
Abstract
Immune system activation has implications for cerebrovascular health, but little is known about the function of the immune system after a major cerebrovascular event, such as cardiac arrest and cardiopulmonary resuscitation (CA/CPR). Cardiac arrest and cardiopulmonary resuscitation damages the hippocampus, an important component of the hypothalamic—pituitary—adrenal (HPA) axis, and alterations in HPA axis activity can affect immune function. We tested the hypothesis that CA/CPR (approximately 8mins) would cause HPA axis dysregulation and alter the delayed type hypersensitivity (DTH) response to antigenic challenge. We also assessed the primary and secondary antibody response of mice exposed to CA/CPR. Of the mice exposed to CA/CPR, half had brains protected by hypothermia to isolate the effects of the CA/CPR procedure from the effects of CA/CPR-induced neuronal damage. Cardiac arrest and cardiopulmonary resuscitation-induced neuronal damage resulted in a persistent elevation of blood corticosterone concentration and a concomitant augmentation of the DTH response to antigenic challenge. Furthermore, immune activation before CA/CPR decreased survival after global ischemia. These data highlight the potential impact of neuronal damage on cell-mediated immune function and the role of humoral immune activation in outcome after global ischemia.
Immune system activation has implications for cerebrovascular health. Nineteen percent of patients recovering from a cardiovascular event report having an upper respiratory infection within the preceding 2 weeks (Meyers, 2003). Furthermore, influenza vaccination protects against cerebrovascular events (Gurfinkel et al, 2004; Lavallee et al, 2002; Nichol et al, 2003; Siscovick et al, 2000); it is associated with a reduction in primary cardiac arrest after adjustment for demographic, clinical, and behavioral differences (Siscovick et al, 2000), and a decrease in the risk of death from cardiac arrest (Gurfinkel et al, 2004). The mechanism through which vaccination decreases cardiovascular risk is not well understood, but may involve decreased exposure to long-term inflammatory processes (Blake and Ridkor, 2002) or prothrombotic states (Macko et al, 1996). Interestingly, influenza vaccination may not be as protective against recurrent cardiovascular events as it is against primary cardiovascular events (Jackson et al, 2002; Naghavi et al, 2000). Whether this discrepancy reflects a shift in the body's response to vaccination after a cardiovascular crisis remains to be determined.
Cerebral ischemia also influences peripheral inflammatory responses. After antigenic challenge, the delayed type hypersensitivity (DTH) immune response is characterized by cutaneous inflammation at the site of challenge caused by an infiltration of monocytes and lymphocytes into the epidermis and dermis. A positive correlation exists between the intensity of the swelling and the immune reaction, thus DTH response is a common in vivo measure of cell-mediated immunity (Janeway et al, 2001). Stroke has a biphasic impact on DTH, such that in the acute recovery phase the DTH response is suppressed; however, over time, the DTH response is augmented compared with age-matched control patients (Tarkowski et al, 1995a). In addition, the severity of the neuronal damage after stroke alters the DTH response in a bimodal fashion; mild damage results in a suppressed DTH response, whereas more severe damage leads to an augmented DTH response (Tarkowski et al, 1995b). The effects of focal cerebral ischemia on the DTH response are correlated with a subcortical lesion (Tarkowski et al, 1995a), and global cerebral ischemia also causes a subcortical lesion (Bottiger et al, 1999; Kofler et al, 2004; Neigh et al, 2004b; Sadowski et al, 1999). Damage to the hippocampus accounts for at least a portion of this subcortical lesion and is the primary site of negative feedback regulation for the hypothalamic—pituitary—adrenal (HPA) axis (Sapolsky, 1994). Alterations in immune function also are mediated by changes in the HPA axis (Dhabhar, 2003; Dhabhar and McEwen, 1999; Tournier et al, 2001), and the hippocampus plays a role in HPA axis modulation of immune function (Maier and Watkins, 2003). We hypothesized that cardiac arrest and cardiopulmonary resuscitation (CA/CPR)-induced hippocampal damage would alter HPA axis reactivity and, subsequently, immune function.
Given the increasing number of individuals surviving CA/CPR, an understanding of the alterations in HPA axis activity and the implications for immune function are of critical importance to best serve this growing patient population. With this in mind, we tested the hypothesis that CA/CPR causes HPA axis dysregulation and alters the DTH response to antigenic challenge. Furthermore, with the exception of the recent work that examined the effects of vaccination on cardiac arrest survival, little is currently known about the effects of CA/CPR on humoral immune function or the effects of an activated humoral immune system on outcome after CA/CPR. To begin to address this topic, we assessed the primary and secondary immune response of mice exposed to CA/CPR.
Materials and methods
Animals
Adult male C57BL/6 mice were housed individually in polycarbonate cages (28 × 17 × 12 cm3) in rooms maintained on a 14:10 light:dark cycle, lights illuminated at 0000 h Eastern Standard Time, (EST) at 20°C ± 4°C and relative humidity of 50% ± 5%. Tap water and food (LabDiet 5001; PMI Nutrition; Brentwood, MO, USA) were available ad libitum throughout the study.
