
Abstract
Recent evidence suggests that matrix metalloproteinases (MMPs) contribute to acute edema and lesion formation following ischemic and traumatic brain injuries (TBI). Experimental and clinical studies have also reported the beneficial effects of posttraumatic hypothermia on histopathological and behavioral outcome. The purpose of this study was to determine whether therapeutic hypothermia would affect the activity of MMPs after TBI. Male Sprague-Dawley rats were traumatized by moderate parasagittal fluid-percussion (F-P) brain injury. Seven groups (n = 5/group) of animals were investigated: sham-operated, TBI with normothermia (37°C), and TBI with hypothermia (33°C). Normothermia animals were killed at 4, 24, 72 h and 5 days, and hypothermia animals at 24 or 72 h. Brain temperature was reduced to target temperature 30 mins after trauma and maintained for 4 h. Ipsilateral and contralateral cortical, hippocampal, and thalamic regions were analyzed by gelatin and in situ zymography. In traumatized normothermic animals, TBI significantly (P<0.005) increased MMP-9 levels in ipsilateral (right) cortical and hippocampal regions, compared with contralateral or sham animals, beginning at 4 h and persisting to 5 days. At 1, 3, and 5 days after TBI, significant increases in MMP-2 levels were observed. In contrast to these findings observed with normothermia, posttraumatic hypothermia significantly reduced MMP-9 levels. Hypothermic treatment, however, did not affect the delayed activation of MMP-2. Clarifying the mechanisms underlying the beneficial effects of posttraumatic hypothermia is an active area of research. Posttraumatic hypothermia may attenuate the deleterious consequences of brain trauma by reducing MMP activation acutely.
Keywords
Introduction
Matrix metalloproteinases (MMPs) are a family of extracellular zinc- and calcium-dependent proteases that degrade the extracellular matrix (ECM) and other extracellular proteins (Campbell and Pagenstecher, 1999; Lukes et al, 1999; Rosenberg, 2002; Yong et al, 2001). While MMPs are essential for the remodeling of the ECM, tissue morphogenesis, and wound healing, excessive proteolytic activity of MMPs may be detrimental (Ding et al, 2004; Gasche et al, 1999; Lenzlinger et al, 2001; Lo et al, 2002; Mun-Bryce and Rosenberg, 1998; Noble et al, 2002; Romanic et al, 1998; Rosenberg, 1995; Rosenberg et al, 1996, 1998, 2001; Yong et al, 1998; Zalewska et al, 2002, 2003). Rosenberg et al (1996) first reported that biphasic increases in blood—brain barrier (BBB) permeability following reperfusion of middle cerebral artery occlusion in rats correlated with peak increases in MMP-9 and MMP-2. Romanic et al (1998) reported that MMP protein content and activity increased early, whereas MMP-2 increased later after cerebral ischemia.
Treatment with a pan-MMP inhibitor was reported to reduce MMP permeability and hemorrhage after ischemic reperfusion (Gasche et al, 2001; Rosenberg and Navratil, 1997; Sumii and Lo, 2002). Recently, the use of transgenic mice has allowed investigators to more critically evaluate cause-and-effect relationships between MMP activity and pathological outcome in models of cerebral ischemia. For example, using MMP knockout mice, Asahi et al (2001b) reported reduced infarct volume in MMP-9 gene knockout mice compared with wild-type. Interestingly, no effect on infarct volume was reported with MMP-2 knockout mice (Asahi et al, 2001a). Most recently, Tang et al (2004) reported that MMP-9 gene deficiency enhanced collagenase-induced intracerebral hemorrhage and brain injury in mutant mice.
Overactivity of MMPs has also been reported to be important in brain and spinal cord trauma (Rosenberg et al, 1992; Szklarczyk et al, 2002; Von Gertten et al, 2003; Wells et al, 2003). Wang et al (2000) showed that after controlled cortical impact injury in mice, MMP-9 activity was increased at 3 h and persisted until the last study period of 7 days. A slight activity of MMP-2 was also reported in this study. In addition, knockout mice deficient in MMP-9 gene expression showed fewer motor deficits and smaller contusion volumes compared with wild type (Wang et al, 2000). In a model of spinal cord trauma, Noble et al (2002) reported increased expression of MMP-9 using immunocytochemical approaches. In addition, MMP-9 null mice exhibited significantly less disruption of the blood—spinal cord barrier, attenuation of neutrophils infiltration, and significant locomotor recovery (Noble et al, 2002). In another spinal cord injury (SCI) study, MMP-12 was increased 189-fold over normal levels and MMP-12 null mice exhibited improved functional recovery compared with wild-type mice (Wells et al, 2003). Taken together, these studies emphasize the importance of MMPs in the pathogenesis of central nervous system (CNS) trauma.
The increased presence of MMP-9 and MMP-2 has also been associated with early inflammatory consequences of injury, including neutrophil and macrophage accumulation after SCI (Noble et al, 2002). Increased nitric oxide (NO) and peroxynitrite after injury has been shown to activate latent MMPs (Rajagopalan et al, 1996). Some inflammatory cytokines also increase MMP production, to a self-perpetuating cycle, whereby injury induces MMPs to release cytokines which, in turn, enhance MMP activity has been suggested (Winkler and Fowlkes, 2002). Some of the inflammatory cytokines targeted by MMP-2 and 9 include IL-1β, TNF-α, TGFβs, and IGF. Matrix metalloproteinase-9 and 2, by different mechanisms, are both involved in TGF-β activation (Bradham et al, 2002). Matrix metalloproteinases 1, 2, 3, 7, and 9 are involved in TNF-α shedding (Parks and Shapiro, 2001). Blocking by the MMP inhibitor Batimasit decreased TNF-α levels in an LPS model of acute lung injury (Corbel et al, 2001).
The beneficial effects of mild to moderate hypothermia in experimental models of traumatic brain injuries (TBI) have been shown in a large number of laboratories (Bramlett et al, 1995; Clifton et al, 1991; Dietrich et al, 1994b; Hayashi and Dietrich, 2004; Koizumi and Povlishock, 1998). Recently, these experimental findings have been supported by clinical data from individual institutions, where treatment with hypothermia has improved outcome in trauma patients after severe brain injury (Jiang et al, 2000; Marion et al, 1997; Polderman et al, 2002; Resnick et al, 1994). Based on these data, a resurgence in the potential use of therapeutic hypothermia in experimental models of CNS has occurred. There is, therefore, a continued necessity to investigate the mechanisms underlying hypothermic protection. Indeed, recent results from the National Multi-Center Brain Injury Study (hypothermia) Clinical Trial were disappointing (Clifton et al, 2001), and more refinement of the clinical application of hypothermia is required.
