
Abstract
To evaluate the effect of body iron stores on the vulnerability of the brain to ischemia, a focal permanent brain ischemia was induced by photothrombotic occlusion of cortical vessels in rats with or without chronic treatment with iron dextran (25 mg iron/kg, every other day for 20 days, intraperitoneally). Iron dextran induced systemic iron overload as evidenced by high ferritin (Ft) (x 5) and total iron levels (x 3) in serum as well as increased Ft expression in the liver and heart. Conversely, neither serum free iron levels nor Ft expression in the brain were changed by iron dextran. Finally, infarct volume was not modified by iron dextran. In addition, induction of ischemia in rats treated with FeCl3 (560 μg iron/kg, intravenously) as a means of increasing serum free iron levels during the ischemic period did not enlarge infarct volume. We then explored the effect of brain ischemia itself on serum Ft by measuring serum Ft before and after induction of brain ischemic insults with different neurologic outcomes in rats (brain embolization with microspheres, photothrombotic occlusion of cortical vessels, four-vessel occlusion). Serum Ft levels were found higher at day 1 after ischemia than before ischemia only in rats subjected to the most severe insult (brain embolization). In conclusion, our study showed that increased body iron stores do not increase the vulnerability of the brain to ischemia and that brain ischemia, if severe, results in the elevation of serum Ft levels.
Introduction
In the mammalian organism, ferritin (Ft) is a complex of iron with the protein apoferritin that is composed of 24 subunits of two types (high and light subunits also named H and L chains, respectively). Ferritin is essentially located within cells and constitutes the main intracellular iron storage protein (Eisenstein, 2000). The principal factor that controls cellular Ft content is the intracellular level of free iron itself (Cairo et al, 1995), the only form of iron able to catalyze radical oxygen species production. Thus, Ft provides a means of storing the metal within cells in a safe and available manner. Ferritin is also present at a very low concentration in blood but the role of circulating Ft is still unknown. However, serum Ft has been used widely in clinical medicine chiefly as an indicator of body iron stores. Thus, high Ft levels are frequent in patients with hereditary hemochromatosis, frequent blood perfusion or parenteral iron infusion.
Although controversial, elevated body iron stores as evidenced by classical markers including serum Ft, serum total iron, transferrin saturation, or iron binding capacity were associated with increased risk of ischemic events (Salonen et al, 1992; Sullivan, 1992; Kiechl et al, 1993). Carriers of the hemochromatosis gene appear to be at increased risk of myocardial infarction and cardiovascular death (Cartwright et al, 1979; Bulaj et al, 1996; Roest et al, 1999; Tuomainen et al, 1999). Recent animal experiments have suggested that iron overload contributes to the development of vascular diseases by promoting thrombosis after arterial injury (Day et al, 2003). The role of iron overload in stroke is poorly documented. However, high serum Ft on admission of acute stroke patients (within 24 to 48 h after stroke onset) was reported to predict a bad prognosis (Davalos et al, 1994, 2000; Erdemoglu and Ozbakir, 2002) suggesting that increased body iron stores before stroke onset can aggravate the cytotoxicity of brain ischemia. However, this interpretation is questionable because it is unknown whether Ft levels were already elevated before stroke onset. In addition, there is a study in which high Ft levels on admission were not associated with deteriorating stroke (Christensen et al, 2002). Ferritin is a positive acute phase protein and stroke is associated with systemic inflammation (Davalos et al, 1997; Vila et al, 2000; Di Napoli et al, 2001; Perini et al, 2001). For these reasons, the hypothesis that high Ft levels on admission were secondary to stroke and Ft levels increased during the interval between stroke onset and blood sampling as a reflection of stroke severity cannot be excluded.
The objectives of our study were (i) to evaluate the effects of increased body iron stores on brain vulnerability to ischemia and (ii) to explore the effects of brain ischemia itself on serum Ft levels as well as the potential association between Ft levels and stroke severity. For this purpose, brain infarct volume was measured in stroke rats with and without systemic iron overload. We then measured serum Ft levels in rats subjected to ischemic stroke models with different histologic and neurologic outcomes.
Materials and methods
The experiments were performed on male Wistar rats (280–350 g, Depré, Saint Doulchard, France) and conducted according to the French Department of Agriculture guidelines (licence number 21CAE035). Surgical interventions were performed under chloral anaesthesia (400 mg/kg, intraperitoneally) (Sigma, Saint Quentin Fallavier, France) and blood withdrawal under halothane anaesthesia (3% for induction and 0.7% for maintenance) (Belamont, Boulogne Billancourt, France).
