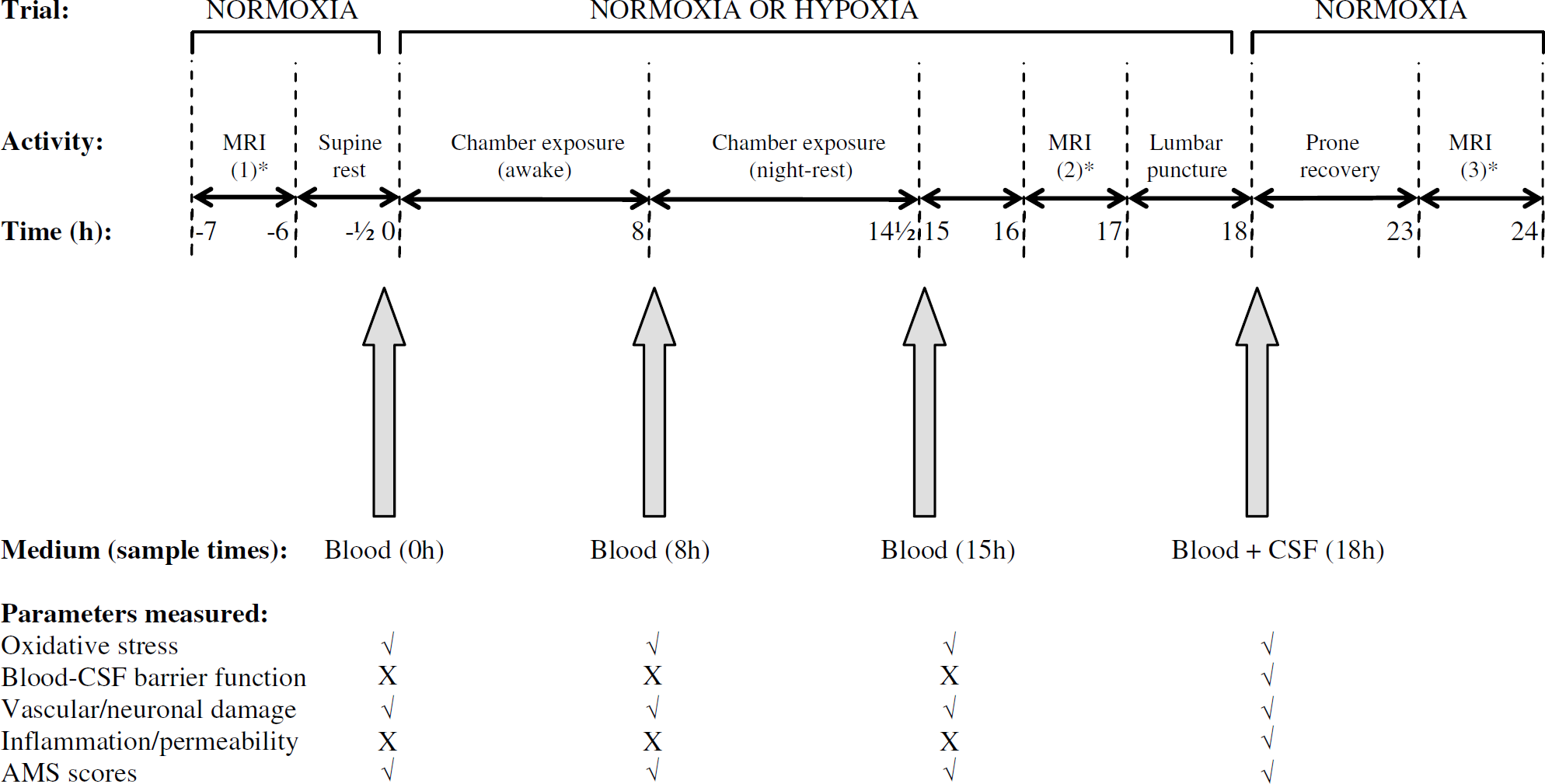
Experimental protocol. Note that baseline blood sampling (0 h) was performed in normoxia for both normoxia and hypoxia trials. *Magnetic resonance imaging scans performed during the hypoxia trial only.
Metabolic Parameters
Blood samples were centrifuged and the serum or plasma supernatant was immediately frozen at −196°C before storage at −70°C. With the exception of spin-trapped samples, lumbar CSF was immediately frozen at −196°C before storage at −70°C. Venous concentrations of hematocrit and hemoglobin were determined using the micro-hematocrit centrifuge and Coulter-counter methods, respectively, to correct metabolic parameters for relative shifts in plasma volume (Dill and Costill, 1974).
Biomarkers of Oxidative Stress
Electron paramagnetic resonance spectroscopy: Ex vivo spin-trapping with a 190mmol/L solution of α-phenyl-tert-butylnitrone (PBN) was incorporated for the EPR spectroscopic detection of free radicals as previously described (Bailey et al, 2004c, 2003). All measurements were performed at 21°C under standard conditions using an EMX spectrometer fitted with a TM110 resonant cavity (Bruker, Karlsruhe, Germany) operating at X-band (9.7 GHz). The average spectral peak-to-trough line height of all peaks (expressed relative to square root peak line width) was considered a measure of the relative spin adduct concentration after conformation of peak-to-trough line-width conformity and double integration on selected samples.
Lipid hydroperoxides (LH): Serum LH was determined using the ferrous iron/xylenol orange (FOX) assay (Wolff, 1994) with modification. The intra/inter-assay coefficients of variation (CVs) were >2% and >4%, respectively.
Antioxidants: For ascorbic acid measurements, plasma was stabilized and deproteinated by adding 900 μL of 5% metaphosphoric acid (Sigma Chemical, Dorset, UK) to 100 μL EDTA plasma. Ascorbic acid was subsequently assayed by fluorimetry based on the condensation of dehydroascorbic acid with 1,2-phenylenediamine (Vuilleumier and Keck, 1993). The intra- and interassay CVs were both >5%. The plasma concentration of α-tocopherol and the carotenoids lycopene, α-carotene and β-carotene were determined using an high-performance liquid chromatography (HPLC) method (Catignani and Bieri, 1983; Thurnham et al, 1988). The intra- and interassay CVs were both >5%.
Biomarkers of Permeability and Inflammation
Skeletal tissue-specific proteins: Serum total creatine phosphokinase (CPK) and lactate dehydrogenase (LDH) activities were determined by reflectance spectrophotometry using a Vitros 950 analyser (Amersham, Bucks, UK). The intra- and interassay CV were both >4%. Serum myoglobin was measured using an automated chemiluminescence immunoassay (ACS 180, Bayer-Chiron Immunodiagnostics). The intra- and interassay CVs were both >4%.
Brain-specific proteins: The serum and CSF concentrations of neuron-specific enolase (NSE) and S100β were measured using commercially available immunoradiometric assays (AB, SANGTEC MEDICAL, Bromma, Sweden). The intra- and interassay CVs were both >5% for NSE and >9%/ >4%, respectively, for S100β.
Barrier function: The CSF concentration of total protein and CSF and serum concentrations of albumin, IgG, IgA, and IgM were determined via nephelometric detection. Subsequent CSF/serum quotients were calculated as described previously (Reiber and Peter, 2001).