Cardiac Arrest Procedure
Mice were anesthetized with 3% halothane in air, intubated, and maintained on 1.5% halothane. A temperature probe was placed in the temporalis muscle on the left side of the head. We have previously shown that cortical temperature and temporalis temperature are highly correlated over a range that encompasses the temperatures experienced during our CA/CPR and SHAM procedures (Neigh et al, 2004b). Thus, temporalis muscle temperature is used as an index of brain temperature. A rectal temperature probe coupled to a heating blanket (Harvard Apparatus, Holliston, MA, USA) was used to maintain body temperature. A PE10 catheter was inserted into the right jugular vein for drug administration. Continuous monitoring of arterial blood pressure was achieved via a blood pressure transducer (Columbus Instruments, Columbus, OH, USA) connected to a right femoral artery cannula (Fine Science, Foster City, CA, USA). Mice were ventilated with a tidal volume of 120μL and a respiratory rate of 160 breaths per minute (Columbus Instruments, Columbus, OH, USA). Mice were allowed to stabilize for 10 mins during which time blood pressure and temperatures were recorded at 1-min intervals. At the conclusion of the acclimation period, body temperature was decreased to 27°C by circulating cold water through a coil system beneath the animal and placement of an alcohol patch on the ventrum. Induction of hypothermia by this method prevents peripheral organ damage in mice (Kofler et al, 2004; Neigh et al, 2004b). Head temperature was manipulated independently of body temperature through the use of a double lumen coil that was placed around the head and filled with circulating water to achieve a brain temperature of either 27°C or 37°C. Maintaining temporalis muscle temperature at 27°C prevents the neuronal damage normally incurred by 8 mins of CA/CPR; however, the animal is still exposed to potassium chloride (KCl), epinephrine (EPI), and the hemodynamics of CA/CPR (Neigh et al, 2004b). To induce cardiac arrest, cold KCl (50.0μL, 0.5 mol/L, 4°C) was injected via the jugular catheter and the mouse was detached from the ventilator. Slow rewarming via heating lamp and thermal blanket began when body temperature reached 27°C after approximately 4mins of arrest. At 7 mins 45secs into the arrest period, the mouse was reattached to the ventilator and ventilated on 100% oxygen with a tidal volume of 120μL and a respiratory rate of 160 breaths per minute. At 8mins after injection of KCl, 8μg of EPI in 0.5 cc saline, warmed to 37°C, was injected via the jugular vein catheter and chest compressions (approximately 300/min) were initiated. Additional EPI was administered in increments of 0.5μg every 30secs in conjunction with continued chest compressions until mice were resuscitated or until 16μg EPI was administered. The total amount of EPI administered and the total duration of CA/CPR time from KCl injection to the first minute of spontaneous mean blood pressure above 60mm Hg were recorded. Mice were maintained on 100% oxygen for 15mins after initiation of spontaneous circulation and then extubated, followed by the removal of catheters and suturing of wounds. An injection of 0.75 cc of prewarmed (37°C) lactated ringers was administered subcutaneously immediately after the conclusion of the procedure. Mice were placed in a clean cage on a thermal barrier for an additional hour before return to the colony.
The surgical preparations, anesthetic exposure, and temperature modulation described above were similar for CA/CPR and SHAM-operated mice, except that SHAM-operated mice received a 50.0μL injection of isotonic saline instead of KCl and a 0.5cc injection of isotonic saline instead of EPI. The SHAM-operated mice did not experience CA/CPR and were not exposed to anoxia, EPI, or chest compressions. At the conclusion of the surgical procedure, a 100μL blood sample was collected from the femoral artery catheter for the CA-normo and SHAM-normo mice. The samples were immediately analyzed using an i-stat blood gas/chemistry analyzer (Heska, Fort Collins, CO, USA) for the concentrations of Na+, K+, Cl−, hematocrit, and hemoglobin. In addition, naïve mice from the same colony conditions were tested at a similar time of day (n = 5).
Experiment 1. Assessment of Effects of Cardiac Arrest and Cardiopulmonary Resuscitation on Delayed Type Hypersensitivity
To assess DTH, mice were randomly assigned to one of five experimental groups: (1) SHAM surgery with head temperature lowered to 27°C (SHAM-hypo; n = 7), (2) SHAM surgery with head temperature maintained at 37°C (SHAM-normo; n = 8), (3) CA/CPR with head temperature lowered to 27°C (CA-hypo; n = 6), (4) CA/CPR with head temperature maintained at 37°C (CA-normo; n 8), and (5) nonsurgical controls (n = 7).
Induction of delayed type hypersensitivity: Delayed type hypersensitivity was induced by application of the antigen, 2,4-dinitro-1-fluorobenzene (DNFB; Sigma, St Louis, MO, USA), to the pinnae of mice after initial immunization to the dorsum. On day 1, all mice were lightly anesthetized at 0800 h EST with isoflurane, and an area of approximately 2 × 3cm2 was shaved on the dorsum. The shaved skin was swabbed with 70% alcohol, and 25μL of DNFB (0.5% (wt/vol) in 4:1, acetone:olive oil vehicle) was applied via pipette on day 1, and again on day 2. The thickness of both pinnae was measured on day 1 before sensitization using a constant-loading dial micrometer (Long Island Indicator Service, Hauppauge, NY, USA). Twenty-one days after sensitization, baseline pinnae thickness was again measured. Next, 20μL of DNFB (0.2% (wt/vol) in 4:1, acetone:olive oil) was applied to the skin of the dorsal surface of the right pinna at 0800 h EST. Left pinna were treated with vehicle and animals were returned to their home cages. Pinnae thickness was measured every 24 h for the next 8 days at 1100 h EST, and all measurements were made on the same relative region of the pinna.