Matrix metalloproteinases are regulated at several levels: gene transcription, posttranslational activation of zymogens, and interaction with TIMPs and other molecules, including cytokines. Self-activation via conformational changes that expose catalytic sites of pro-MMPs can also serve as activators. Therefore, the inter-relationships of inflammatory, ECM, and hypothermic mechanisms may be very important. For example, mild to moderate hypothermia has recently been reported to reduce MMP-2 and MMP-9 after experimental focal ischemia (Hamann et al, 2004). Recently, Suehiro et al (2004) reported that induced hypothermia (32°C to 35°C) in severe TBI patients reduced levels of MMP-9 measured in arterial and jugular venous blood samples. Because of the potential for excessive MMP activation to participate in the pathogenesis of TBI, the present study was undertaken to determine whether therapeutic hypothermia would have an effect on MMP activation after fluid-percussion (F-P) brain injury in rats. To this end, a TBI model that is sensitive to posttraumatic hypothermic therapy was used (Dietrich et al, 1994b). Quantitative end points, including gelatin zymography and in situ zymography were utilized to assess regional and temporal profiles of MMP activation.
Materials and methods
Animals
Male Sprague—Dawley rats (n = 5/group) weighing between 270 to 300 g obtained from Charles River Breeders were used for all experiments. Animal care was in accordance with the guidelines set by the University of Miami Care and Use Committee, and all animal protocols were approved by the above committee before study initiation. All animals were kept at a constant temperature (23°C to 25°C) in an air-conditioned room for at least 7 days before the study and exposed to a 12 h light-dark cycle. Rats were allowed free access to water, but food was withheld overnight before surgery.
Temperature manipulation was initiated 15 mins after placement of plastic injury tubing (sham animals) or TBI. Temporalis muscle and rectal (core) temperature probes were used to measure temperature. Animals were placed in a plastic container 15 mins after injury, with target hypothermic temperature (33°C to 34°C) achieved 30 mins after TBI and maintained using cooled air and heating lamps for a period of 4 h. Animals were next allowed to rewarm slowly (1.5 h) and returned to their cages. At the time of killings, animals were deeply reanesthetized using halothane and killed.
Surgical Preparation
Anesthesia was induced using 3.0% halothane in a gas mixture of 70% nitrous oxide and a balance of oxygen, to achieve deep sedation. The animals were then endotracheally intubated and mechanically ventilated with a Harvard rodent ventilator, on a mixture of 70% nitrous oxide, 0.5% to 1.5% halothane, with a balance of oxygen adjusted as described below. Pancuronium bromide 0.5 mg/kg, intravenous was administered every hour during the surgical procedure to facilitate mechanical ventilation. Femoral artery and vein were cannulated with PE-50 tubing for purposes of drug administration, blood sampling for serum glucose and hematocrit measurements, arterial blood gas determination and continuous arterial blood pressure monitoring. Anesthetic concentrations and ventilatory settings were adjusted to maintain mean arterial pressure 100 mm Hg, Pa02 125 to 135 mm Hg and PaC02 35 to 45 mm Hg. Rectal and temporalis muscle thermometers were placed, and self-adjusting feedback external warming lamps were used to maintain desired temperature. Arterial blood gases, blood glucose, and hematocrit were measured 15 mins before and after TBI, and 1, 2, and 3 h after TBI. Animals were then placed in a stereotactic frame for introduction of a right parietal craniotomy 3.8 mm posterior to bregma and 2.5 mm lateral to midline (Paxinos and Watson, 1982). In TBI animals, an F-P injury device (Clifton et al, 1991) was used to induce moderate parasagittal TBI as previously described (Dietrich et al, 1994; Kinoshita et al, 2002a, b; Vitarbo et al, 2004).
Gel Zymography
At either 4, 24, 72 h, or 5 days after TBI, animals (n = 4 to 5) were reanesthetized and killed. Ipsilateral and contralateral cortex, hippocampus, and thalamus were dissected and frozen in liquid nitrogen. At the time of assay, the tissues were thawed on ice and minced in 1 × zymogram sample buffer (Biorad®, Hercules, CA, USA) containing 4% SDS, 25% glycerol, 62.5 mmol/L Tris (6.8), and 0.01% bromophenol blue. Equal volumes of each sample were electrophoresed on 12% Tris-glycine PAGE gels with 0.1% gelatin at 70 V. Gels were renatured in 2.7% Triton X-100 for 40 mins and then incubated at 37°C for 48 h in development buffer (50 mmol/L Tris, 200 mmol/L NaCl, 5 mmol/L CaCl2, 0.2% Brij35). After development, gels were stained in 0.5% Coomassie blue R-250 for 30 mins and destained in 50% methanol, 10% acetic acid. Matrix metalloproteinase-2 and MMP-9 appear as clear bands against the blue-stained background. Both the pro (uncleaved) and active (cleaved) forms of MMP-9 were visible on the gels. Based on molecular weight, the bands corresponding to the cleaved (active) form of the enzymes were identified and used for analysis. Band intensities were analyzed using the standard computer-assisted image analysis software (UVP®, Upland, CA). To correct for evenness of loading, equal amounts of sample were added to 2% SDS, boiled 5 mins and run on a parallel gel for Western analysis of β-actin protein levels. Data were expressed as mean ± s.d. One-way analysis of variance (ANOVA) was used to compare groups with post hoc comparisons conducted using Fisher LSD.
In Situ Zymography
At 24 or 72 h after TBI, animals were killed and brains were removed and frozen. Coronal sections were cryosectioned at 12 μm at bregma levels 1.2, −1.8, −3.3, and −5.3. Sections were then processed for in situ zymography by methods recently described (Lee et al, 2004). The sections were thawed and incubated with reaction buffer (50 mmol/L Tris-HCl, 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.2 mmol/L NaN3, pH 7.6) containing 285 μg/mL DQ™ gelatin, fluorescein conjugate (Molecular Probes®, Eugene, OR, USA) overnight in a humid chamber at room temperature. The FITC labeled gelatin is quenched until degraded by the gelatinase activity, whereupon it fluoresces at 520 nm. The localization of fluorescence by fluorescent microscopy indicates the regions and cells exhibiting gelatinase activity. After incubation, the slides were quickly rinsed in 1× PB, and without fixation, photographed by fluorescent microscopy.