Ischemic Stroke Models
To evaluate the effect of brain ischemia itself on serum Ft levels and the potential link between changes in Ft levels and the severity of ischemia in terms of neurologic deficit, rats were subjected to ischemic stroke models with different histologic outcomes assuming that the location and lesion size are the main determinants of stroke neurologic outcome (Reith et al, 1996; Rogers et al, 1997). Three models routinely performed in our laboratory were used. Multiple infarcts scattered within the right hemisphere were induced by cerebral embolization with calibrated (50 μm) microspheres (3 M, Cergy-Pontoise, France). The injection of microspheres into the right common carotid artery was performed in rats with or without previous occlusion of the homolateral external carotid artery to modulate the degree of embolization. In high embolization protocol, the external carotid was occluded whereas it was not in the low embolization protocol. A right cortical infarct was induced by the photothrombotic ischemic stroke model using Rose Bengal as the photosensitizer agent (20 mg/kg, intravenously) (Sigma Aldrich, Saint Quentin Fallavier, France) and a diode-pumped solid-state laser working at 532 nm as the illumination source. Emerging power of the optic fiber was 90 mW for inducing a large infarct (around 70 mm3, infarct involving the hindlimb and the forelimb sensorimotor cortex) and 30 mW for inducing a smaller infarct (around 25 mm3). The small infarct is more appropriate than the large infarct to evaluate potential cytotoxic interventions. Finally, the selective and bilateral neuronal loss of the CA1 subfield of the hippocampus was induced by the four-vessel occlusion (4-VO) model. The occlusion of the two common carotid arteries and the vertebral arteries was maintained for 10 min. Rats that did not lose consciousness during 4-VO were excluded. The histologic outcomes from these models as well as detailed protocols were previously reported by our laboratory (Bralet et al (1987) and Demougeot et al (2001) for brain embolization; Demougeot et al (2001, 2003) for photothrombotic occlusion and Marie et al (1994) for 4-VO).
Neurologic Evaluation
Popular simple tests (De Ryck et al, 1989; Schallert et al, 2000; Peeling et al, 2001) were performed to assess the general sensorimotor function after (1, 3, and 7 days) brain ischemia induction. The beam walking test measured the ability to move along a 150-cm long × 2.5-cm wide elevated (1 m from floor) wood beam (graded 0 for a rat unable to move or to retain on the beam to 4 for normal behavior). The vibrissae-elicited forelimb placing test was performed in rats held by their torsos allowing forelimbs to hang free. Independent testing of each forelimb was induced by brushing the respective vibrissae on the edge of the table top (graded 0 for a rat unable to place the forelimb quickly onto the countertop and 1 for a rat with normal placing). In the limb placing test, the limbs were tested in rats put along the edge of the platform for placing in response to their sudden release away from the platform (graded 0 for a rat unable to place the forelimb quickly onto the countertop and 1 for a rat with normal placing). Finally, a test of sensory neglect was performed by recording the time taken to remove a small adhesive label placed on the distal aspect of each forelimb (graded 0 for a rat unable to remove the label within 2 min and 1 for rat without neglect). In the last three tests, only the left limbs were scored. Rats without deficit have a neurologic score of 8. Rats that have not been successful in doing one of the four tests before ischemia induction were excluded from the study.
Biochemical Analysis
Sera were kept at -80°C until analysis. Serum Ft was blindly assayed using an immunoturbidimetry kit (FERRITIN GEN.2, Cobas INTEGRA 400, Roche Diagnostics, Meylan, France). Iron was measured by atomic absorption spectrometry (Hitachi Z7000) using polarized Zeeman background correction for matrix interference (Demougeot et al, 2000). The detection limit of the method was 0.001 mg/L. For determination of serum free iron (unbound protein iron) concentration, sera were previously passed through a centrifugal filter device with a membrane cutoff of 10,000 Da (Microcon YM-10, Millipore, Bedford, USA).
Ferritin expression in the heart, liver, and brain was analyzed by Western blotting. Ferritin was detected in protein extracts using rabbit antibodies against human H-Ft and L-Ft (Alpha Diagnostic International, San Antonio, USA) (1:1,000 dilution) and peroxidase-conjugated anti-rabbit IgG (Jackson, ImmunoResearch Laboratories Interchim, Montluçon, France) (1:27,000 dilution). High Ft and L-Ft bands were evaluated by densitometry. Purified recombinant human Ft for the L-chain and for the H-chain was used as positive control.
Measurement of Infarct Volume
Infarct volume induced by the photothrombotic ischemic stroke model was measured from coronal brain sections (50-μm thick) stained with cresyl violet (0.4%) (Sigma, Saint Quentin Fallavier, France). Injured cortical areas (unstained tissue) were blindly measured using a computer image analysis system (Scion Image, NIH, Bethesda, MD, USA) and the distances between respective sections were used to calculate a linear integration for the lesion volume.
Treatment
Iron dextran and FeCl3 purchased from Sigma (Saint Quentin Fallavier, France) were dissolved in saline. After dissolution, the pH of the saline solutions were 7 and 2.7, respectively. Chronic administration of iron dextran (25 mg iron/kg, intraperitoneally) every other day for 20 days (10 injections) was used to increase body iron stores. Administration of FeCl3 was used to increase serum free iron levels during the ischemic period. Injection of a dose of 560 μg iron/kg (10 μmol iron/kg) was calculated to be required for increasing serum free iron to 100 μmol/L. FeCl3 (insoluble at neutral pH) was preferred to ferric ammonium citrate (soluble at neutral pH) because we found that ferric ammonium citrate but not FeCl3 induced mortality when given at high dose (50 μmol iron/kg). Preliminary experiments (n = 6) showed that serum free iron levels increased from undetectable levels to 11.8±5.2 μmol/L (0.66±0.29 mg/L) after (1 h) a single injection (intravenously) of FeCl3 (10 μmol iron/kg). Thus, free iron in serum was lower (12 versus 100 μmol/L) than that expected from the calculation likely as the result of iron binding to albumin (Hider, 2002). The role of serum free iron on infarction processes was investigated in rats injected with FeCl3 (560 μg iron/kg, intravenously) just before and 1 h after photothrombosis. Indeed, regarding the toxicity of iron, two injections of FeCl3 rather than injection of a higher single dose of FeCl3 were preferred. The effects of iron dextran and FeCl3 were compared with that of saline and saline acidified with HCl to pH 2.7, respectively.