Determination of serum corticosterone concentration: A blood sample for the determination of basal corticosterone concentrations was obtained under light isoflurane anesthesia (< 2 mins) from the retro-orbital sinus between 15 and 20 days after either SHAM surgery or CA/CPR. Three days later, mice were placed in adequately ventilated clear polypropylene restrainers (50mL conical tubes measuring 9.7 cm in length and an internal diameter of 2.8 cm) for 1 h; the tubes were of sufficient size to allow postural adjustments, but restrict head to tail turns. Immediately on the termination of restraint, a blood sample was collected under light isoflurane anesthesia (< 2 mins) from the retro-orbital sinus. The samples were centrifuged for 30 mins at 1250g. Serum was collected and stored at —80°C. Serum corticosterone was assessed by radioimmunoassay (RIA; ICN, Costa Mesa, CA, USA). The RIA is highly specific, crossreacting at less than 1% with other hormones.
Immunohistochemistry: A separate cohort of animals was divided into the treatment groups described earlier. Brains were collected for analysis of corticotrophin-releasing hormone (CRH) protein expression in the paraventricular nucleus (PVN) of the hypothalamus 7 days after either SHAM or CA/CPR. Mice were injected with an overdose of sodium pentobarbital and transcardially perfused with 0.9% saline (4°C) followed by 4% paraformaldehyde (4°C). Brains were removed, placed in 4% paraformaldehyde for 24 h at 4°C, and then transferred into 30% sucrose in phosphate-buffered saline (PBS) solution for 24 h at 4°C. Brains were frozen on dry ice and stored at −80°C until processing. Brains were sectioned into 30mm sections on a cryostat and thaw mounted onto slides. Slides were rinsed in PBS (pH = 7.2) and then placed in anti-CRH antibody (diluted 1:2,000 in PBS; courtesy of Dr W Vale) for 72 h at 4°C on a rotator. Slides were again rinsed in PBS, and then placed in cy3 secondary antibody (1:2,000 in PBS; Jackson Immuno-Research, West Grove, PA, USA) for 2 h at room temperature on a rotator. Slides were again rinsed in PBS, and then coverslipped with immunomount (Thermo Shandon, Pittsburgh, PA, USA). Corticotrophin-releasing hormone expression was semi-quantitatively assessed with Inquiry Autoradiography software (Loates Associates, Westminster, MD, USA). Median OD was determined from three to four sections from each brain region and the median value for each group was calculated.
Experiment 2: Assessment of Cardiac Arrest and Cardiopulmonary Resuscitation on Humoral Immune Function
To assess humoral immunity, mice were injected subcutaneously with the antigen, keyhole limpet hemocyanin (KLH), to which they were previously naïve. Keyhole limpet hemocyanin evokes an acute immune response, but does not replicate or cause long-term fever or inflammation (Dixon et al, 1966). Both the primary and secondary antibody responses were assessed after CA/CPR. At the beginning of the experiment, mice were randomly assigned to one of ten experimental groups. The first five groups were first exposed to KLH after CA/CPR to assess the primary humoral immune response: (1) SHAM surgery with head temperature lowered to 27°C (SHAM-hypo; n = 5), (2) SHAM surgery with head temperature maintained at 37°C (SHAM-normo; n = 6), (3) CA/CPR with head temperature lowered to 27°C (CA-hypo; n =8), (4) CA/CPR with head temperature maintained at 37°C (CA-normo; n = 7), and (5) nonsurgical controls (n = 6). The second set of five groups were first exposed to KLH before the CA/CPR procedure and then exposed a second time after the procedure to assess the secondary humoral immune response: (6) SHAM surgery with head temperature lowered to 27°C (SHAM-hypo; n = 4), (7) SHAM surgery with head temperature maintained at 37°C (SHAM-normo; n = 6), (8) CA/CPR with head temperature lowered to 27°C (CA-hypo; n = 5), (9) CA/CPR with head temperature maintained at 37°C (CA-normo; n = 10), and (10) nonsurgical controls (n = 6).
Determination of anti-KLH IgG: To assess the primary antibody response to KLH, mice were injected subcutaneously with the antigen, KLH (150μg suspended in 0.1mL sterile 0.9%. saline), to which they were previously naïve, 10 days after undergoing the CA/CPR or SHAM surgery. Serum samples were obtained 4, 7, 14, 21, and 28 days after immunization. For the assessment of the secondary immune response to KLH, mice were first exposed to KLH (150mg suspended in 0.1mL sterile 0.9% saline) 16 days before the CA/CPR procedure. Serum samples were obtained 4, 7, and 14 days after the primary exposure. Mice were then given a secondary exposure 10 days after the CA/CPR or SHAM surgery (30mg suspended in 0.1mL sterile 0.9% saline) and serum samples were collected on days 4, 7, 14, 21, and 28 days after immunization.
Diluted serum samples (1:50) were added to antigen-coated plates that had been treated with 0.5% milk in PBS to reduce nonspecific binding. After incubation, secondary antibody (AP-conjugated anti-mouse IgG, 1:1,000; MP Biomedicals, Irvine, CA, USA) was added, and after further incubation, the enzyme substrate p-nitrophenyl phosphate was added. The absorbance of each well was determined using a BioRad plate reader (405 nm; Bio-Rad, Benchmark, Hercules, CA, USA). In addition, the samples were diluted six-fold and the titer was determined by graphing the average absorbance for each dilution and determining the asymptotic point. The titer was the inverse of this point. Titers were then log transformed for further analysis.