Results
Physiologic Parameters
Physiologic parameters were assessed for each experimental group 15 mins before and 15 mins, 1, 2, and 3 h after TBI or sham procedures (15 mins pre- and post-values are shown in Table 1). No significant differences were observed between normothermic groups with respect to temporalis muscle temperature, arterial pH, arterial p02, pC02, or arterial blood pressure. Physiologic variables were within normal ranges throughout the study periods. Temporalis muscle temperatures differed after TBI between the normothermic and hypothermic groups (bold values, P<0.001), as expected.
Physiological data
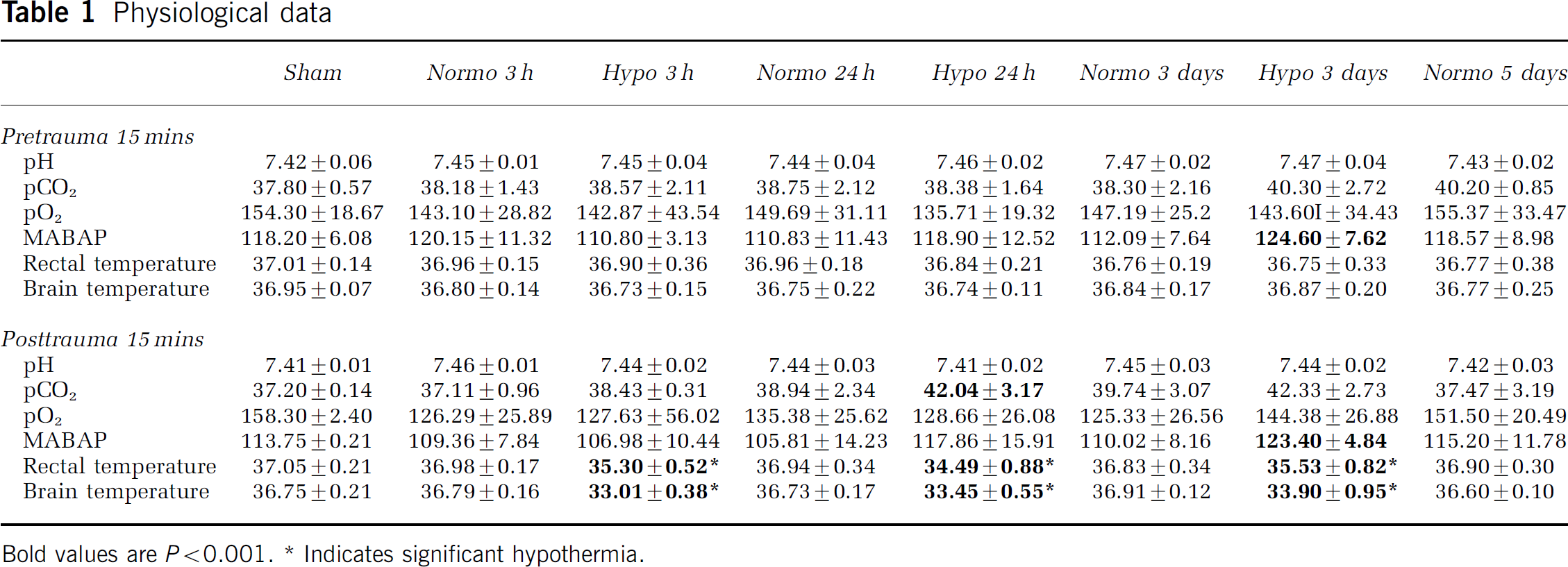
BoldvaluesareP<0.001.
Indicatessignificanthypothermia.
Matrix Metalloproteinase Activity Time course
To determine the temporal profile of MMP-2 and MMP-9 activation after TBI, animals were subjected to moderate TBI and allowed to survive for 4 h, 24 h, 3 days, and 5 days. Gelatinase activity was assessed by gelatin zymography for each of these groups and sham-operated animals. Zymography gels showed bands corresponding to the pro and cleaved forms of MMP-2 and MMP-9, although the pro forms were present at much lower levels. Bands representing other gelatinases were not observed. The active form of MMP-9 was observed in sham animals, as well as all TBI groups (Figure 1A). Matrix metalloproteinase-9 activity was elevated significantly over sham starting at 4 h after TBI, reaching a maximum at 24 h, and was still elevated at 3 and 5 days after TBI. Matrix metalloproteinase-2 was observed at very low levels in the sham, 4 h, and 24 h groups (Figure 1B). Matrix metalloproteinase-2 activity increased significantly at 3 days over sham, and remained elevated at 5 days. The increases in MMP-2 and MMP-9 observed were limited to the ipsilateral hemisphere in both cortex and hippocampus (Figure 1C). Matrix metalloproteinase-9 was also elevated at 24 h in the ipsilateral thalamus (Figure 1C).