Cytokines and vascular endothelial growth factor (VEGF): Plasma and CSF concentrations of tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, and VEGF were determined using high-sensitivity sandwich ELISA kits (Amersham Biosciences, UK and R&D Systems, USA). The applicability of these assays for CSF was verified by recovery tests with spiked CSF standards and dilution series within the dynamic range of the assays. The intra- and interassay CVs were both >5%, respectively.
Leucocytes: Plasma total leucocyte count was determined using a COULTER®GEN S™ automated hematology analyzer (Coulter Corporation, Miami, USA).
Endotoxin: Plasma bacterial lipopolysaccharide concentration (endotoxin) derived from pyrogen-free vacutainers was measured using the Limulus amebocyte lysate assay (KQCL, Bio-Whittaker, UK) as decribed previously (Novitsky, 1994).
Magnetic resonance imaging: MR images were acquired on a 1.5-T whole-body scanner (Edge, Picker) using a standard head coil. For volumetric measurements, T1-weighted gradient-echo sequences were obtained covering the whole brain to the level of the foramen magnum incorporating the following parameters: TE 4.4 ms, TR 30 ms, flip angle 30°, field of view (FOV) 256 mm, 85 slices, 2.0 mm thickness. SIENAX software was used to determine baseline brain volume and subsequent changes were compared using SIENA software. The same software was used to measure intracranial volume and intracranial cerebrospinal fluid volume. For the calculation of T2 relaxation times, conventional spin-echo (dual-echo) sequences were used consisting of proton density and T2-weighted images incorporating the following parameters: TE 20/90; TR 2248; Flip 90; FOV 220 mm. T2 relaxation time calculation was performed on a workstation (EASY VISION) and calculated for six regions of interest (ROI) focusing on the left, middle and right hemispheres of the white matter (frontal and supraventricular), corpus callosum (genu and splenium), basal ganglia (lentiform nucleus) and cerebellum. For all scans, subjects were connected to either a normoxic or hypoxic inspirate as described previously. The inclusion of a normoxic inspirate was considered important since pilot studies identified that the respiratory apparatus induced a hyperventilatory-hypocapneic response that might have influenced cerebral blood flow and thus, by consequence, morphology.
Neurological Symptoms
Acute mountain sickness: Neurological symptoms typically ascribed to AMS were comprehensively examined using the Lake Louise (LL) (Roach et al, 1993) and Environmental Symptoms Questionnaires (ESQ) (Sampson et al, 1983). Clinical AMS was defined if a subject presented with a total LL score (self assessment + clinical scores) of ≥5 points in the presence of a headache and an ESQ cerebral symptoms (ESQ-C) score ≥0.7 points at the point when the LP was performed during the hypoxic trial (18 h exposure).
Headache: Subjects were asked to rate their cephalgia using a clinically validated visual analogue scale (0 to 100 mm) incorporating hedonic descriptors (0 = no headache, 10mm = mild headache including a sensation of pressing or throbbing, 50mm = moderate intensity headache and 100mm = worst possible headache) (Iversen et al, 1989).
Safety Measures
Chamber exposure: Though subjects were informed that the experimental procedures would result in considerable discomfort, principally headache and emesis, they were made aware that they could terminate the study at their own free will and at any stage without prejudice. For persistent, particularly debilitating AMS often resulting in emesis that challenged their continued participation, subjects received an oral dose of paracetamol and/or metoclopramide hydrochloride. Supplemental O2 was also made available during the hypoxic exposure.
Lumbar puncture: Of the first 17 LPs performed (50% of total), 10 (6 in hypoxia, 4 in normoxia) were complicated by symptoms consistent with post puncture syndrome (PPS). In an attempt to reduce the incidence and severity of PPS, the remaining subjects (17 additional LPs) received 400 mg of ibuprofen immediately after the LP and final venous sample (after 18 h exposure to hypoxia). A second dose (400 mg) was administered after 7 h recovery in normoxia (after the final MRI during the hypoxia trial) and supplementation was enforced three times/day for 2 days after completion of the study.
In total, PPS was recorded in 13 separate cases (4 in normoxia, 9 in hypoxia) within 2 days of the initial LP with symptoms lasting for 7 days. Six subjects subsequently refused a follow-up LP and 1 additional CSF sample was discarded due to contamination with blood. One subject failed to attend the normoxic follow-up trial.
Statistical Analyses
A Shapiro—Wilks test was applied to each dependent variable to mathematically assess distribution normality. Parametric and nonparametric equivalents of a two-factor trial: normoxia versus hypoxia and exposure time: 0 h versus 8 h versus 15 h) repeated-measures analysis of variance (ANOVA) and two-way mixed ANOVA with one between (state: non-AMS versus AMS) and one within (exposure time) factor were incorporated to examine the effects of trial, time and state on selected variables. After a simple main effect and interaction, Bonferroni-corrected paired samples t-tests were employed to make a posteriori comparisons at each level of the within-subjects factor of interest. Bonferroni-corrected Wilcoxon matched pairs signed ranks tests served as the nonparametric equivalents. Between-state comparisons were assessed using independent samples t-tests or Mann—Whitney U-tests applied to each level of the exposure time factor. Significance for all two tailed tests was established at an alpha level of P > 0.05 and data are expressed as a mean ± standard deviation (s.d.).
Results
Subject Compliance
Due to subject dropout (see Materials and methods), a maximum of 21 complete data sets for blood and 13 for CSF were available for comparative analyses between normoxia and hypoxia. A maximum of 22 data sets for blood (non-AMS, n = 11 versus AMS, n = 11) and 19 for CSF (non-AMS, n = 11 versus AMS, n = 8) were available for comparative analyses between non-AMS and AMS.
Rescue Medication
Seven subjects with severe headache and emesis requested medication after the third venous sample after ≈15 h exposure to hypoxia consistent with sound ethical practice and ensuring continued participation in the study. They received an acute oral dose of metoclopramide hydrochloride (21 to 32 mg) in combination with paracetamol (500 to 1000 mg). Despite medication, all subjects subsequently developed clinical AMS.
Clinical Findings
Acute mountain sickness and headache: A marked increase in AMS scores was observed during the hypoxic trial and 11 subjects (50% of the group) were diagnosed with clinical AMS after 18 h exposure (Table 1). Corresponding headache scores were clearly elevated in hypoxia and AMS relative to normoxic and non-AMS counterparts.