Statistical Analysis
Blood pressure, temporalis muscle temperature, body temperature, pinnae thickness, serum corticosterone concentrations, and anti-KLH IgG concentrations were compared using two-way repeated measures ANOVAs assessing the effects of time and group. Total surgery time, total time in cardiac arrest, amount of EPI administered, and blood chemistry end points were assessed using one-way ANOVAs. Nonparametric statistics were used for the statistical analysis of the semiquantitative measure of CRH protein in the PVN. When statistically appropriate (P>0.05; Keppel, 1991), SHAM-hypo, SHAM-normo, and nonsurgical values were collapsed into one group for further analyses and this group was designated as ‘control.’ For the mice exposed to CA/CPR with a normothermic head temperature, a Kaplan—Meier survival analysis was conducted between the mice that received the primary dose of KLH before the CA/CPR procedure and those that received the primary dose after the CA/CPR procedure. Post hoc analysis was used to further distinguish among groups, and all differences were considered statistically significant if P < 0.05.
Results
Surgical End Points
The effects of CA/CPR on blood pressure were consistent with previous results (Neigh et al, 2004a, b). Briefly, cardiac arrest reduced blood pressure in both the CA-hypo and CA-normo groups as compared with controls (P < 0.01), and resuscitation restored blood pressure. Temperature manipulations of the head resulted in a temporalis muscle temperature that differed between groups in the normo conditions (head maintained at 37°C) and those in the hypo conditions (head cooled to 27°C; F3,27 = 496.5, P < 0.05), as intended. Temperature manipulations of the body resulted in a decline in the rectal temperature for all groups (F18,486 = 874.2, P < 0.05).
Mean surgical time was 68.2 × 7.7 mins and did not vary as a function of group. The time in cardiopulmonary arrest was 8 mins 56 × 23.1 secs (n = 14). The mean of EPI administered was 8.0 × 0.6 μg (n = 14). The cohort of mice used for analysis of CRH expression in the PVN and those used for the assessment of humoral immune function exhibited similar surgical parameters (data not shown).
Concentrations of Na+ and Cl− in the blood were significantly elevated after both the SHAM and CA/CPR procedures relative to nonsurgical controls; the CA/CPR values also were significantly greater than the SHAM values (F2,20 = 17.4, P < 0.05; F2,20 = 28.9, P < 0.05, respectively; Table 1). K+ declines to a similar extent in both SHAM-operated and CA/CPR mice relative to nonsurgical controls (F2,20 = 8.0, P < 0.05). Hematocrit and hemoglobin measures were significantly lower in CA/CPR animals than the nonsurgical and SHAM groups (F2,20 = 12.1, P < 0.05; F2,20 = 12.3, P < 0.05, respectively).
Both SHAM surgery and CA/CPR alter blood chemistry

Values = mean ± s.d.
Values in the same row with the different superscript letters are significantly different at P< 0.05.
Delayed Type Hypersensitivity Response
Analysis of the DTH response to challenge revealed a main effect of group (F4,31 = 7.5, P < 0.05), a main effect of time (F8,248 = 5.9, P < 0.05), and an interaction between group and time (F32,248 = 2.15, P < 0.05). Neuronal alterations after CA/CPR augmented the DTH response to pinnae challenge to a subclinical-threshold dose of DNFB (P < 0.05; Figure 1). Mice in the CA-normo group showed enhanced edema as compared with mice in the CA-hypo group, which were afforded neuronal protection (Neigh et al, 2004b). Neither SHAM surgery (in either temperature condition) nor CA-hypo elicited a DTH response to the subthreshold dose of DNFB, as compared with nonsurgical control mice (P>0.05).
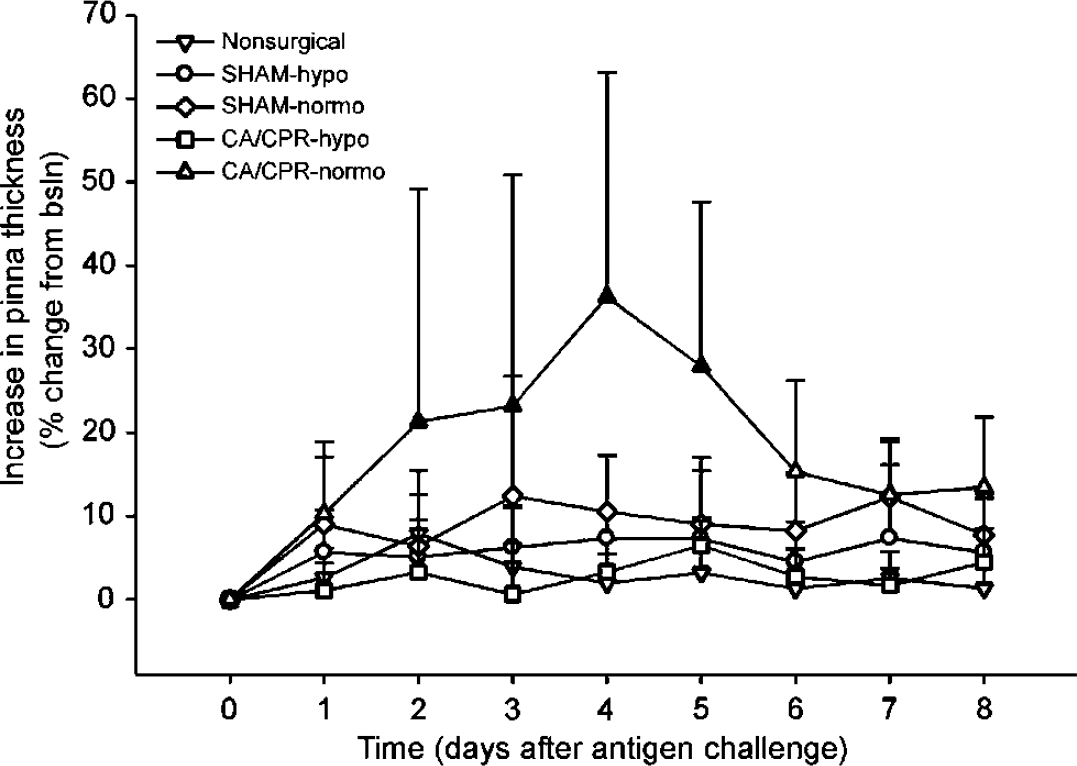
Exposure to 8mins of cardiac arrest followed by reperfusion, while the head was maintained at 37°C (CA-normo), resulted in an enhanced response to the antigen DNFB. This augmentation of the delayed type hypersensitivity (DTH) response was specific to mice with neuronal damage, such that mice with a hypothermic head during the cardiac arrest and cardiopulmonary resuscitation (CA/CPR) (CA-hypo) do not show alterations in the DTH response. In addition, the SHAM surgery with either head temperature condition did not alter the DTH response. Filled in symbols indicate a difference from the CA-hypo group (P < 0.05). The data are presented as mean×s.d.