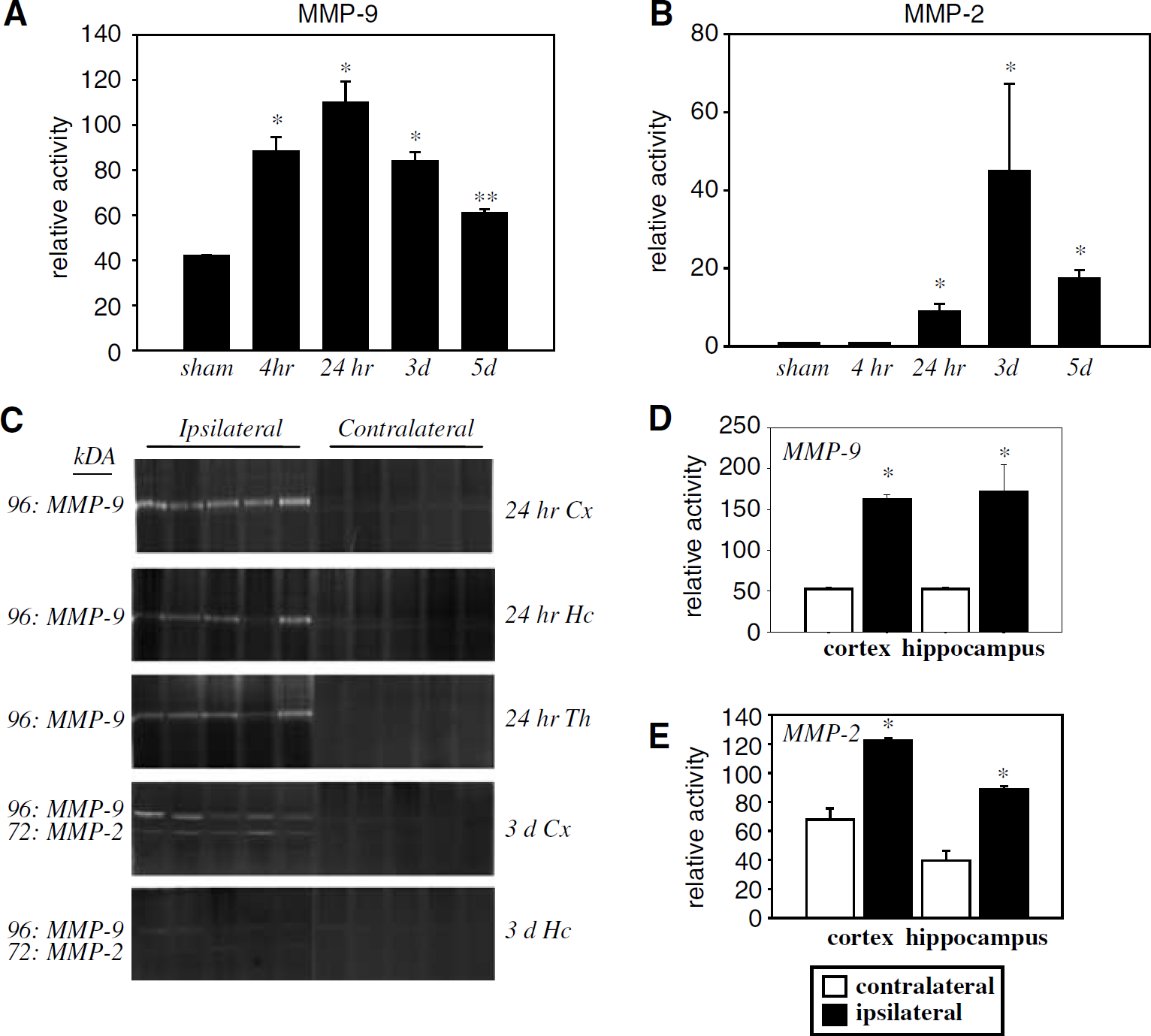
Gelatinase activity after traumatic brain injuries (TBI). (
Effects of Hypothermia on Matrix Metalloproteinase Activity
Based on the results of the time course data, the hypothermia animals were assessed at the time when activity was highest after TBI: 24 h for MMP-9 and 3 days for MMP-2 (Figure 2). Matrix metalloproteinase-9 activity (Figure 2A) at 24 h after TBI was increased in the normothermic group over sham values (P<0.005). Posttraumatic hypothermia reduced this activation (P<0.005) to near sham levels. Matrix metalloproteinase-2 activity (Figure 2B), was increased after TBI at 3 days in both normothermia and hypothermia groups (P<0.001) compared with sham. There was no difference in MMP-2 activity between the normothermic and hypothermic groups.
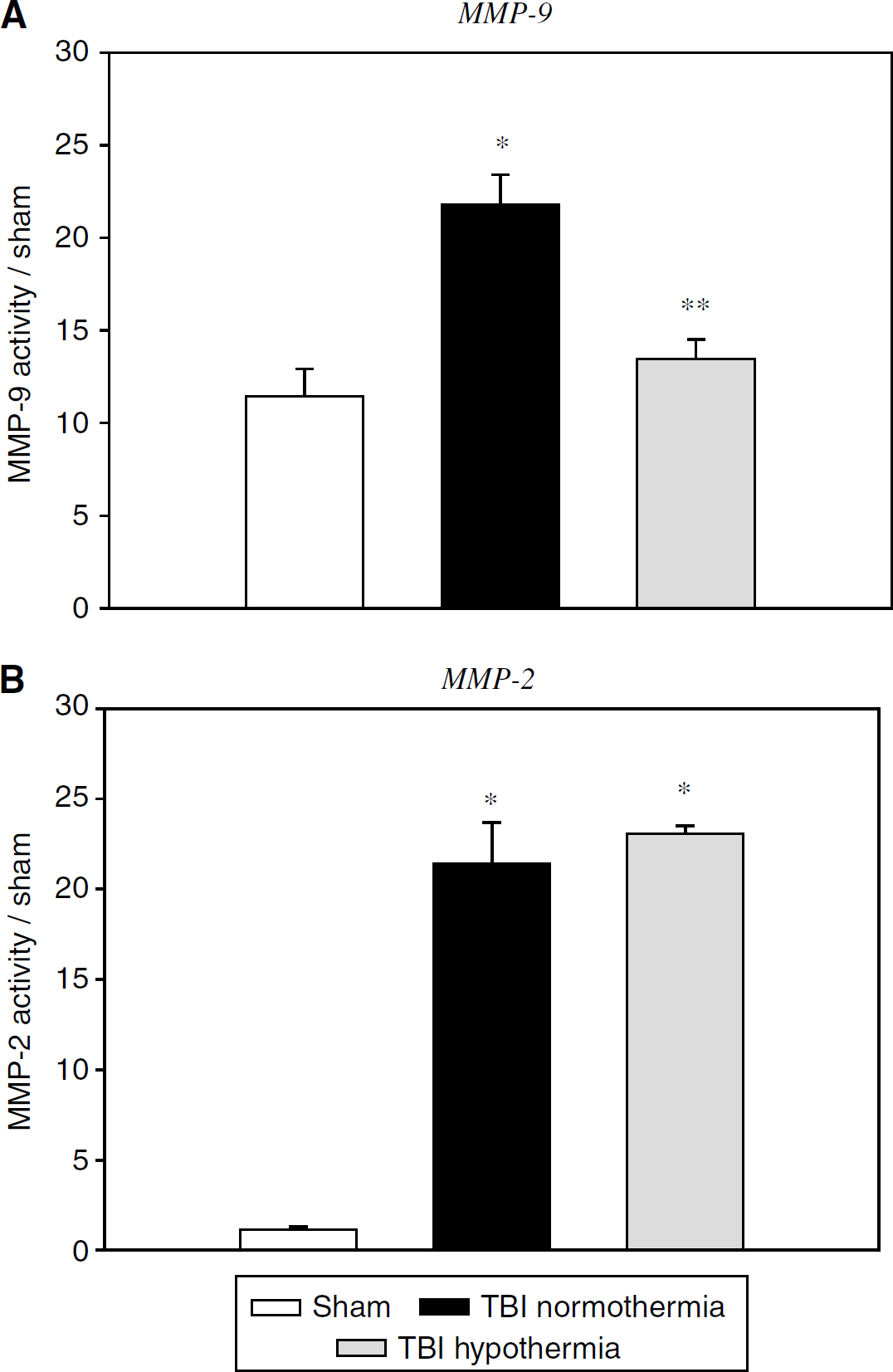
Effect of hypothermia on gelatinase activity as assessed by gelatin zymography. (
In Situ Zymography
The regional and cellular localization of gelatinase activity in the brain was studied by incubating the tissue with a masked fluorescent-tagged gelatin substrate. In regions or cells where the activity was present, fluorescence was observed. The activity of MMP-9 and MMP-2 could not be distinguished by this method because they are both gelatinases. However, based on the time course data, it can be inferred that activity seen at 24 h is most likely because of MMP-9 alone, whereas at 3 days, a combination of both MMP-2 and MMP-9 is observed. In all animals, including sham operated, blood vessels throughout the brains were positive for MMPs.