Statistical Analyses
Except for neurologic scores that were expressed as median, all values were expressed as mean±s.d. Values were analyzed by nonparametric statistical tests (Kruskall—Wallis or Mann—Whitney U-test for independent measurements and Friedman or Wilcoxon for pairwise comparisons). Statistical significance was set at P<0.05.
Results
Effect of Iron Dextran and FeCl3 on Body Iron Stores and Infarct Volume
The repeated injections of iron dextran led to a time-dependent increase in serum Ft levels (Figure 1). They also increased by 3.3-fold the amount of total iron in serum when collected the day after the last injection of iron dextran (1.22±0.14 mg/L in control group versus 4.01±1.44 mg/L in the iron dextran group, P<0.05, data not shown). By contrast, serum free iron remained under detectable levels after iron dextran administration. Figure 2 shows tissue Ft expression in control and iron dextran-treated rats. Light Ft expression in control rats was five-fold higher in the liver (1.35±0.42) than in the heart (0.27±0.08, P<0.05, significance not shown) whereas H-Ft expression between the liver (1.50±1.67) and the heart (0.28±0.22) was not significantly different. Treatment with iron dextran significantly increased Ft expression in the liver and the heart but not in the brain (Figure 2), indicating increased delivery of free iron to peripheral organs but not to the brain. Ferritin expression in the liver was increased by a factor of 17 and 10 for H-Ft and L-Ft, respectively. The corresponding values for Ft expression in the heart were 3 and 4. Table 1 shows infarct volume induced by the photothrombotic ischemic stroke model (30 mW for the emerging power) in rats receiving iron dextran or FeCl3 and in their corresponding controls. No difference was observed between treated and control groups. These results indicated that neither systemic iron stores nor the levels of circulating free iron during the ischemic period influenced infarction processes.
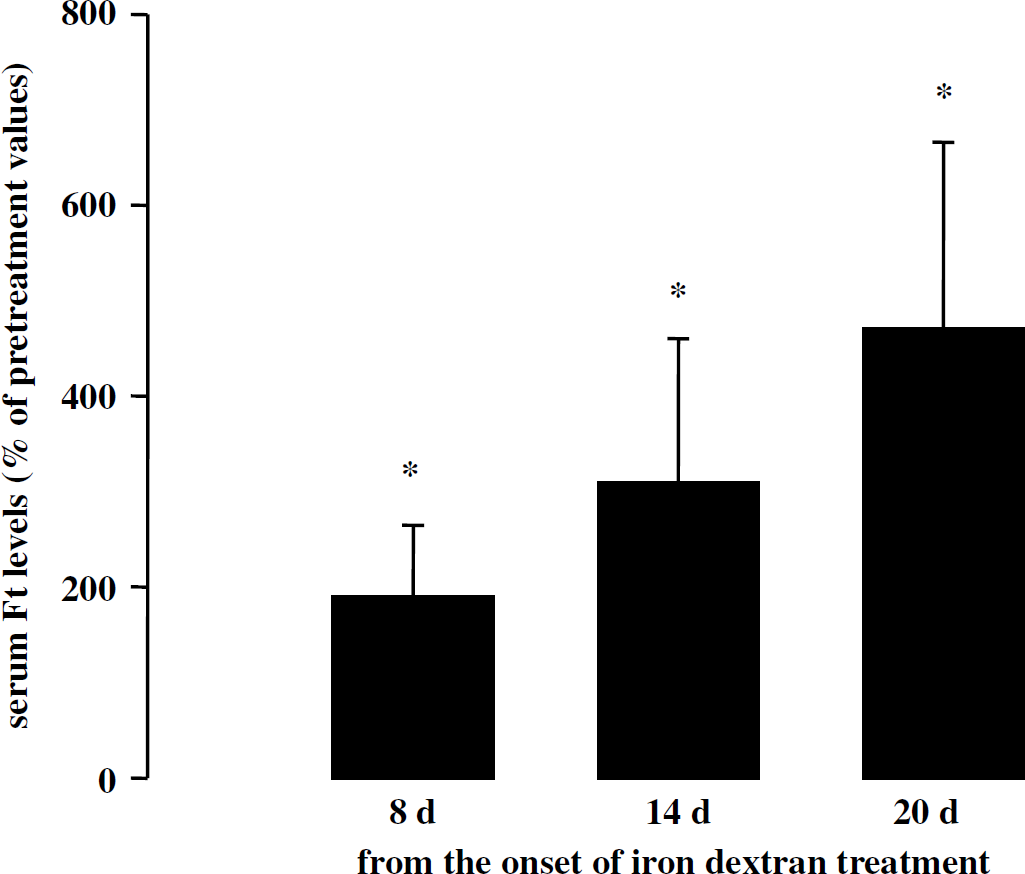
Effect of iron dextran on serum ferritin (Ft) levels. Iron dextran was injected (25 mg iron/kg, intraperitoneally) every other day for 20 days in 7 rats. For each rat, blood was collected before and during treatment. Ferritin levels were expressed as percentage of pretreatment values (49.1±23.0 ng/mL). *P<0.05 different from pretreatment values.