Acute mountain sickness and headache scores
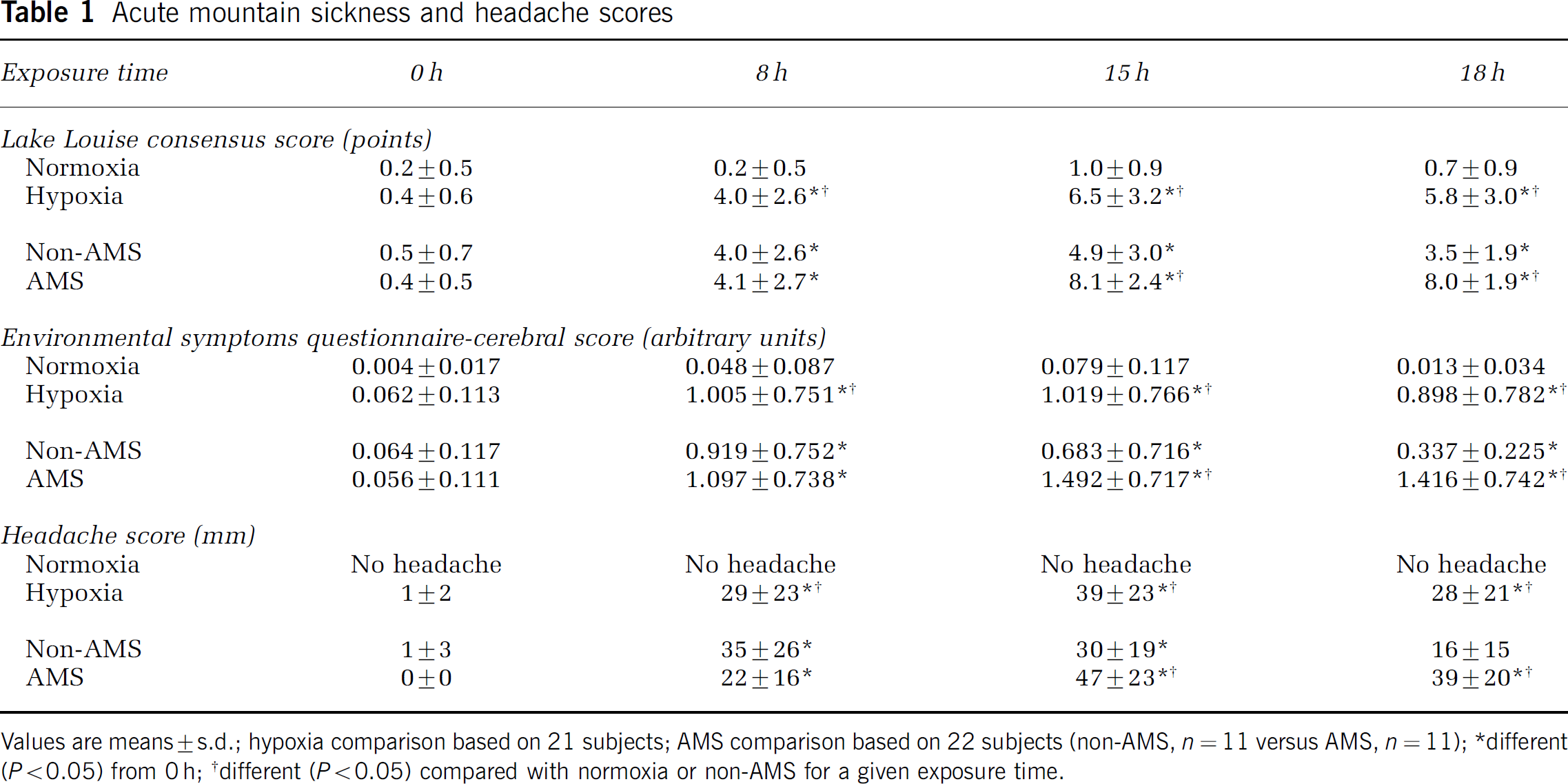
Values are means ± s.d.; hypoxia comparison based on 21 subjects; AMS comparison based on 22 subjects (non-AMS, n = 11 versus AMS, n = 11);
different (P > 0.05) from 0 h;
different (P > 0.05) compared with normoxia or non-AMS for a given exposure time.
Metabolic Findings
Oxidative stress: A progressive increase in the blood and CSF concentration of PBN adducts and LH was observed during hypoxia (Table 2, Figure 2). All PBN adducts detected in blood and CSF exhibited nitrogen coupling constants (aN) of ≈ 13.6G and a ≈1.9 G. Computer simulation later confirmed the presence of an additional adduct albeit of low signal intensity exhibiting aN ≈ 14.0 G and a≈ 4.1 G. Venous ascorbate and α-tocopherol were comparatively lower during hypoxia (Table 2), whereas changes in the carotenoids were unremarkable. There was no relationship between the CSF concentration or changes (15 minus 0 h) in the venous concentration of PBN adducts, LH and increase in brain volume during hypoxia.
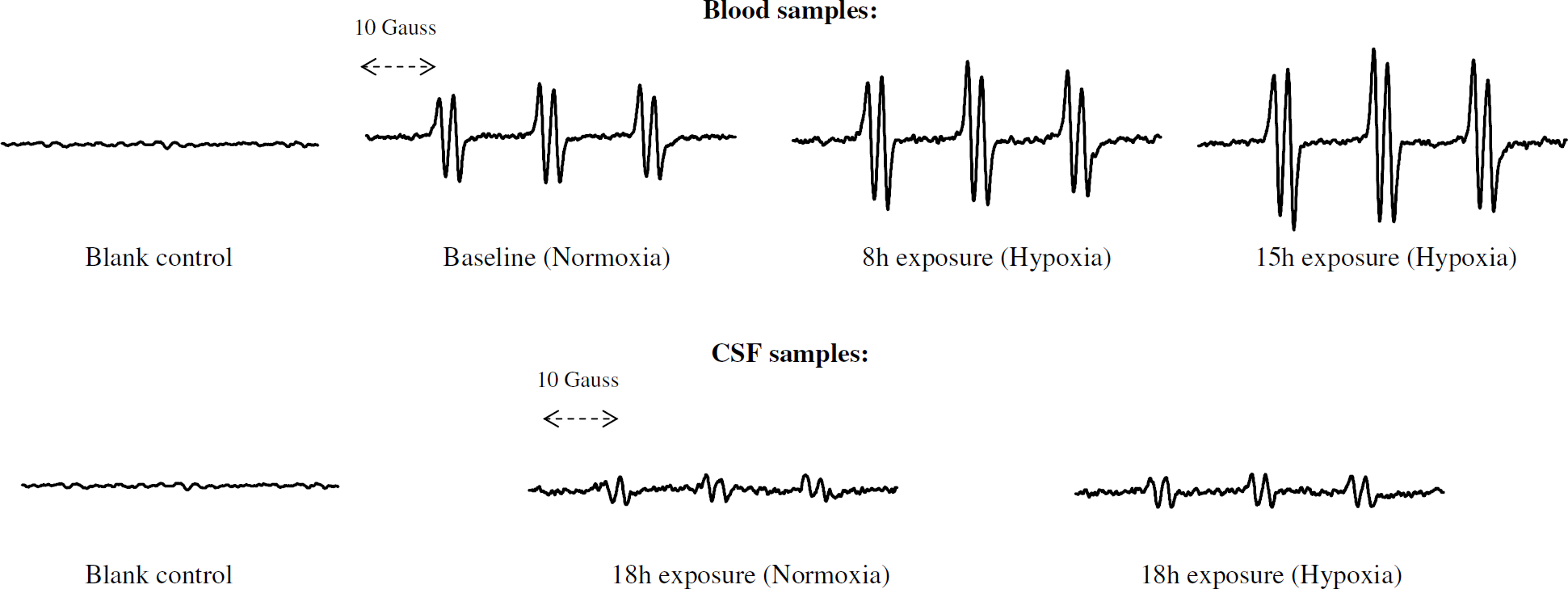
Typical EPR spectra showing the progressive increase in PBN adduct signal intensity during the hypoxia trial. All blood and CSF spectra are derived from the same subject. Respective ordinates for all PBN adducts are identically scaled. Blank control spectra are based on degassed toluene + PBN (excludes serum or CSF).