Serum Corticosterone
Corticosterone concentrations varied as a function of group (F4,25 = 15.0, P < 0.05), time (F1,25 = 470.4, P < 0.05), and the interaction between group and time of sample (F4,25 = 18.8, P < 0.05). Basal corticosterone concentrations were elevated in mice in the CA-normo group as compared with control mice (P < 0.05; Figure 2). Exposure to 1 h of restraint resulted in increased corticosterone concentrations in all groups; however, the mice in the CA-normo group exhibited a blunted increase as compared with mice in the CA-hypo group (P < 0.05). This difference was further illustrated by a robust attenuation of the percent increase in corticosterone concentration elicited by exposure to restraint in the CA-normo group as compared with change exhibited by the control mice (F2,27 = 3.7, P < 0.05).
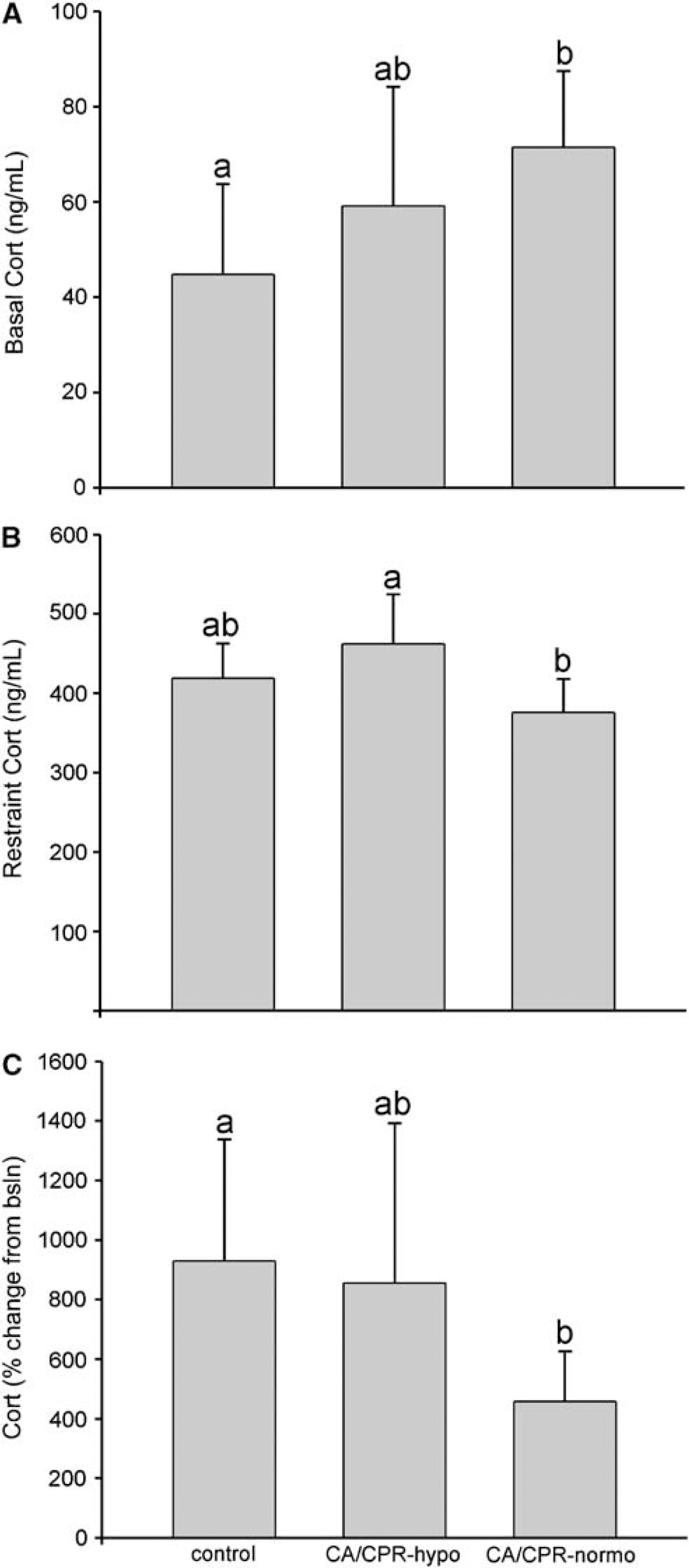
(
Corticotropin Releasing Hormone
Semiquantitative analysis of CRH expression in the PVN of the hypothalamus did not reveal an effect of either CA/CPR condition as compared with control mice (H2 = 1.9, P>0.05; data not shown). Median values of OD for the groups were as follows: control, 0.503; CA-hypo, 0.513; CA-normo, 0.550.