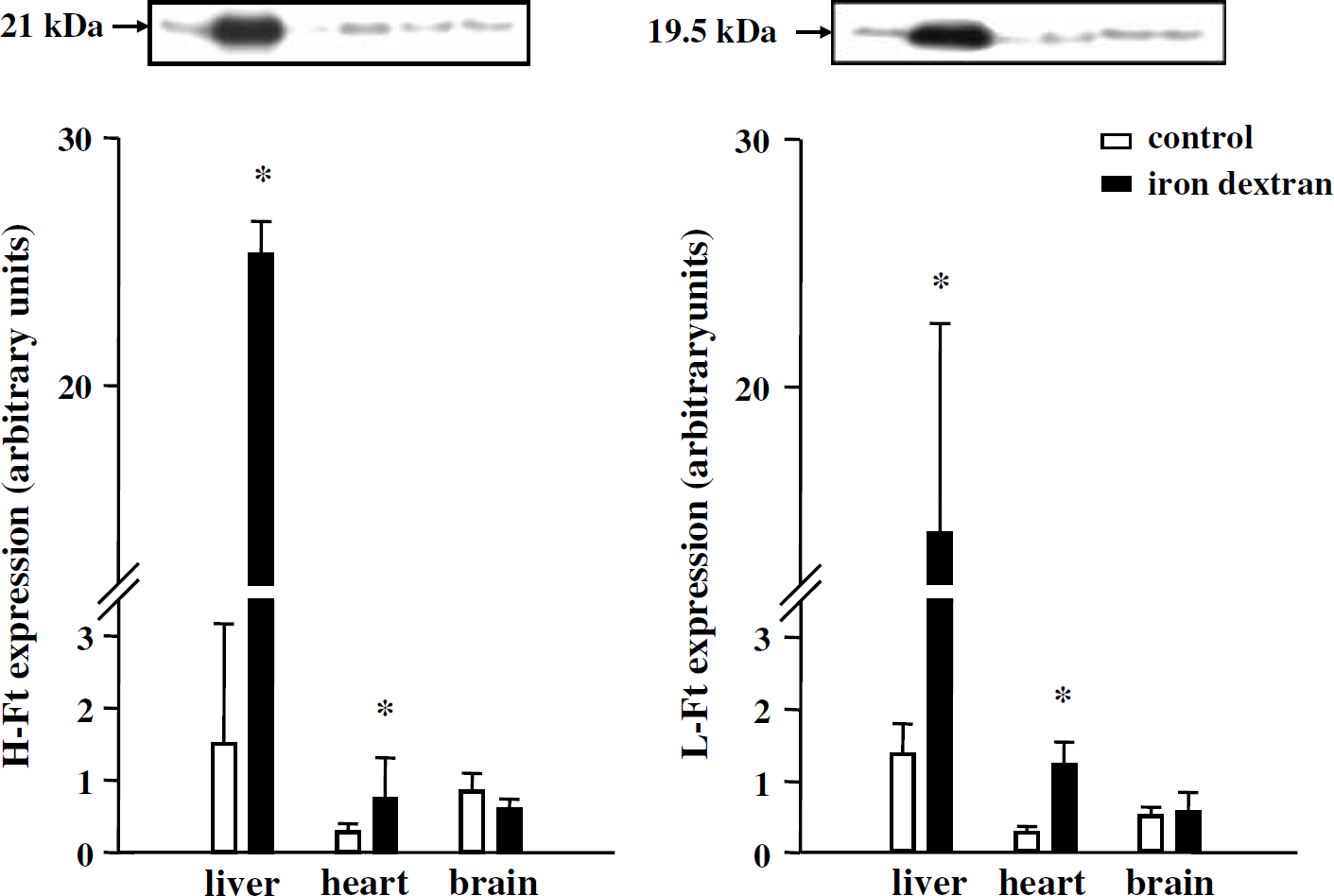
Effect of iron dextran on the expression of high ferritin (H-Ft) and light ferritin (L-Ft) in homogenates from the liver, heart, and brain. Iron dextran was injected (25 mg iron/kg, intraperitoneally) every other day for 20 days. Western blot analysis was performed on control and iron dextran-treated rats (three animals per group). *P<0.05 different from control.