Oxidants and antioxidants
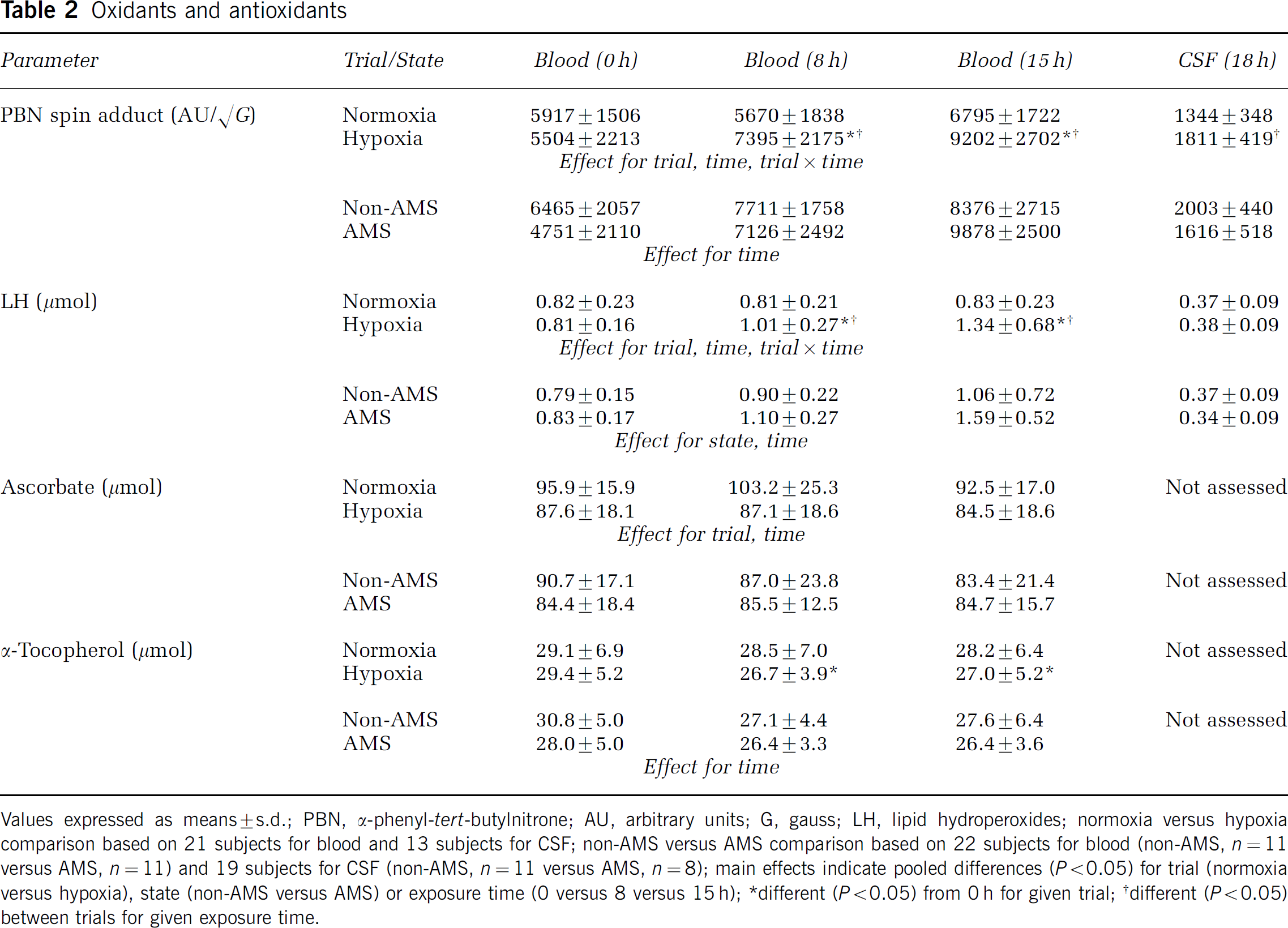
Values expressed as means ± s.d.; PBN, α-phenyl-tert-butylnitrone; AU, arbitrary units; G, gauss; LH, lipid hydroperoxides; normoxia versus hypoxia comparison based on 21 subjects for blood and 13 subjects for CSF; non-AMS versus AMS comparison based on 22 subjects for blood (non-AMS, n = 11 versus AMS, n = 11) and 19 subjects for CSF (non-AMS, n = 11 versus AMS, n = 8); main effects indicate pooled differences (P > 0.05) for trial (normoxia versus hypoxia), state (non-AMS versus AMS) or exposure time (0 versus 8 versus 15 h);
different (P > 0.05) from 0 h for given trial;
different (P > 0.05) between trials for given exposure time.
Vascular and neuronal damage: Hypoxia was associated with a selective increase in serum CPK and LDH, whereas no changes were observed for serum or CSF concentrations of S100β and NSE (Table 3). These parameters were unaffected by AMS.
Vascular and neuronal damage
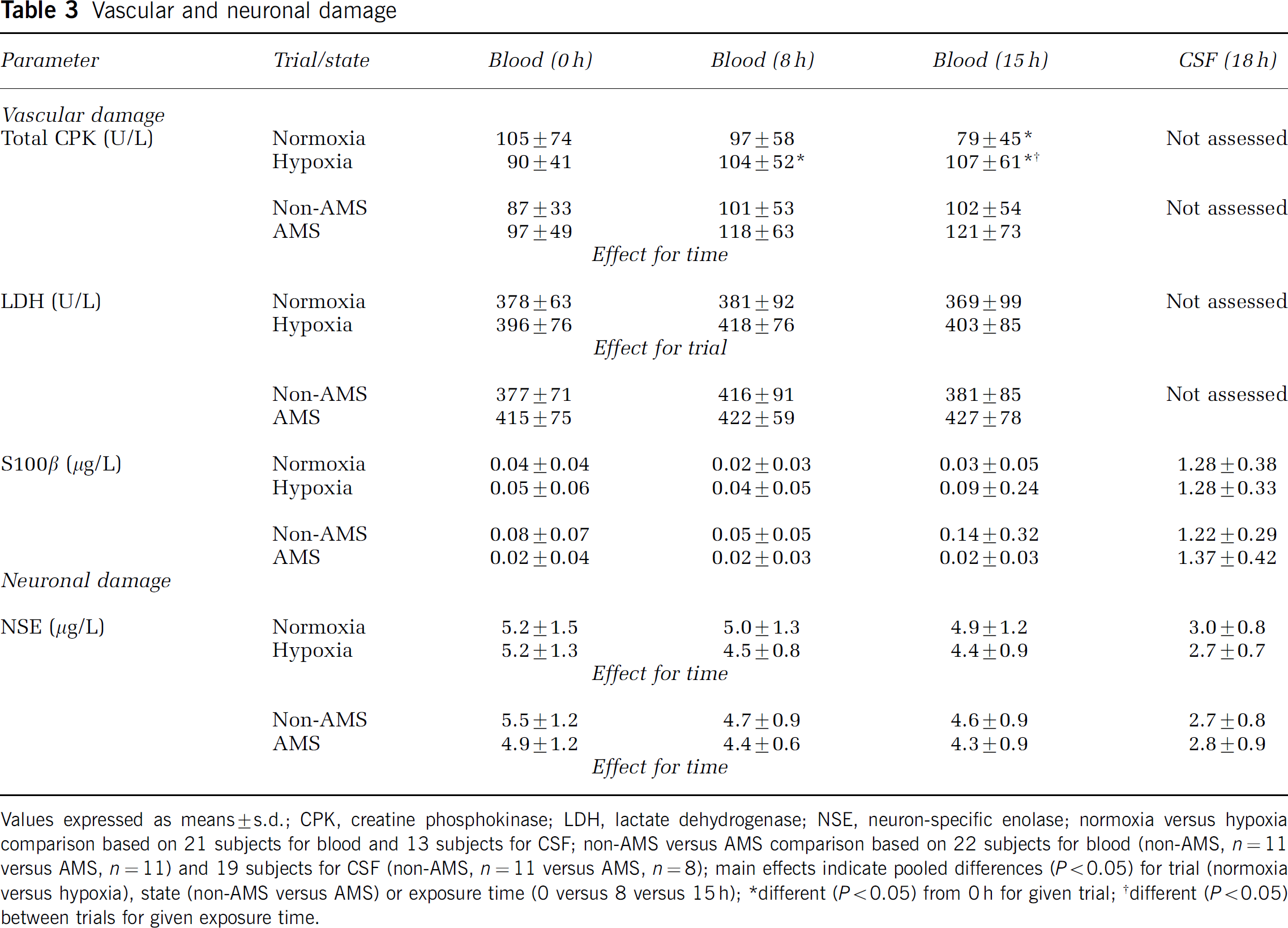
Values expressed as means ± s.d.; CPK, creatine phosphokinase; LDH, lactate dehydrogenase; NSE, neuron-specific enolase; normoxia versus hypoxia comparison based on 21 subjects for blood and 13 subjects for CSF; non-AMS versus AMS comparison based on 22 subjects for blood (non-AMS, n = 11 versus AMS, n = 11) and 19 subjects for CSF (non-AMS, n = 11 versus AMS, n = 8); main effects indicate pooled differences (P > 0.05) for trial (normoxia versus hypoxia), state (non-AMS versus AMS) or exposure time (0 versus 8 versus 15 h); *different (P > 0.05) from 0 h for given trial; †different (P > 0.05) between trials for given exposure time.