Anti-KLH IgG
Exposure to CA/CPR did not alter the primary IgG response (F4,26 = 0.41; P>0.05); however, all groups show an increase in anti-KLH IgG concentrations over time (F4,104 = 38.3, P < 0.05; Figure 3A). Likewise, there were no differences among the primary immune responses of those mice later exposed to the secondary dose of KLH (F3,21 = 0.52; P>0.05; Figure 3B), and the same increase in anti-KLH IgG across days was showed (F2,42 = 6.3; P < 0.05). There was no effect of CA/CPR on the secondary IgG response (F2,21 = 0.92; P>0.05), but there was an effect of time (F4,84 = 2.987; P < 0.05), such that there was an increase in the concentration of anti-KLH IgG across days. Post hoc assessment of the day 21 time point indicated that those mice exposed to CA/CPR while the head was maintained at 37°C had a lower concentration of anti-KLH IgG than the collapsed control group (t23 = 55; P < 0.05).
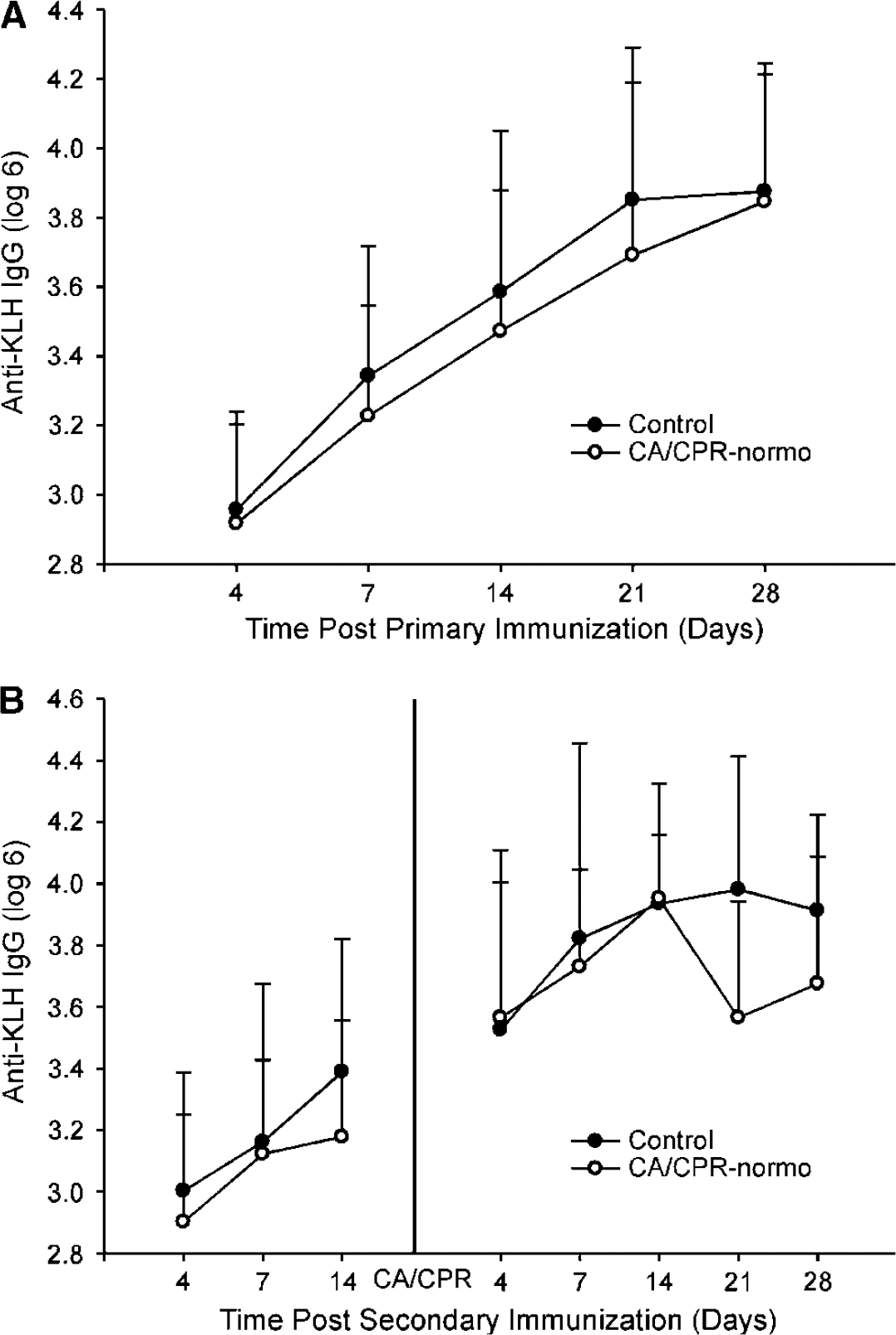
(
Kaplan—Meier Survival Analysis
Only 2 out of the 21 mice in the CA/CPR hypo groups died after the procedure (91% survival). Of those mice exposed to CA/CPR with a normothermic head temperature, 72% survived in both the DTH and primary response KLH experiments. In contrast, 48% of the mice exposed to the first dose of KLH before the CA/CPR-normothermic procedure survived. The Kaplan—Meier survival analysis of mice with and without previous exposure to KLH before CA/CPR with a normothermic head verified that KLH exposure significantly affected the survival curves of the mice (P < 0.05; Figure 4).