Effect of iron dextran and FeCl3 on the vulnerability of the brain to infarction
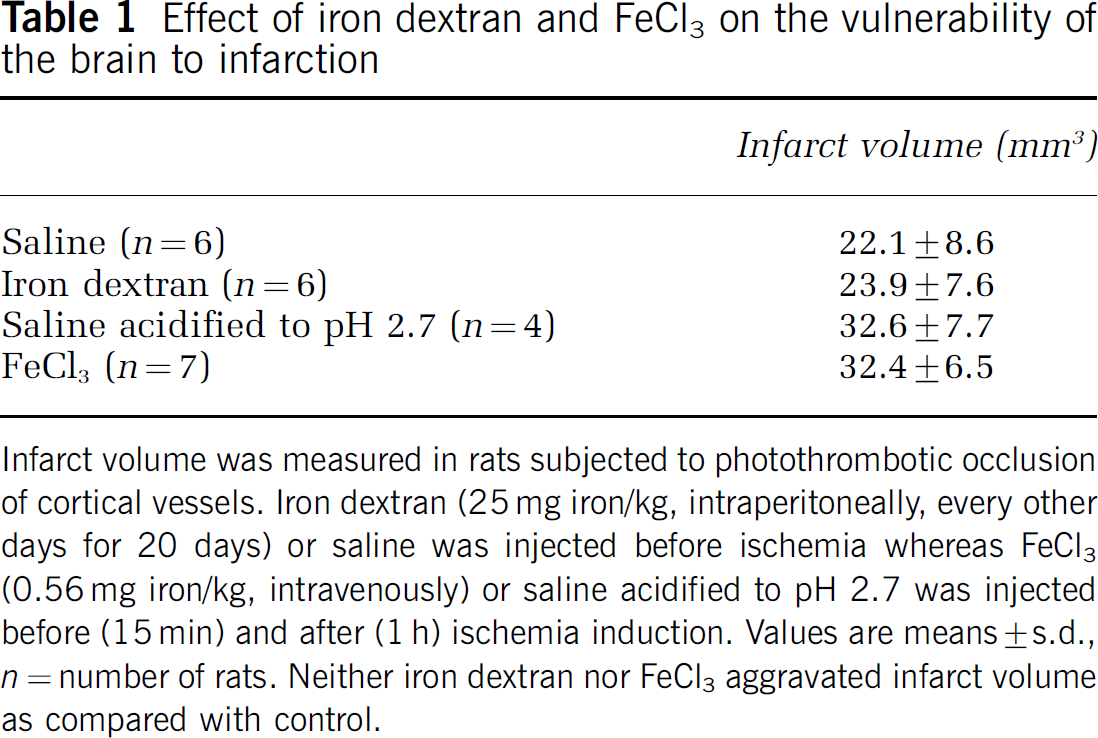
Infarct volume was measured in rats subjected to photothrombotic occlusion of cortical vessels. Iron dextran (25 mg iron/kg, intraperitoneally, every other days for 20 days) or saline was injected before ischemia whereas FeCl3 (0.56 mg iron/kg, intravenously) or saline acidified to pH 2.7 was injected before (15 min) and after (1 h) ischemia induction. Values are means±s.d., n = number of rats. Neither iron dextran nor FeCl3 aggravated infarct volume as compared with control.
Effect of Brain Ischemia on Neurologic Status
The experiments were conducted in rats subjected to 4-VO, photothrombosis (90 mW for the emerging power), and high embolization protocol as well as in sham-embolized rats (occlusion of the common and external carotids but no injection of microspheres). Embolized rats exhibited a degree of embolization of 427.5±47.9 microspheres. The effects of brain ischemia on neurologic scores are shown in Figure 3. Brain embolization was the most severe stroke model in terms of neurologic deficit. While all rats subjected to 4-VO recovered a normal neurologic score as soon as 24 h after the ischemic insult, all rats subjected to photothrombotic occlusion and cerebral embolization exhibited a neurologic deficit at that time. However, the neurologic score was lower in embolized rats than in rats subjected to photothrombosis (P<0.05, not indicated in Figure 3). In addition, only embolized rats still exhibited neurologic deficit on days 3 and 7. The neurologic score after surgical intervention was normal in sham-embolized rats (data not shown).
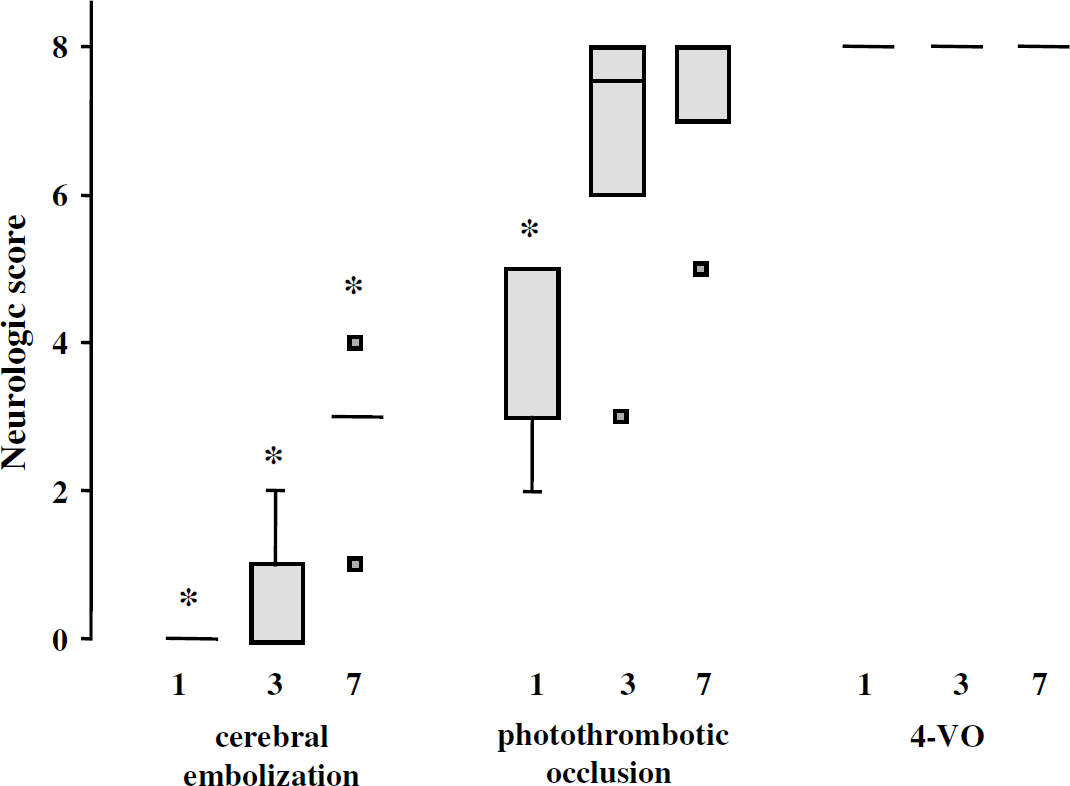
Effect of brain ischemia on neurologic status. Rats were subjected to cerebral embolization (n = 6), photothrombotic occlusion (n = 6), and four-vessel occlusion (4-VO) (n = 5). The neurologic scores were determined before and after (1, 3, and 7 days) ischemia. The neurologic score in rats without deficit (before ischemia) was 8. Data are the medians and percentiles. *P<0.05 different from preischemic values.