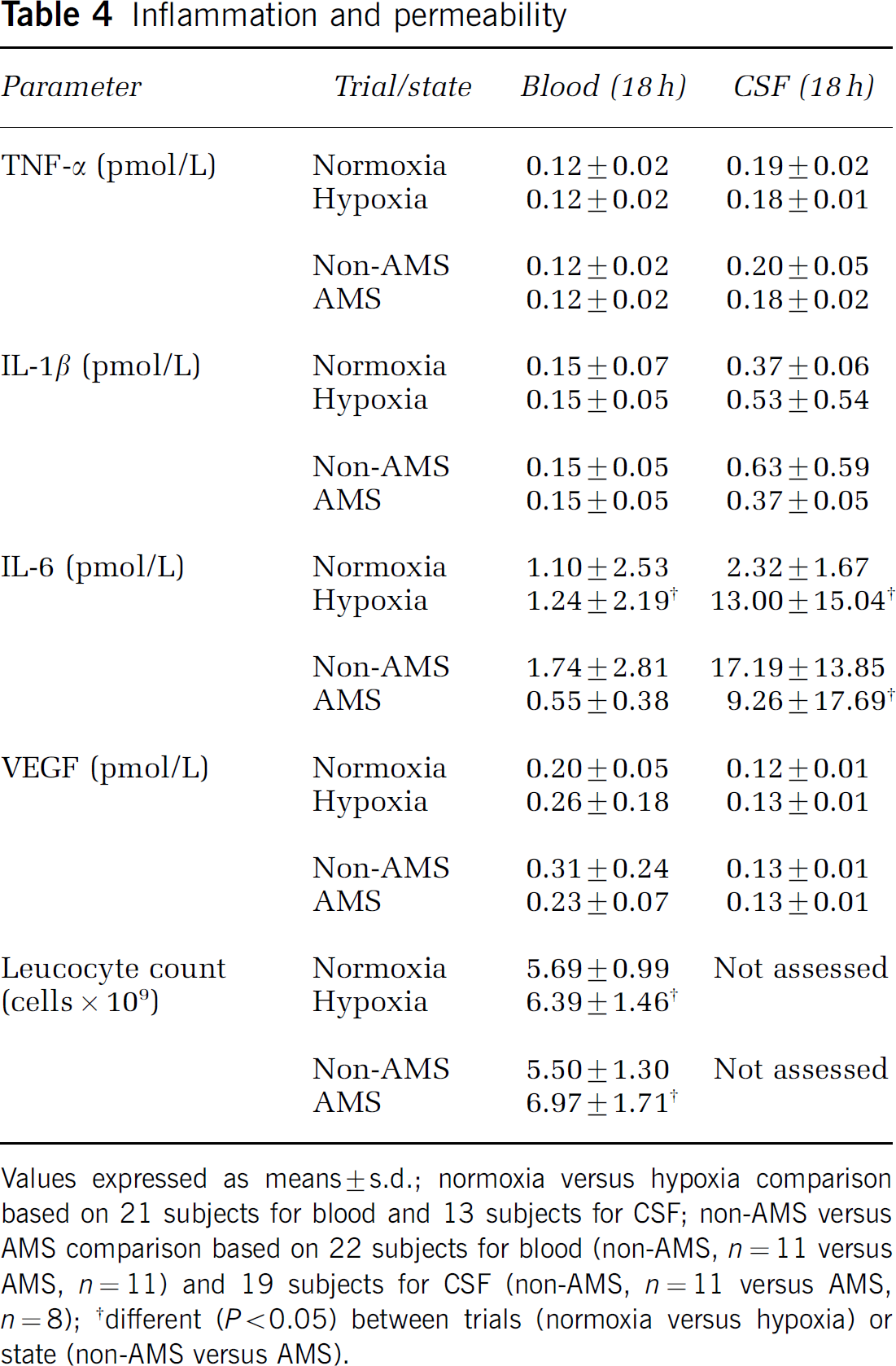
Inflammation and permeability: Plasma and CSF concentrations of TNF-α, IL-1β and VEGF were unaffected by hypoxia or AMS (Table 4) and endotoxin was below detection limits. In contrast, the plasma and CSF concentrations of IL-6 and plasma leucocyte counts were elevated in hypoxia. Acute mountain sickness was characterized by a lower CSF IL-6 and elevated plasma leucocyte count. Oral temperature increased more markedly between baseline (0 h) and 15 h in hypoxia (+ 0.6°C ± 0.7°C versus normoxia: + 0.0°C ± 0.4°C, P > 0.05) and AMS (+ 0.9°C ± 0.6°C versus non-AMS: + 0.3°C ± 0.8°C, P > 0.05).
Inflammation and permeability
Barrier function: Blood—CSF barrier function was not altered by hypoxia (Figure 3A) or AMS (Figure 3B) as indicated by the stable CSF-serum quotients of total protein, albumin, IgG, IgA, and IgM.