Gelatinase activity at 24 h after TBI was seen in the traumatized hemispheres of all animals. In the cerebral cortex (Figures 3B–3D), numerous discrete cells showed high levels of activity that were not present in the sham (Figure 3A) animals. Consistent with the gelatin zymography data, animals treated with posttraumatic hypothermia (Figures 3E and 3F) showed many fewer cells that were positive for excessive MMP activity.
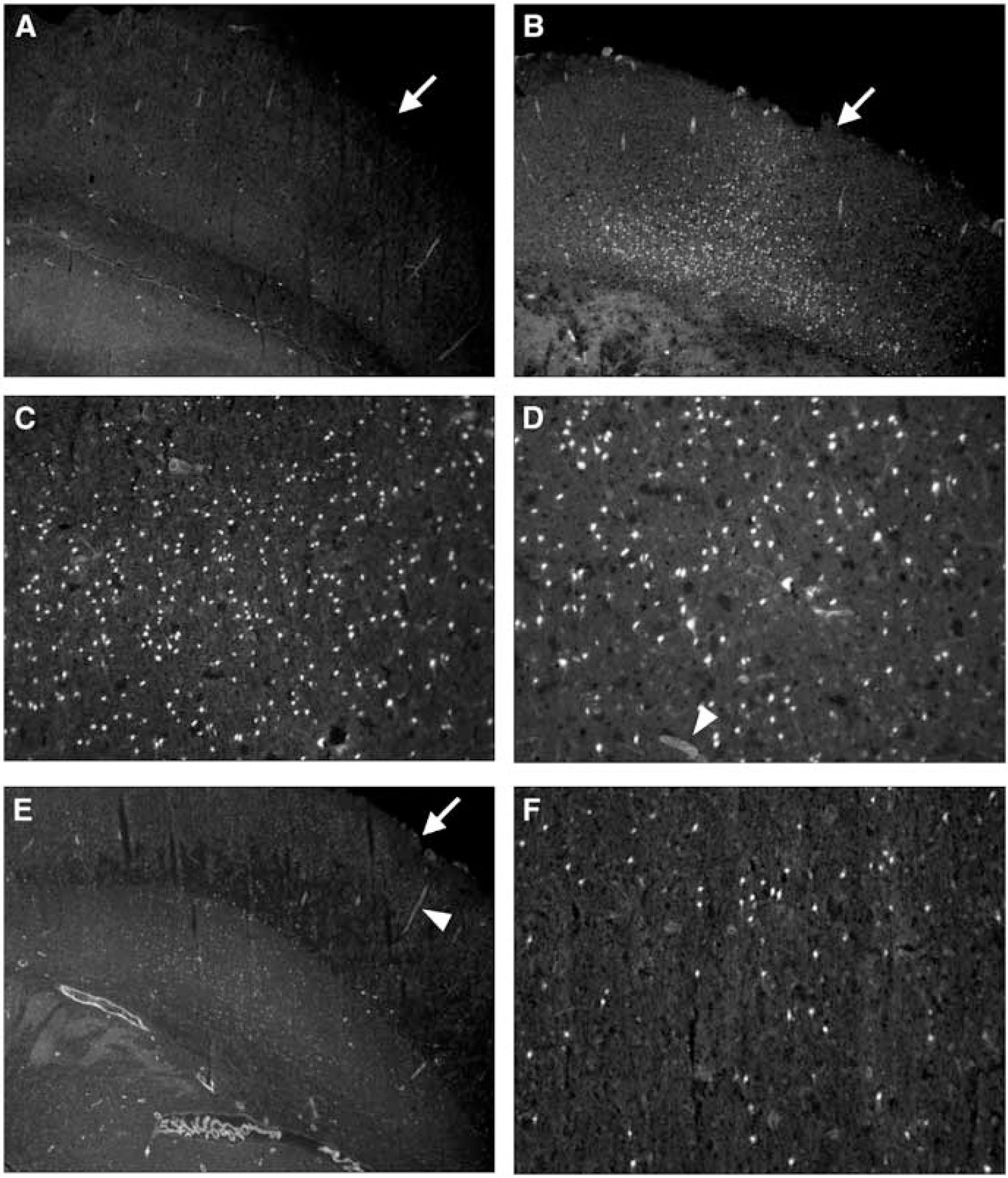
Gelatinase activity in the ipsilateral cortex 24 h after traumatic brain injuries (TBI) as visualized by in situ zymography. (
In the ipsilateral hippocampus 24 h after TBI (Figure 4) discrete neurons were positive for gelatinase activity. No activity was observed in the sham (Figure 4A) or contralateral (data not shown) hippocampus. Traumatic brain injuries resulted in an increase of signal in neurons of the CA3 region (Figures 4B and 4C). Posttraumatic hypothermia attenuated this response (Figure 4D). In the ipsilateral dentate gyrus and hilar neurons, there was also a mild increase after TBI (Figure 4E). This response was also decreased by hypothermia (Figure 4F). Gelatinase activity was observed in select cells of the ipsilateral thalamus as well, but in smaller numbers (data not shown).