Effect of Brain Ischemia on Serum Ft Levels
Additive experiments were conducted to assess the effects of ischemia on serum Ft levels. Ferritin levels were measured in each rat before (1 day) and after (4 h, 24 h) induction of 4-VO, photothrombosis (90 mW for the emerging power), and embolization (both high and low embolization protocols). Rats used in these experiments are different from those used for the determination of neurologic status. The mortality rate of embolized rats was null (0/20) for the low embolized protocol and reached 53% for the high embolized protocol (10/19). The corresponding degree of embolization of surviving embolized rats (in which serum Ft levels were measured) was 397±129 (n = 20) and 560±217 microspheres (n = 9). No mortality was observed in rats subjected to photothrombotic occlusion or 4-VO.
The mean serum Ft level before ischemia was 41.2±15.2 ng/mL (n = 47). As shown in Table 2, only brain embolization, the most severe ischemic stroke model, resulted in high Ft levels. Thus, postischemic Ft levels at 24 h were 143% (P<0.05) and 200% (P<0.001) of preischemic values in high and low embolized groups, respectively, whereas no significant changes were observed in sham-animals. By contrast, serum Ft levels at this time were 97% and 81% of preischemic values after photothrombotic occlusion and 4-VO, respectively (NS). Ferritin levels at 4 h postischemia were not modified by embolization or 4-VO.
Effect of brain ischemia on serum Ft levels
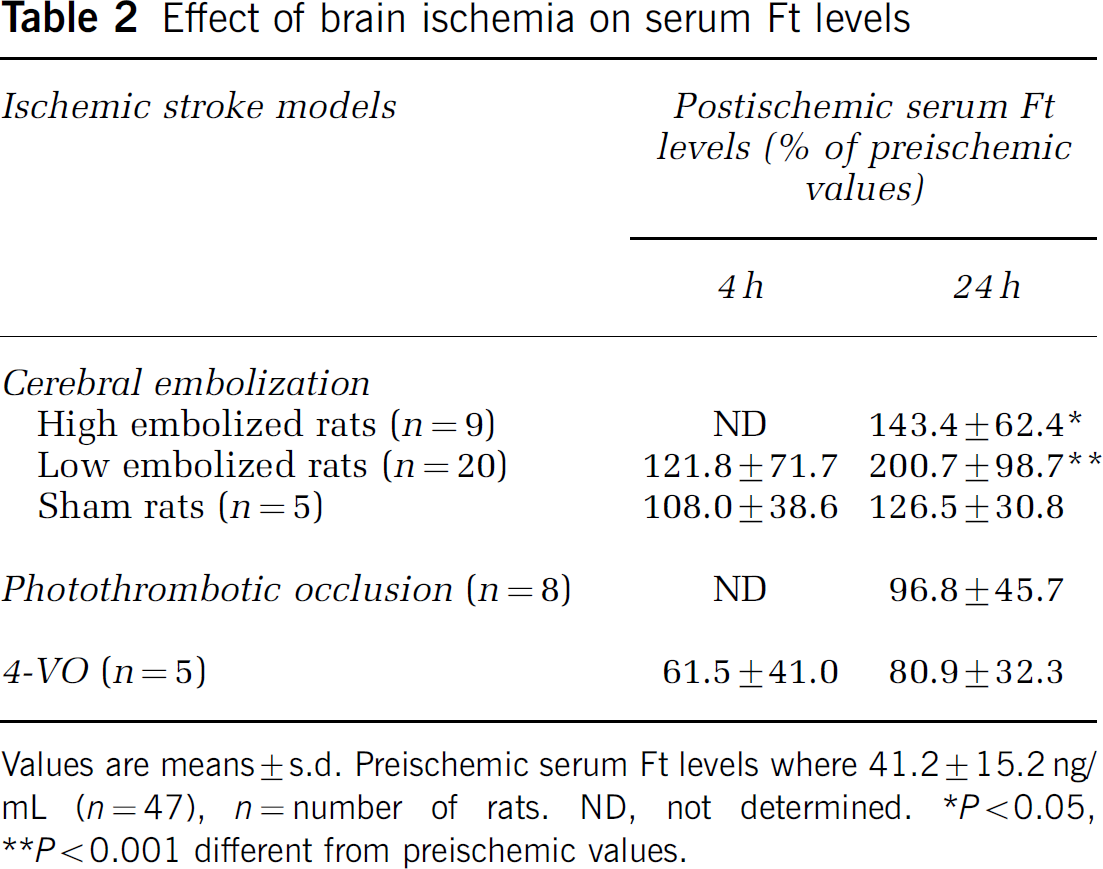
Values are means±s.d. Preischemic serum Ft levels where 41.2±15.2 ng/mL (n = 47), n = number of rats. ND, not determined. *P<0.05, **P<0.001 different from preischemic values.
