
Abstract
Cortical spreading depression (CSD) is thought to be a neuronal mechanism that expands the penumbra zone after focal brain ischemia and that causes migraine aura. Both adrenergic agonists and antagonists significantly influence the size of the penumbra zone and decline the frequency of migraine. To study whether these compounds act by influencing CSD, we applied different drugs topically to an area of the exposed cortex of anesthetized adult rats and observed the migration of CSD-related DC potential deflections across the treated area. The adrenergic agonist norepinephrine (1 mmol/L) and the α2-agonist clonidine (0.56 mmol/L) blocked reversibly the migration of CSD. The β-blocker propranolol (250 μmol/L to 1 mmol/L) dose-dependently diminished migration velocity or even blocked migration of CSD. The CSD blockade by the α2-antagonist yohimbine (1.75 mmol/L) was because of its action on inhibitory 5-HT1A receptors. None of the substances in the concentrations used had influence on regional cerebral blood flow or on systemic arterial blood pressure. The data suggest that the interference of these compounds with CSD may contribute to their beneficial therapeutic effect. The effect of β-receptor antagonists in human migraine needs further exploration, since these drugs also work in migraine without aura.
Introduction
α- and β-adrenergic agonists and antagonists are successfully used in the prophylaxis of human migraine, and they support neuronal recovery after focal ischemic stroke in animal experiments. For example, the administration of the α2-adrenoceptoragonist tizanidine has reduced headache intensity and duration in chronic headache patients (Lake and Saper, 2003; Saper et al, 2002). Other α2-adrenoceptor agonists were efficient in improving motor ability after stroke or in decreasing the infarct volume in animal experiments after occlusion of the middle cerebral artery (Jolkkonen et al, 1999; Kulinskii, 2000). The β-adrenoceptor antagonist propranolol is widely used in the prophylaxis of human migraine with and without aura (Diener et al, 2000). The beneficial effect of propranolol, that is, reduction of migraine frequency, was observed by chance more than 30 years ago in patients who received β-adrenoceptor antagonists for prophylaxis of angina pectoris (Rabkin et al, 1966). Up to now no experimental data have shown how propranolol decreases migraine frequency (Limmroth and Michel, 2001; Gobel and Heinze, 2003; Diener et al, 2003).
In the pathophysiology of both migraine and the development of the penumbra zone after stroke, a neuronal mechanism, namely the cortical spreading depression (CSD), is thought to play an important role. It is possible, therefore, that the beneficial effects of the β-adrenoceptor antagonists and α2-adrenoceptor agonists are because of a suppression of CSD. Cortical spreading depression is a slowly moving depolarization wave that can be elicited in neuronal tissue, for example, in the cortical gray matter (Leão, 1944). Cortical spreading depression waves can be elicited in animal experiments by a local depolarization, for example, by pricking or the application of KCl (Bureš et al, 1974) and they may develop spontaneously in human brain in vivo after severe head injury (Strong et al, 2002). Cortical spreading depression is thought to be a putative neuronal mechanism underlying migraine aura (Lauritzen, 1994). Whether CSD itself is initiating the pain phase of migraine is controversially disputed (Ebersberger et al, 2001; Bolay et al, 2002; Spierings, 2004).
Cortical spreading depression waves are characterized by transient negative DC potential shifts accompanied by rises in extracellular potassium concentration ([K+]e) to 60 to 70 mmol/L, whereas sodium, calcium, and chloride ions move into neurons and glial cells together with water causing a temporary swelling (Bureš et al, 1974; Kraig and Nicholson, 1978; Phillips and Nicholson, 1979; Somjen, 2001). Usually these transients recover within 90 to 120 secs in adult animals. A CSD wave propagates across the cortical surface at a velocity of 2 to 5 mm/min. The spread or spillover of excitatory amino acids (e.g., glutamate) into the extracellular space after release is thought to be a possible mechanism for CSD propagation. Other proposed mechanisms of CSD propagation are the elevation in [K+]e, and neuronal and/or glial communication processes via gap junctions (Somjen, 2001).