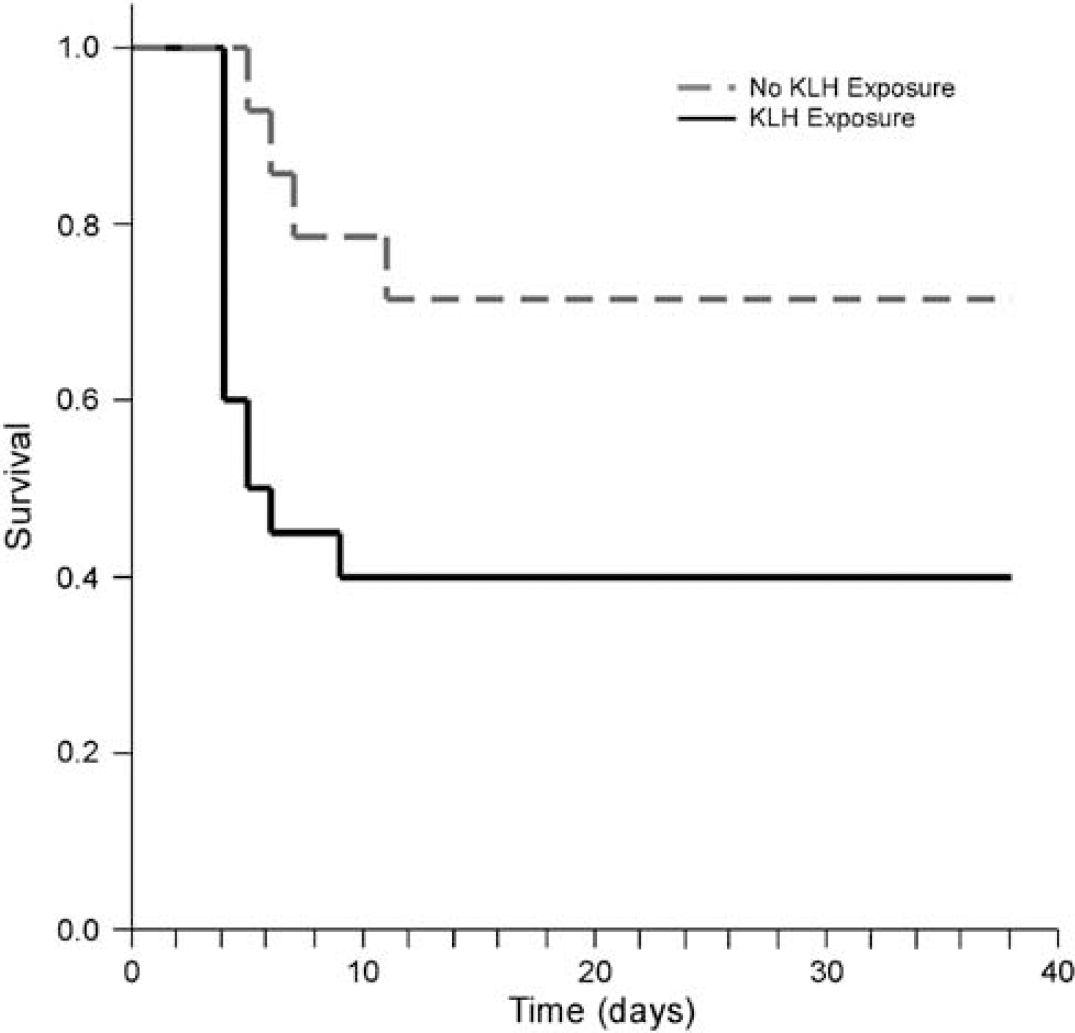
Previous exposure to keyhole limpet hemocyanin (KLH) decreases survival from cardiac arrest and cardiopulmonary resuscitation (CA/CPR) from 72% to 48%. This Kaplan—Meier survival curve shows this change in survival after previous KLH exposure.
Discussion
Cardiac arrest and cardiopulmonary resuscitation resulted in a dysregulation of the HPA axis such that basal corticosterone concentrations were elevated, but responsivity to restraint was blunted relative to SHAM-operated mice (Figure 2). This dysregulation was not attributable to peripheral organ damage or the effects of KCl and EPI administration, as evidenced by similar basal and stress-induced corticosteroid concentrations between mice with a hypothermic head during CA/CPR and SHAM-operated mice. Previously, we showed that maintenance of the head at hypothermic temperatures during CA/CPR prevents CA/CPR-induced neuronal damage (Neigh et al, 2004b). Thus, these data suggest that post-CA/CPR neuronal damage leads to HPA axis dysregulation. In humans, increases in norepinephrine concentrations in cerebrospinal fluid during cardiac arrest are dependent on increased cortisol release (Wortsman et al, 1987). In addition, open heart surgery, a procedure that requires circulatory arrest, causes phasic changes in corticosteroid-binding globulin and cortisol concentrations in humans (Tinnikov et al, 1996). Focal cerebral ischemia also has been linked to altered HPA axis activity in humans and rodents (DeVries et al, 2001; Slowik et al, 2002).