From the results obtained in all embolized rats, changes in Ft levels at 24 h after ischemia (abscissa) and the degree of embolization (ordinate) were plotted. As shown in Figure 4, the two parameters did not correlate (r = 0.01). Finally, total protein levels in serum were not modified by brain embolization (data not shown), indicating that high Ft levels after embolization did not result from hemoconcentration.
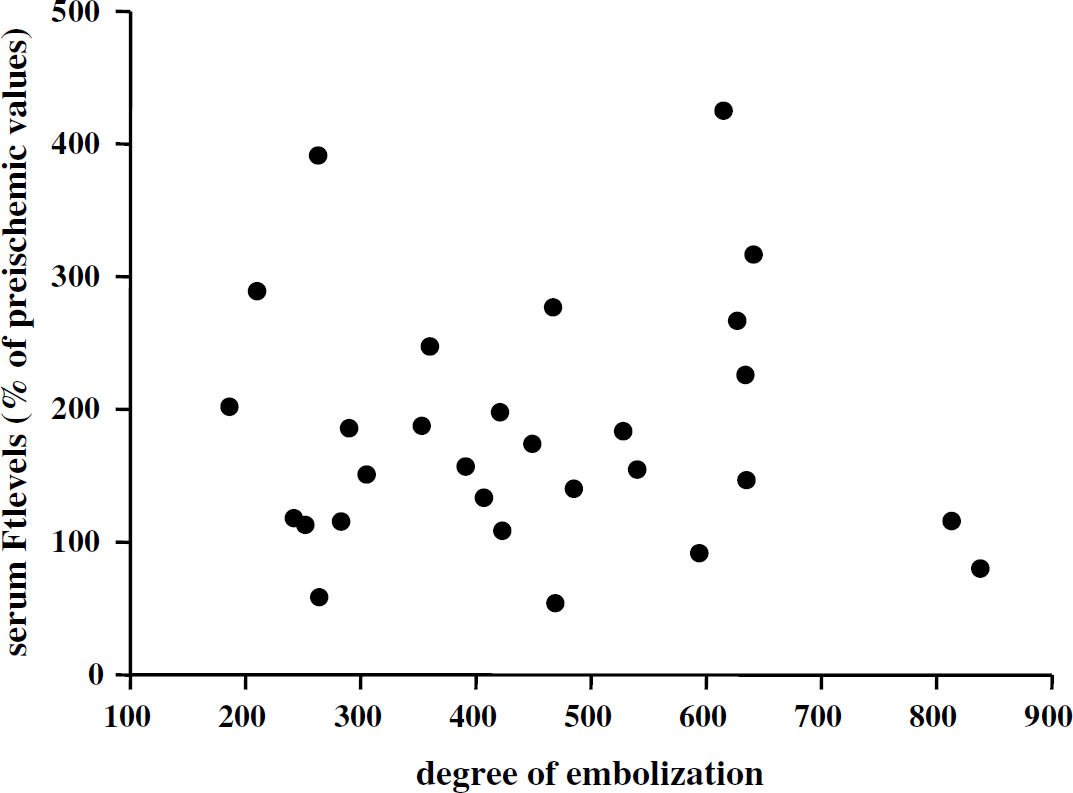
Lack of correlation between serum ferritin (Ft) levels and degree of embolization.
Discussion
The main results of the present study are that increased body iron stores before brain ischemia do not aggravate infarct volume and that brain ischemia, only if severe, results in high Ft levels within the 24 h after brain ischemia induction.