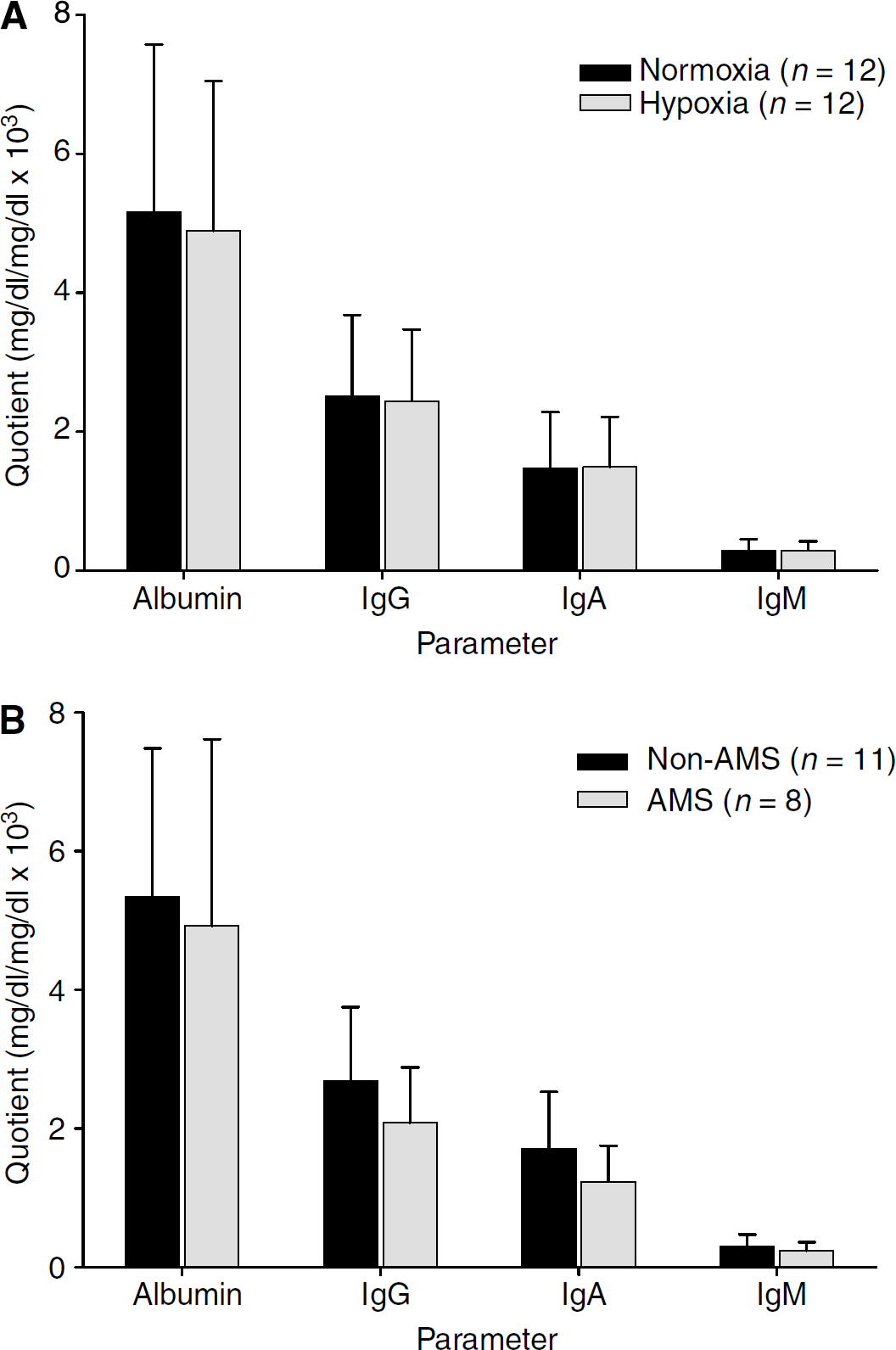
Barrier function in hypoxia (
Morphological Findings
Magnetic resonance imaging: Visual inspection of the MR images did not show any clear evidence for brain swelling. However, calculations identified that hypoxia resulted in an absolute increase in brain volume of 7.0 ± 4.8 mL (0.6% ± 0.4%) that returned towards baseline control values after 6 h normoxic recovery (Figure 4A). The same qualitative outcome was observed when brain volumes were expressed relative to stature (data not shown). A moderate association was observed between the increase in brain volume and ESQ-C scores in hypoxia (r =0.50, P > 0.05). However, the increase in brain volume was not different in AMS during the overall group analysis (Figure 4B) and only became apparent when comparing the extremes of distribution (see below). Though a selective increase in T2 signal intensity was observed within the splenium of the corpus callosum (SCC) during hypoxia (P > 0.05 versus preexposure), average values (mean of 6 ROI) were not different in either hypoxia (Figure 4C) or AMS (Figure 4D). Lumbar pressure was also unaffected by hypoxia (hypoxia: 12.4 ± 5.0 versus normoxia: 13.2 ± 5.2cm/H2O, n = 13) and AMS (AMS, n = 8: 11.8 ± 3.7 versus non-AMS, n = 11: 12.2 ± 5.1cm H2O).
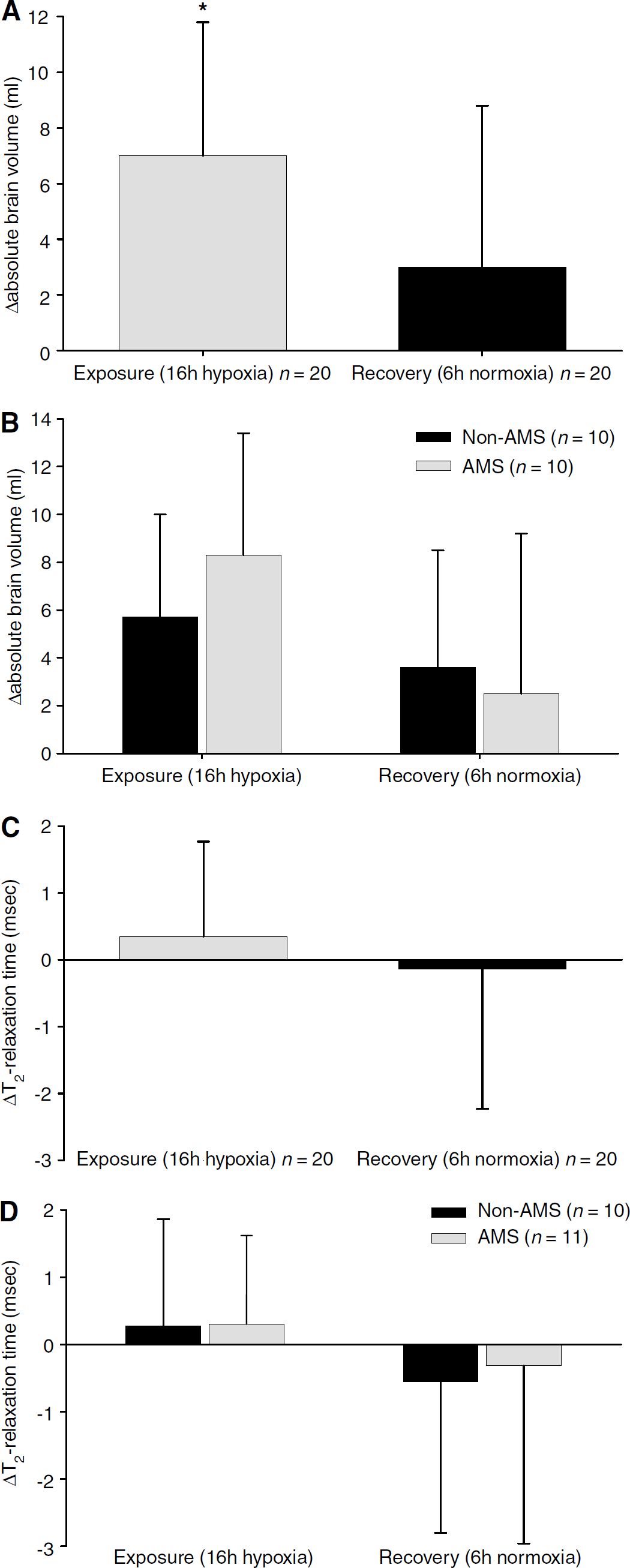
Brain morphology. Changes (Δ) in brain volume in hypoxia (
Ibuprofen: The change in brain volume during normoxic recovery (relative to 16 h exposure to hypoxia) was not different in subjects who received ibuprofen (−3.6 ± 2.6mL, n = 8) compared with those without (−4.7 ± 6.5mL, n = 10). A similar finding was observed for the change in mean T2 relaxation time (with ibuprofen: −1.43 ± 1.99 ms, n = 8 versus without ibuprofen: 0.21 ± 1.51ms, n = 10, P > 0.05).