Because the hippocampus is an extensively damaged region of the brain in our model of cardiac arrest (Neigh et al, 2004a, b), and the hippocampus is an important source of negative feedback inhibition for the HPA axis (reviewed in Brown et al, 1999), we expected CA/CPR to produce increased CRH immunoreactivity in the PVN of the hypothalamus (see Herman et al, 1989). However, there was no significant difference in CRH expression between experimental groups, which suggests that the elevated corticosteroid concentrations and immune alterations in CA/CPR mice were not mediated by increased CRH expression. However, CRH expression in the current study was assessed only under basal conditions, at one time point after surgery. The changes that mediate altered HPA axis reactivity and altered immune function could occur at the receptor level, may only be prevalent after a stressor (e.g. a change in synthesis or release), or may occur in an area of the brain other than the PVN of the hypothalamus (e.g. amydgala or hippocampus). Further studies are required to determine the exact mechanism through which CA/CPR alters HPA axis activity.
In addition to altering circulating corticosterone concentrations, CA/CPR enhanced the cutaneous DTH response to an antigen challenge (Figure 1). Elevated cortisol concentrations documented after focal ischemia correlate with inflammatory markers (Slowik et al, 2002), including elevated IL-6 concentrations (Johansson et al, 1997), which can increase the intensity of a DTH response (Jayaraman et al, 1990; Marcinkiewicz, 1990). In addition, survivors of CA/CPR show a ‘systemic inflammatory response syndrome’ that is characterized by increased production of cytokines and cytokine receptors (Adrie et al, 2002). Cardiopulmonary bypass, a procedure that reproduces some of the same hemodynamic properties as CA/CPR, also results in elevated cytokine and adhesion molecule expression in humans (Grunenfelder et al, 2000), and causes changes in T-cell levels that persist for at least several days after surgery (Tinnikov et al, 1996). Furthermore, in the absence of hypothermia, the mouse model of CA/CPR used in this study elicits alterations in T-cell activation in the kidney (Burne-Taney et al, 2003).
In the current study, there was no effect of CA/CPR on the primary or secondary IgG response to a novel antigen (Figures 3A and 3B, respectively), although IgG was significantly lower at one time point occurring 21 days after the secondary exposure (Figure 3B). These data suggest that mice exposed to CA/CPR initially mount the same secondary IgG response as mice exposed to no surgery, SHAM surgery, or CA/CPR with a hypothermic head; however, CA/CPR mice may have problems maintaining the response.
Assessment of survival after previous exposure to KLH suggests exposure to even an innocuous novel protein can alter survival after a cerebrovascular event. Survival was not affected in mice that were exposed to CA/CPR while the head was maintained at a hypothermic temperature, suggesting that the neuronal damage after the procedure is the key variable that interacts with antigen exposure to alter survival. These data appear to be in contrast to the recent reports that suggest that vaccination is protective against cardiac arrest (Gurfinkel et al, 2004; Lavallee et al, 2002; Nichol et al, 2003; Siscovick et al, 2000), but it is likely that the active immune response to vaccination had resolved before the ischemic episodes experienced in these studies. Likewise, if the mice in the current experiment had undergone the CA/CPR procedure after anti-KLH IgG concentrations had declined, there may not have been a change in survival after the ischemic event.
The interaction of the humoral immune system with outcome from CA/CPR, in combination with the demonstration of an enhanced response of mice exposed to CA/CPR to a subthreshold antigen challenge, may indicate a source of concern for CA/CPR survivors. Recent epidemiological studies show that influenza vaccination may reduce the risk of cerebrovascular events (Gurfinkel et al, 2004; Lavallee et al, 2002; Nichol et al, 2003; Siscovick et al, 2000), but does not reduce the risk of recurrent coronary events after myocardial infarction (Jackson et al, 2002). The effectiveness and possible risks of immunization after CA/CPR have not yet been fully investigated. Also of concern are the possible implications for autoimmune disorders after neuronal damage. Cardiac surgery results in an imbalance in T-helper cells such that there is a suppression of the Th1-induced cell-mediated immune response but Th2 responses remain intact (Markewitz et al, 1996). However, the current results suggest that, in the long term, a cerebrovascular event, such as CA/CPR, may lead to an increase in the Th1 response. Although the mechanism underlying this change is not yet clear, the current findings suggest that CA/CPR leads to a sensitization of the Th1 response. Given that disruption of regulatory T cells can lead to allergic disease and asthma (Umetsu et al, 2003) and that T-cell regulation plays a pivotal role in the development of autoimmune disorders (Bach, 2003), the changes in T-cell responsivity after CA/CPR-induced neuronal damage may have important implications for autoimmune disease after insult. To this end, survivors of traumatic brain injury, the classification of patients in which CA/CPR survivors had been placed until recently, report a higher incidence of autoimmune disorders, such as arthritis (Hibbard et al, 1998).
In conclusion, CA/CPR-induced neuronal damage results in a dysregulation of the HPA axis and a concomitant augmentation of the DTH response to antigenic challenge. Furthermore, immune activation before CA/CPR decreases survival from global ischemia. These data highlight the potential impact of neuronal damage on cell-mediated immune function, and the role of humoral immune activation in outcome after global ischemia.
Footnotes
Acknowledgements
We thank Stephanie L Bowers for technical support, Dr Wylie Vale for CRH antibody, and Tricia Uhor for animal care.