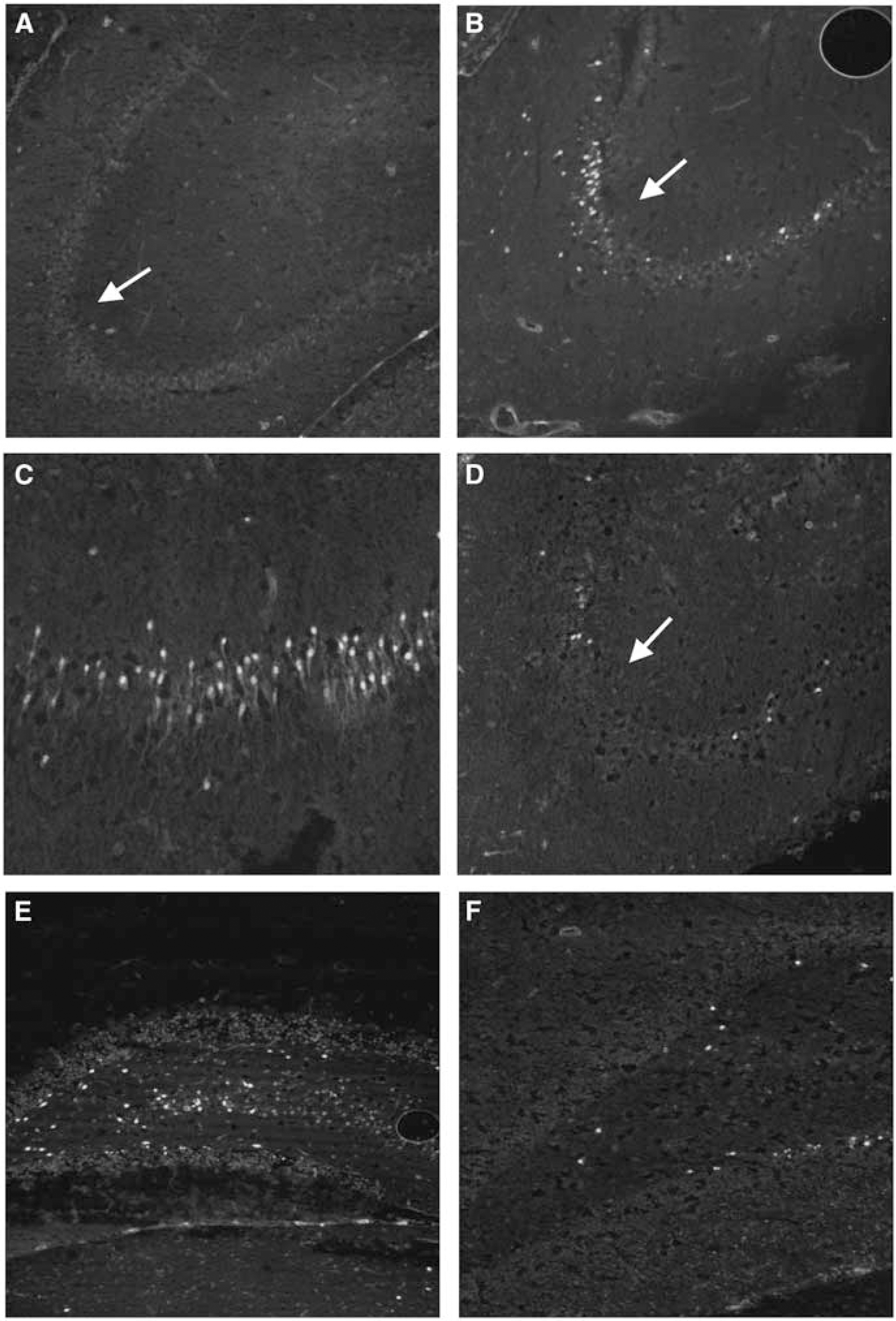
In situ zymography of gelatinase activity in the hippocampus 24 h after traumatic brain injuries (TBI). (
At 3 days after TBI, in situ zymography revealed a very different pattern of fluorescence (Figure 5). Whereas the activity at 24 h was restricted to very discrete cells, at 3 days, in addition to this discrete cellular localization, there was a very robust, diffuse pattern observed as well in the traumatized cerebral cortex (Figures 5C and 5D). This diffuse pattern was restricted to the traumatized cerebral cortex and was likely because of a combination of both excessive MMP-2 and 9 activity. In the injured cortex, gelatinase activity was observed in the contused areas in both the normothermic (Figure 5A) and the hypothermic (Figure 5B) animals with little to no difference in activity levels. Matrix metalloproteinase activity was also seen in discrete cells in the ipsilateral thalamus and CA3 hippocampus similar to the 24 h patterns observed.