After a severe disturbance of cerebral blood supply with declined oxygen delivery to the brain, for example, ischemic stroke, an ischemic core zone develops that is surrounded by an area of constrained blood supply in which the energy metabolism is preserved (Hossmann, 1994). In this penumbra zone, spreading depression-like depolarizations occur spontaneously, triggered probably by the efflux of glutamate and potassium from injured cells (Hossmann, 1996; Back, 1998; Selman et al, 2004). The ionic pumping functions during CSD increase the energy consumption in this area, therefore leading to energy deterioration in the penumbra zone and finally to cell death (Back, 1998; Selman et al, 2004), whereas in normal brain tissue, CSD is of little consequence for neuronal metabolism and does not lead to metabolic deficiency (Nedergaard and Hansen, 1988).
α- and β-adrenergic agonists and antagonists may influence CSD. Wiedemann et al (1996) showed a dose-dependent inhibitory effect of propranolol on the propagation of retinal spreading depression (SD) and a small reduction of the SD amplitudes after the α2-adrenoceptor agonist clonidine. Because the retina has no vasculature, the authors supposed that the inhibition of SD must be of neuronal origin. The particular mechanism, through which propranolol and the other substances exert their inhibitory effects, is unknown. In the present work, we tested whether sympathetic agonists or antagonists such as norepinephrine (NE) and propranolol may have an influence on spreading depression in the cortical gray matter (CSD) when they are applied topically to the cortical surface.
Materials and methods
The experiments were approved by the animal protection committee and the regional government of Thueringen (reg. 02-09/01). Adult male Wistar rats (350 to 450 g) were used for the experiments. The rats were deeply anesthetized with sodium thiopental (Trapanal, Byk Gulden, Konstanz, Germany; initially 100 mg/kg intraperitoneally). During dissection animals were not paralyzed, and depth of anesthesia was checked by testing for nociceptive reflexes to squeezing the tip of the tail and by testing the corneal blink reflex. During the experiments, absence of the corneal blink reflex was maintained by supplemental doses of 20 mg/kg intraperitoneally. The trachea, the left femoral vein, and the artery were cannulated. The mean arterial blood pressure was continuously monitored. Body temperature was kept at 37°C by a feedback controlled heating system.
The head of the animal was fixed stereotactically and the skin above the skull was removed. A rectangular trephination was made over the left hemisphere (from midline spanning 4 to 6 mm laterally, 2 to 3 mm wide) using a mini-drill under saline cooling and the underlying dura was cut and removed. For DC recording and CSD elicitation, two further openings (diameter 1.5 to 2 mm each) were made at 2 to 3 mm anterior and posterior from this opening, and 2 and 4 mm lateral from the midline, respectively, on the left hemisphere. Underneath these openings, the dura mater was incised. The exposed cortex in the large trephination was sealed against the skull bone and dura mater edges with vacuum fat. Dental acrylic was used to build a wall on the skull around the exposed cortex surface forming a trough to prevent accidental overflow of the solutions (see also Figure 1).
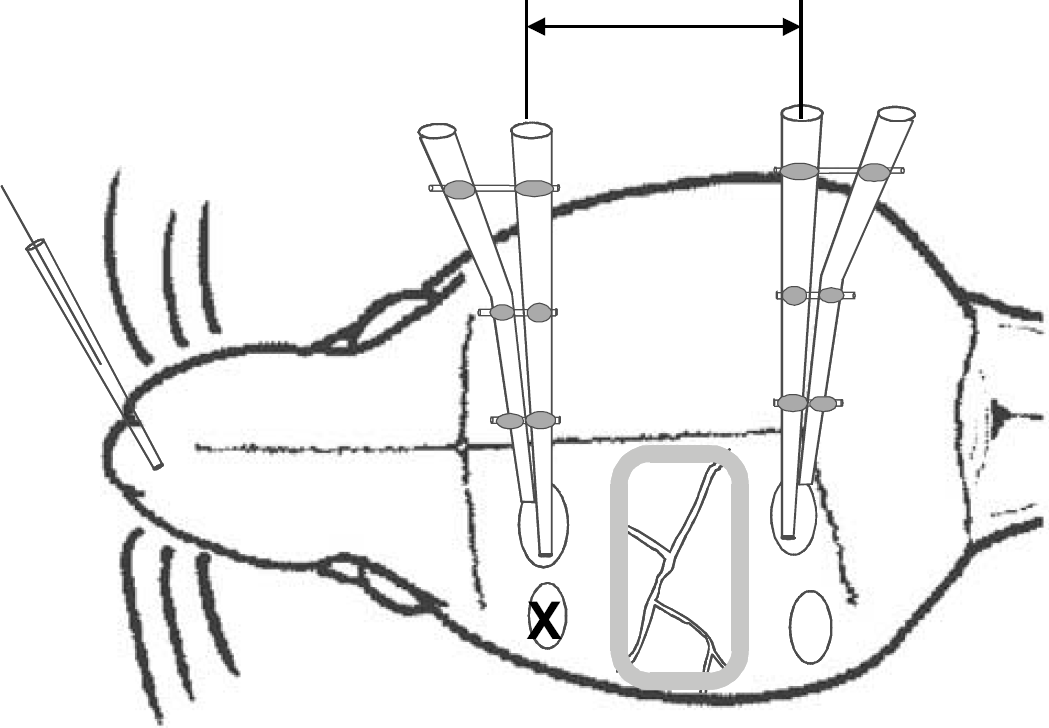
Schematic drawing (not to scale) of the rat skull with the trephinations and the exposed cortical surface surrounded by a wall made from dental acrylic (gray shaded border). The arrays consisting of two glass microelectrodes each are inserted into the gray matter, the reference electrode is located at the nasal bone. The site where pricks were set to elicit cortical spreading depression (CSD) or KCl was applied is marked by ‘X'. The arrow at the top side labels the distance between the frontal and caudal electrodes (9 to 11 mm). The migration period was measured between the CSD peak at the frontal and the peak at the caudal electrode.