Extremes of Acute Mountain Sickness
To examine if physiological extremes of AMS were characterized by any defining metabolic or morphologic changes, we compared five subjects with the highest AMS scores (calculated as the mean LL and ESQ-C score at 18 h) in the AMS group against five subjects with the lowest AMS scores in the non-AMS group during the hypoxic trial. Though differences in AMS (LL: 9.6 ± 1.3 versus 1.8 71.0 points, P > 0.05; ESQ-C: 1.80 ± 1.10 versus 0.14 ± 0.12 arbitrary units, P > 0.05) and headache (48 ± 19 versus 11 ± 15 mm, P > 0.05) scores in hypoxia were more pronounced, metabolic findings were qualitatively similar. In contrast, severe AMS was associated with a selective increase in brain volume during hypoxia (P > 0.05 versus preexposure) though T2 relaxation times, barrier function, and lumbar pressures were not different. Furthermore, night-time SaO2 was lower in the AMS group (mean of 15 recordings: 70% ± 2% versus non-AMS: 77% ± 2%, P > 0.05).
Discussion
This study tested the hypothesis that free radical-mediated damage to barrier function in normobaric hypoxia would cause vasogenic edema and that altered intracranial dynamics would initiate headache and associated neurological symptoms collectively known as AMS. Passive exposure to inspiratory normobaric hypoxia proved an effective paradigm for the induction of AMS in 50% of subjects and evoked distinct metabolic and morphological changes to the brain. This incidence of AMS fits very well with the predicted 30% to 60% (depending on individual susceptibility) recently identified in a large epidemiologic study conducted at 4559m (Schneider et al, 2002).
The concentration of LH- and O2/C-centered free radicals increased progressively in blood and CSF, providing the first direct evidence for increased peroxidative stress. These changes were associated with altered sarcolemmal membrane permeability and inflammation, though molecular damage was confined to the skeletal vasculature since there was no evidence for neuronal or astroglial damage. A mild increase in brain volume was observed though the lack of evidence for gross barrier dysfunction, increased lumbar pressure, or marked T2 prolongation mitigated against a vasogenic mechanism. In contrast, a comparative analysis of subjects with AMS against their apparently healthy non-AMS counterparts was less revealing, though evidence for increased lipid peroxidation was detectable in blood. A greater increase in brain volume was observed in the most severe cases consistent with previous observations (Fischer et al, 2004), though this was slight, not associated with biomarkers of oxidative stress, barrier dysfunction, vascular damage, or cerebral edema. In conclusion, these findings do not support our original hypotheses and exclude a free-radical-mediated vasogenic mechanism as an important pathophysiological event that contributes to the mild brain swelling observed in AMS during acute exposure to normobaric hypoxia.
Seven subjects received an acute dose of analgesics and/or antiemetics consistent with sound ethical practice owing to the severity of neurological symptoms. While this might be considered a potentially confounding factor, two lines of evidence suggest that it did not affect the experimental outcome. First, despite receiving medication, subjects still presented with AMS at the end of the study, thus having no effect on the qualitative aspects of comparative analyses. Second, a follow-up study with 5 subjects showed that an acute bolus dose of 1000 mg paracetamol in combination with 32 mg metoclopramide hydrochloride (i.e. worse-case scenario) 3 h before a maximal exercise challenge (an established paradigm for the induction of oxidative stress) did not attenuate the exercise-induced increase in the venous concentration of PBN adducts (data not shown) compared with a placebo. Thus, we consider it highly unlikely that the prescribed medication would have influenced the metabolic or morphological findings observed in the present study.
Free Radicals; Identity and Origins
While EPR is considered the most direct, specific and sensitive analytical technique sine qua non for the molecular detection and subsequent identification of free radicals (Jackson, 1999), the spin-trapping approach still relies on ex vivo detection of resonance-stabilized reactants formed ‘downstream’ of the primary oxidant cascade that we assume reflects events initiated in vivo. However, the increase in LH, one of the major initial reactants of lipid peroxidation, complements the observed increase in PBN adducts and, for the first time, provides clear evidence that hypoxia induced oxidative stress in both the vascular and CSF compartments.
Though a definitive assignment was beyond the scope of the present study, the molecular characteristics of the primary adduct were consistent with the trapping of an O2-centered hydroxyl (OH•-) (Baker et al, 1994) and/or lipid-derived alkoxyl (LO•) species (Buettner, 1987). Our previous research has suggested that these species might have evolved during the metal-catalyzed (Fe2+) reductive decomposition of extracellular LH formed distal to primary radical (possibly OH•- initiated by Fenton or Haber—Weiss chemistry)-mediated attack to membrane phospholipids (Bailey et al, 2004c). The minor signal was consistent with a lipid-derived carbon-centered species (LC•) such as a longer-lived short-chain ethyl, pentyl or pentenyl radical that might have evolved during β-scission of LO• (Gardner, 1989). The selective decrease in ascorbate and α-tocopherol identifies that these chain-breaking donor antioxidants were attempting to slow chain initiation and propagation by these secondary radicals in hypoxia.
It is unlikely that the gut was a primary source or target for these radicals since there was no detectable evidence of bacterial LPS in the peripheral circulation. We have previously shown that a decrease in intracellular PO2 may prove the unifying mechanism that initiates lipid-derived radical generation by increasing the extracellular availability of ‘free’ catalytic iron (Bailey et al, 2004c). This might prove of particular significance for the brain since surrounding structures of the splenium including the basal ganglia, substantia nigra, globus pallidus, caudate nucleus and putamen are particularly rich in catalytic iron (Bartsch et al, 2004; Gutteridge, 1992). Furthermore, unlike human plasma, the CSF has no significant iron-binding capacity since transferrin content is remarkably low and is thus considered close to saturation (Bartsch et al, 2004).