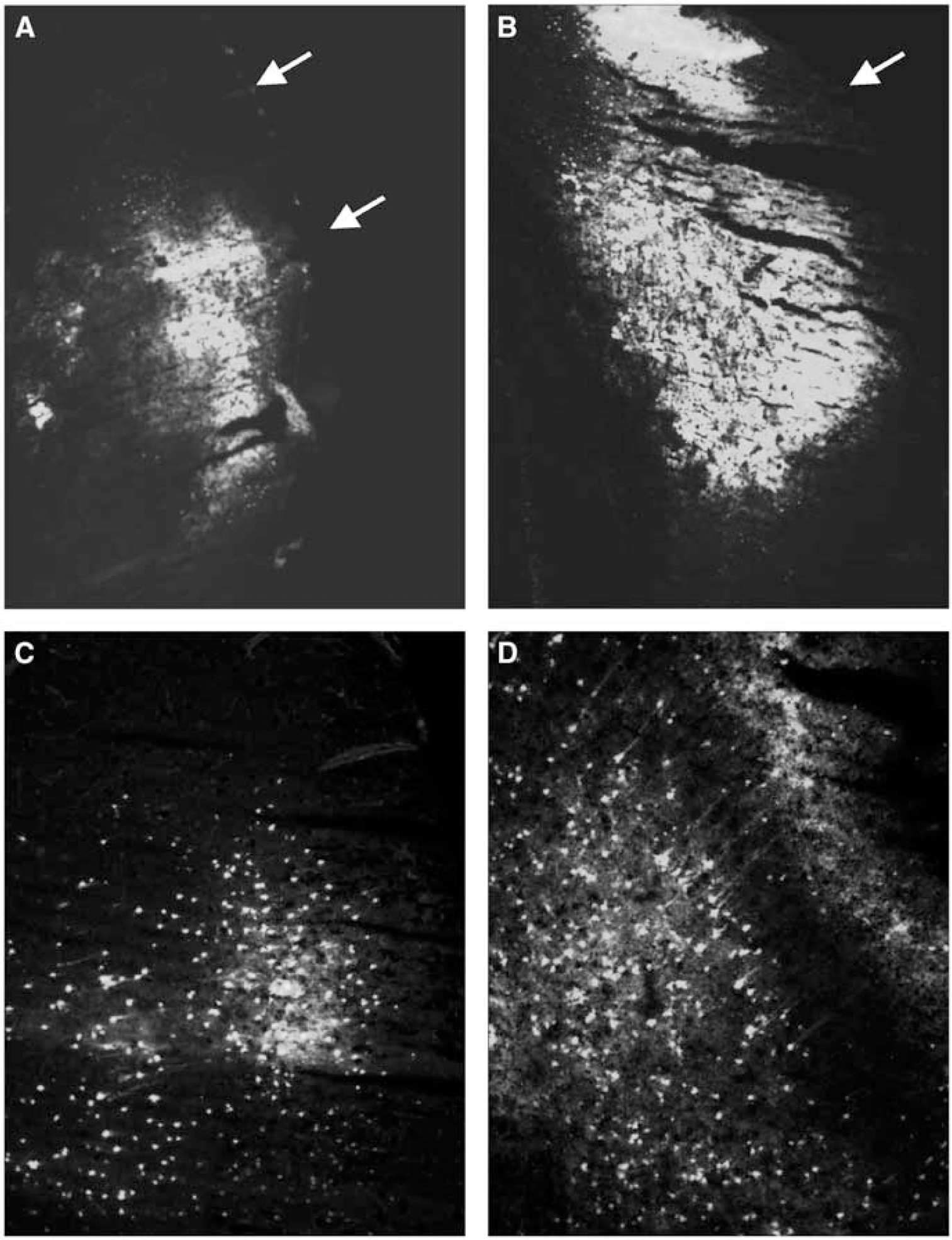
In situ zymography 3 days after traumatic brain injuries (TBI) shows increased gelatinase activity in the contusion area. Ipsilateral cortex in a normothermic animal (
Discussion
We report that moderate F-P brain injury upregulates MMP-9 and MMP-2 and that posttraumatic hypothermia reduces MMP-9 expression. Gelatin gel zymography showed quantitative elevations in gelatinase activity at 4 h, 1, 3, and 5 days after TBI. Additionally, in situ zymography showed gelatinase activity localized in ipsilateral cortical, hippocampal, and thalamic neurons 24 h after injury. At 3 days after injury, there was an additional diffuse pattern of activation, primarily in the contused cerebral cortex. Moderate posttraumatic hypothermia induced 30 mins after the insult and maintained for a 4-h period significantly reduced MMP-9 levels at 24 h, compared with normothermic traumatized rats. Taken together, these findings emphasize that posttraumatic hypothermia may improve structural and functional outcome by reducing the deleterious effects of acute uncontrolled MMP activation.
Data support the possibility that inflammatory cells and mediators are important contributing factors to cell death after brain trauma (Lotocki et al, 2004; McIntosh et al, 1998; Morganti-Kossmann et al, 2002). In particular, proinflammatory cytokines, BBB permeability, leukocyte adhesion and extravasation, and NO have all been implicated in the pathogenesis of brain injury (del Zoppo and Mabuchi, 2003; Dirnagl et al, 1999). With respect to therapeutic hypothermia, it has been reported that mild cooling attenuates many of these microvascular and inflammatory cascades (Chatzipanteli et al, 1999, 2000; Jiang et al, 1992; Kinoshita et al, 2002a, b; Smith and Hall, 1996; Sutcliffe et al, 2001). For example, in models of cerebral ischemia and trauma, moderate hypothermia significantly reduces early polymorphonuclear leukocyte (PMNL) accumulation and protects against BBB dysfunction (Chatzipanteli et al, 2000; Dietrich et al, 1990; Jiang et al, 1992; Smith and Hall, 1996). In addition, moderate hypothermia has been reported to attenuate trauma-induced increases in inducible NOS activity (Chatzipanteli et al, 1999; Han et al, 2002).
As previously discussed, the activation of MMPs has been studied in various models of cerebral ischemia and inflammation. In models of global and focal ischemia, MMPs are up-regulated in both cortical and hippocampal areas (Lee et al, 2004; Romanic et al, 1998; Rosenberg et al, 1998; Zalewska et al, 2002). Using knockout mice, various investigations showed the importance of MMP activation in ischemic cell death, BBB permeability, and lesion formation (Asahi et al, 2001a, b; Wang et al, 2000). Because MMPs degrade components of the extracellular matrix, investigations have concentrated on the importance of MMP activation on progressive microvascular damage including hemorrhage (Gasche et al, 2001; Lo et al, 2002; Power et al, 2003; Tang et al, 2004).
Blood—brain barder breakdown as a consequence of mechanical damage to blood vessels is an acute consequence of TBI (Cortez et al, 1989; Dietrich et al, 1994a). Altered BBB integrity leads to the activation of inflammatory processes and subsequent edema formation. These acute mechanical events can then lead to the stimulation of neurons, astrocytes, oligodendrocytes, microglia, and endothelial cells to secrete MMPs (Hanemaaijer et al, 1993; Jiang et al, 1992; Pagenstecher et al, 1998; Rosenberg, 2002). The pharmacologic inhibition of MMPs has been reported to protect against BBB dysfunction, infarction, and the incidence of hemorrhage (Romanic et al, 1998). In a model of ischemic reperfusion, Sumii and Lo (2002) reported that treatment with a broad-spectrum MMP inhibitor, BB-94 reduced hemorrhagic volumes in rats that had received the thrombolytic tissue plasminigen activator. Matrix metalloproteinases, particularly MMP-9, play a significant role in inflammatory cell recruitment after brain and spinal cord injury and are released from vascular endothelium and leukocytes during the inflammatory phase of ischemic stroke (Opedenakker et al, 2001). Noble et al (2002) reported reduced numbers of neutrophils after SCI in MMP-9-depleted mice compared with wild type. Thus, an important role of MMP-9 activation in the transmigration of PMNLs has been emphasized.