In the brain as in other organs, the levels of intracellular free iron are tightly controlled and the scenario elaborated by cells to maintain free iron level under toxic threshold is now well understood (Cairo et al, 1995). Conversely, the mechanisms by which circulating iron enters brain parenchyma are still not completely understood. The paucity of studies aimed at elucidating these mechanisms contrasts with the increasing evidence that iron accumulation in the brain can cause a vast array of CNS disorders (Zecca et al, 2004). Of course, the blood—brain barrier limits the delivery of plasma iron to brain cells. Thus, conversely to peripheral organs, the brain does not seem to accumulate iron when body iron stores are increased, as suggested by studies in which a diet supplemented with iron was reported to not induce change in cerebral total iron levels (Vayenas et al, 1998; Castellanos et al, 2002). One finding of our study is that systemic iron overload increased the delivery of free iron to peripheral organs but not to the brain as evidenced by increased Ft expression in the liver and heart but not in the brain. These results are consistent with the rapid sequestration and inactivation of circulating iron by cerebral endothelial cells (Deane et al, 2004) as well as the recent hypothesis that the brain but not systemic iron status is important in the control of iron delivery to cerebral cells (Burdo et al, 2003). The protection of the brain against changes in circulating iron levels likely explains why patients with hereditary or secondary hemochromatosis are liable to cardiac and hepatic failure, while neurologic symptoms are not usually observed in these patients (Eaton and Qian, 2002). The lack of changes in cerebral free iron levels after iron dextran likely explains the lack of difference in infarct volume between iron dextran-treated rats and rats with normal iron status. Indeed, the amounts of free iron within cells determine the vulnerability of tissue to ischemia (Lesnefsky and Ye, 1994). Moreover, the inactivation of intracellular free iron in ischemic brain by a liposoluble iron chelator was recently reported to decrease oxidative stress and infarct volume (Demougeot et al, 2004). The inefficiency of iron dextran-induced iron overload to aggravate infarct volume suggests that patients with increased body iron stores have not an increased risk for stroke. Although this hypothesis remains to be explored, carriers of the hemochromatosis gene were reported to be not related to stroke (Njajou et al, 2002). However, it is not clear in this study whether patients with positive HFE testing do or do not exhibit iron overload.
To the best of our knowledge, only one previous experimental study has assessed the role of increased body iron stores in brain ischemia (Castellanos et al, 2002). In this study, rats fed a diet supplemented with iron for 9 weeks were reported to exhibit large infarct volume as compared with rats fed a normal diet. Surprisingly, the increase in serum Ft and the liver total iron levels induced by the iron diet was not associated with elevated total iron levels in serum as observed after treatment with iron dextran. Besides, the effect of the iron diet on serum free iron was not investigated. Free iron that does not exist in normal serum was reported in patients with hereditary hemochromatosis (De Valk et al, 2000). Moreover, circulating free iron can enter the cerebral ischemic areas through the disrupted blood—brain barrier and perhaps enhance infarction processes. Therefore, we tested the hypothesis that the lack of effect of iron dextran on infarct volume was related to its incapacity to increase serum free iron levels. Our results with FeCl3 did not favor this hypothesis either. Although speculative, alternative hypothesis to explain the divergent results between iron dextran and the iron diet may be differences in the duration of the iron overload (9 weeks versus 3 weeks for iron dextran), in the ischemic stroke model (permanent middle cerebral artery occlusion versus photothrombotic cortical occlusion) or in iron overload-induced myocardial dysfunction.
High serum Ft levels on admission of acute stroke patients have been found to be predictive of poor outcome (Davalos et al, 1994, 2000; Erdemoglu and Ozbakir, 2002). However, the origin of high Ft levels in stroke is unknown. It is unknown whether Ft levels were already elevated before stroke or whether high Ft levels are secondary to stroke. Regarding the conflicting results on the effects of the diet supplemented with iron and the long-term administration of iron dextran on infarct volume in rats, we are unable to eliminate conclusively the possibility that high Ft levels observed in stroke patients reflected excess body iron stores. However, if Ft levels really reflect systemic iron overload before stroke, we think that serum Ft levels in stroke patients would have been higher than that reported. Indeed, whereas serum Ft levels increase in proportion with serum body iron stores, differences in Ft levels were rather modest (x 2) between stroke patients with bad outcome and that with good outcome. These changes are more compatible with the hypothesis that high Ft levels in stroke patients with bad outcome reflect stroke severity. In accordance with this hypothesis, we demonstrated for the first time that brain ischemia, only if severe, resulted in high serum Ft levels in rats. These results support previous clinical studies in which a positive association was observed in stroke patients between the extent of the tissue damage or neurologic deficit and the strength of the systemic inflammation response when evaluated by serum C-reactive protein or interleukin-6 levels (Fassbender et al, 1994; Vila et al, 2000, Perini et al, 2001). Another argument in favor of serum Ft in acute stroke as a marker of systemic inflammation is the clinical study in which deteriorating stroke without high levels of circulating interleukin-6 on admission was found to be not associated with high levels of Ft (Christensen et al, 2002). However, Ft levels in rats subjected to cerebral embolization did not correlate with the degree of embolization, an indirect marker of the amount of infarcted brain tissue (Figure 4). These data do not question the positive association between the lesion size and the systemic inflammation response to stroke but rather confirm the poor sensitivity of circulating Ft to quantify the acute phase response.
In conclusion, we reported that increased body iron stores before ischemic stroke as evidenced by high Ft levels did not aggravate infarct volume in rats subjected to the photothrombotic ischemic stroke model. Based on this observation, it is unlikely that the vulnerability of brain to stroke was increased in patients with increased body iron stores. We also demonstrated that ischemic stroke, if severe, resulted in the elevation of serum Ft levels. These data are in accordance with a positive association between the lesion size or the neurologic deficit and the systemic inflammation response to stroke. It remains to be determined whether systemic inflammation response to stroke is harmful or just an epiphenomenon in stroke.
Footnotes
Acknowledgements
The authors thank Dr Lequeu and Dr Bidan (LABM, Point Medical, Dijon, France) for their help in serum ferritin assay.