This may explain why the SCC is particularly vulnerable to focal edema as the enhanced T2 signal intensity in the present study, though not a universal finding (Mórocz et al, 2001), clearly suggests. It has also been suggested that the predilection for the SCC albeit in the clinical setting of HACE may prove an anatomical idiosyncrasy since it consists of densely packed fibers that lack adrenergic tone, thus increasing susceptibility to hypoxic cerebral vasodilation, autoregulation failure (Hackett, 1999a; Hackett et al, 1998) and increase in cerebral capillary pressure (Sutton and Lassen, 1979). However, the notion that mild edema selectively confined to the SCC has any clinical bearing on the symptoms of AMS is, contrary to recent opinion (Roach and Hackett, 2001), quite remote. Edema was not sufficient to elevate lumbar pressures and there were no measurable differences detected in AMS, even in the most severe cases. Furthermore, headache is not a defining feature of acute callosal damage, as indicated by studies in patients with stroke and even under extreme conditions requiring elective division of the corpus callosum (CC) for seizure control (Devinsky and Laff, 2003). Since the CC is the primary neocortical commissure that integrates interhemispheric perceptual information at early stages of cortical processing (Seymour et al, 1994), future studies need to consider incorporating specific tests that directly measure changes in callosal dynamics per se to more directly ascertain the neurocognitive consequences of mild edema in hypoxia.
The fact that there were no major differences in the oxidative stress response between those who developed AMS compared with those free of neurological symptoms agrees with one of our previous studies conducted at high altitude (Bailey et al, 2004a), yet contradicts two previous findings (Bailey and Davies, 2001; Bailey et al, 2001c). However, it is difficult to resolve these discrepancies since these studies have incorporated differences in physical activity and ascent rates which are established prooxidant stimuli. It is conceivable that AMS might be initiated by some, as of yet unidentified mediators generated downstream of the oxidizing radical species. For example, symptoms have been shown to coincide with altered eicosanoid metabolism at high altitude (Richalet et al, 1991), which is subject to redox regulation. Thus, it has been suggested that complementary supplementation with n-3 polyunsaturated fatty acids may provide more effective prophylaxis through selective inhibition of arachidonic acid-derived eicosanoid biosynthesis (Bailey et al, 2004a).
Barrier Function and Vascular Damage
Though the radicals detected represent secondary species, they are still thermodynamically capable of initiating and propagating peroxidative damage. In vitro studies have since shown increased paracellular permeability of cerebral microvessel endothelial cells in hypoxia due to functional alterations in actin and tight-junction protein distribution (Mark and Davies, 2002), responses that might have a free radical basis.
However, despite clear evidence of oxidative stress, we could not detect any evidence of ‘gross’ barrier dysfunction or neuroglial damage in hypoxia or AMS as indicated by the lack of change in S100β, NSE, CSF—blood protein concentration quotients, and general failure to detect any signs consistent with cerebral edema or raised lumbar pressure. We chose to incorporate S100β as a specific marker of BBB function since recent studies incorporating osmotic opening of the BBB have identified that extravasation of this astrocytic protein in blood can occur in the absence of or preceding neuroglial damage (Kanner et al, 2003; Kapural et al, 2002; Marchi et al, 2003b). On reflection, it was unfortunate that we did not perform a complementary examination of serum transthyretin, a low-molecular-weight protein that has recently been identified to be a peripheral marker of blood—CSF barrier function (Marchi et al, 2003a).
However, we cannot unequivocally exclude ‘subtle’ damage to barrier function since we have focused on proteins with a relatively large molecular mass ranging from ∼8 to 900 kDa. The accumulation of small molecules in CSF and brain interstitial fluid might have proven irritating or toxic to the brain, thus initiating headache. The CSF—blood quotient of small molecules not synthesized by the brain combined with contrast-enhanced computed tomography (CT)-MRI with guadolinium and localized measurements of neuro-oxidant exchange across the ‘healthy’ and ‘AMS-ill’ brain need to be considered in future studies.
The marked increase in the CSF concentration of IL-6 is intriguing, though clearly independent of structural damage to nervous tissue. This may represent a neuroprotective adaptation since nitric oxide-mediated IL-6 release by glia has been shown to activate the matrix metalloproteinase inhibitor α2-macroglobulin to maintain barrier function (Cucullo et al, 2003). Other factors might have been implicated in the apparent preservation of ‘global’ barrier function in hypoxia. Cerebral vasodilatation and concomitant increase in blood flow initiated by the decrease in PO2 of arterial inflow might have effectively ‘defended’ oxygen delivery to the cerebral endothelium. Local activation of neurotrophic factors capable of stabilizing cellular ion homeostasis and promoting structural preservation of the BBB (Mattson, 1997) might have also provided some defence against barrier failure. Astrocytes might have also contributed to the maintenance of barrier integrity by activating cytoprotective antioxidant enzymes including superoxide dismutase, catalase, and GSH peroxidase in the endothelium to quench local free radical generation (Schroeter et al, 1999). The lack of increase in the permeability cytokine VEGF, capable of altering tight junction proteins and destabilizing barrier integrity (Heo et al, 2005), provides additional evidence against vascular leakage in the human AMS-brain in contrast to other findings obtained in animals (Schoch et al, 2002; Xu and Severinghaus, 1998). Whatever neuroprotective mechanism was operant, our in vivo findings contrast with those generally observed in vitro (Abbott, 2000; Ballabh et al, 2004) and highlight the relative insensitivity of cerebral endothelial cells to low ambient PO2. However, it is important to emphasize that in vitro experiments are typically conducted at subphysiological PO2's and subtle barrier dysfunction remains to be excluded.
Unlike the CNS, skeletal tissue was more susceptible to peroxidative damage and might have further compounded extracellular radical formation through the liberation of intracellular iron (Bailey et al, 2004b; Bolli et al, 1990). Unlike previous high-altitude studies that have demanded a substantial exercise component and hypobaria (Bailey et al, 2004b, 2001c), tissue damage was independent of mechanical trauma since subjects remained essentially passive. Clinical AMS was characterized by increased lipid peroxidation, though this was confined to venous blood and not the CSF, the pathophysiological significance of which remains to be elucidated. The notion that a peripheral redox sensitive mechanism may initiate or contribute to what is essentially a neurogenic syndrome is intriguing and contrary to current opinion (Roach and Hackett, 2001). Unlike previous studies albeit at terrestrial high altitude, we could not confirm a selective increase in sarcolemmal membrane permeability, in part due to limitations associated with insufficient statistical power. Nonetheless, it is conceivable that peripheral peroxidation may alter the subjective perception of headache pain since subjects with AMS have previously been shown to present with more severe myalgia and correspondingly lower pain thresholds (Bailey et al, 2001c).
In conclusion, the present findings indicate that, despite direct evidence for an increase in free radicals during acute exposure to normobaric hypoxia, we could not detect alterations in barrier function or increased lumbar pressure and only a minimal increase in brain volume, the latter finding consistent with previous studies (Fischer et al, 2004; Matsuzawa et al, 1992; Roach and Hackett, 2001). There were no major differences between subjects with and without AMS, though future studies need to address the pathophysiological significance of mild brain swelling and peripheral oxidative stress. In the most severe cases, brain swelling was slightly more pronounced and is consistent with the clinical observation that AMS, if left untreated, may progress to HACE.
Footnotes
Acknowledgements
The authors express their sincere gratitude to Martyna Hasselmayr and Elmar Menold for their expert technical assistance and Professors Joe McCord, Joan Abbott and David Begley for stimulating discourse. This manuscript is dedicated to the late Professor ‘Jack’ T Reeves.