The control of MMPs is regulated at several levels including transcriptional control, the secretion of an inactive zymogen subject to proteolytic activation as well as inhibition by endogenous inhibitors (Rosenberg, 1995). It is possible that after TBI, each one of these regulatory mechanisms may be involved in the observed increased expression of MMP-2 and 9 after trauma. As previously noted, mechanisms underlying the beneficial effects of hypothermia are multifactorial and include glutamate release, stabilization of the BBB, oxygen radical production, intercellular signal conduction, protein synthesis, ischemic depolarization, reduced metabolism, membrane stabilization, inflammation, activation of protein kinases, and early gene expression (Dietrich et al, 1996). Because the pathophysiology of TBI is complex, the fact that multiple injury mechanisms are temperature sensitive may account for the dramatic effects of temperature on traumatic outcome. In the present study, hypothermia significantly reduced MMP-9 activation at 24 h after TBI. This temperature effect on MMP activation may therefore underlie some of the previous observations made with therapeutic hypothermia in multiple CNS injury models.
For example, Dietrich et al (1994b) first reported significant reductions in contusion volume and cortical neuronal damage with posttraumatic hypothermia. In another study, Kinoshita et al (2002a) reported that posttraumatic hypothermia reduced hemoglobin extravasation compared with normothermic traumatized rats. As previously described, excessive MMP activation has been implicated in hemorrhagic transformation of ischemic reperfusion (Horstmann et al, 2003; Lee and Lo, 2004; Power et al, 2003). In a recent study, hypothermia reduced basal lamina collagen type IV loss and hemorrhage after transient focal ischemia in rats (Hamann et al, 2004). The present data indicate that hypothermia may therefore be reducing the amount of hemorrhage and BBB disruption after TBI by attenuating MMP activation.
Posttraumatic hypothermia has also been reported to reduce the inflammatory consequence of TBI. Chatzipanteli et al (2000) reported that posttraumatic hypothermia reduced PMNL accumulation at 1 and 3 days after F-P injury. Because MMPs play a dominant role in the transmigration of neutrophils, hypothermia may also be reducing PMNL accumulation and subsequent degranulation by its effects on MMP activation. The synthesis and release of a variety of proinflammatory cytokines has been implicated in the pathogenesis of TBI (McIntosh et al, 1998; Morganti-Kossmann et al, 2002). Interleukin-1β and TNFα have each been shown to be upregulated early after moderate TBI (Shohami et al, 1994; Knoblach et al, 1999). In models of F-P brain injury, posttraumatic hypothermia has been reported to reduce protein levels of IL-1β and TNFα (Goss et al, 1995; Kinoshita et al, 2002b; Vitarbo et al, 2004). The transcriptional control of MMPs is mediated by activator Protein-1, NFκB, and TNF-alpha-responsive regulatory element (TRE) sites in the promoter region of MMPs (Knittel et al, 1999; Mori et al, 2002). In this regard, it is important to note that therapeutic hypothermia has recently been reported to reduce the activation of NFκB after cerebral ischemia (Han et al, 2003). Thus, posttraumatic hypothermia may be altering MMP activation by reducing upstream signaling cascades as well as transcriptional factors involved in MMP gene expression. It is also possible that various pathomechanisms with corresponding signaling pathways and transcriptional factors have significant crosstalk that allow injury pathways to interact (Fowlkes and Winkler, 2002; Levkau et al, 2002; Neumar et al, 2003). Our data emphasize the effects of TBI on MMP elevations, which would be predicted from the literature to affect BBB alterations, hemorrhage, edema formation, inflammatory mediators, and cellular infiltration.
As previously described, hypothermia has been reported to protect vulnerable neuronal populations from injury. Recently, MMP activation has been reported to directly trigger cellular death by interrupting cell-matrix survival signaling (Chen and Strickland, 1997). In a model of endothelial cell hypoxia, MMP-9 was reported to activate endogenous apoptotic machinery after the hypoxic insult (Lee and Lo, 2004). In the present study, gelatinolytic activity was shown to be present within vulnerable neuronal populations and visualized in the walls of blood vessels using in situ zymographyy. In a model of cortical spreading depression, Gursoy-Ozdemma et al (2004) reported gelatinolytic activity in neurons and the pia-arachnoid. Thus, in addition to hemorrhagic contusion formation, excessive MMP activation may also be involved in selective neuronal vulnerability after TBI (Frisch and Francis, 1994; Levkau et al, 2002). It will be important in future studies to determine how hypothermia-induced MMP reductions affect necrotic and apoptotic neuronal cell death mechanisms after TBI.
In the present study, a 4 h period of posttraumatic hypothermia reduced MMP-9 levels, while not significantly affecting more delayed elevations in MMP-2. In this regard, it is important to note that this level and duration of hypothermia has previously been shown to be neuroprotective in this TBI model (Suzuki et al, 2003). Possibly a longer duration of hypothermia may also have affected MMP-2, since the duration of hypothermic treatment is a critical factor in determining treatment outcome (Hayashi and Dietrich, 2004). However, because late-occurring events such as matrix remodeling may be reparative, it may not be advantageous to extend hypothermic treatments unnecessarily. Based on published data showing that MMP-2 knockouts are not protected against focal ischemia (Asahi et al, 2001a), it may be reasonable to conclude that some of the benefits of hypothermic therapy are causally related to the reduction of acute MMP-9 elevations after trauma. Obviously more work is required to investigate the full effect of temperature alterations on this very complicated family of proteases.
In summary, we report that moderate F-P brain injury leads to temporal and regionally specific increases in MMP-2 and MMP-9 activation. Importantly, these alterations in gelatinase activity occurred in vulnerable brain regions. Post-traumatic hypothermia significantly reduced MMP-9 activation in both cortical and hippocampal areas. Taken together, these findings and previously published data indicate that the effects of hypothermia on MMP activation may be an important mechanism underlying the benefits of therapeutic hypothermia.
Footnotes
Acknowledgements
The authors thank Charlaine Rowlette for expert editorial assistance and manuscript preparation.