An Ag/AgCl reference electrode containing 2 mol/L KCl was placed on the nasal bone. Intracortical DC potentials were recorded using glass micropipettes (tip diameter approximately 5 μm, resistance <10 MΩ) filled with 150 mmol/L NaCl. A pair of pipettes was glued together with a vertical tip separation of 400 μm. The electrode pairs were introduced into the cortex through the small openings in front of and behind the exposed cortex area to a depth of 1200 μm for the deepest electrode. The signals were recorded using a four-channel high-impedance amplifier (Meyer, Munich, Germany) and stored on PC. According to widely used experimental procedures (Somjen, 2001), single CSD was elicited by a pinprick into the cortex with a needle (diameter 0.5 mm) at the frontal opening (see Figure 1). To avoid failures in CSD elicitation on repetition, efforts were made to vary the site of the pricks within the stimulation area. In part of the experiments a small (approximately 0.05 mg weight) KCl crystal was applied to the surface of the untreated cortex to evoke self-regenerating repetitive CSD.
An initial test CSD was elicited by prick to confirm a positive response that was compatible in amplitude and propagation with data found in previous experiments (Richter and Lehmenkühler, 1993). When the negative DC deflection migrated over the whole recording area, the control was accepted. Additionally, we accepted only those waves as CSD that had a steep onset and exceeded amplitudes of ≥5 mV. Thereafter, the test substance was applied for 15 to 20 mins, and then a prick was set into the gray matter to elicit another CSD. When the prick did not elicit a CSD in the untreated cortex, pricking was repeated after a waiting period of 5 mins. If the CSD still propagated through the treated area, the application of the substance was repeated for 15 to 20 mins and another trial to elicit CSD followed. If the CSD did not propagate to the caudal electrodes or if no CSD was elicited, the cortex was thoroughly washed with Tyrode and a waiting period of at least 30 mins was allowed for recovery. Then a prick was set and if a CSD occurred again the application of the compound was repeated (with the same concentration) to confirm the effect of the substance. At the end of an experiment, the animals were killed by an overdose of the anesthetic.
During the control period (cortex superfused with Tyrode), we measured CSD amplitudes and propagation times between the frontal and caudal electrodes to check the parameters of the CSD. To evaluate the effects of substances tested, the number of pricks resulting in propagating CSD was counted for each application. To evaluate effects of compounds on KCl-elicited CSD, the number of repetitive migrating CSDs during the control period of 20 mins was counted and subtracted from the number of migrating repetitive CSD after application of the substances. This standardization allowed a comparison between animals with different repetition rates during controls.
The following substances were applied topically to the cortical surface: the α-adrenoceptor agonist NE (L-norepinephrine-HCl, SIGMA, Germany; 10−-4 or 10−-3 mol/L, dissolved in 10% DMSO in Tyrode; Ferraro et al, 1994) was tested in 6 rats. The α2-adrenoceptor antagonist yohimbine (SIGMA, Germany; 1.75 mmol/L, dissolved in Tyrode; Kovács and Hernádi, 2003) was tested in 11 rats. To test whether yohimbine inhibited the CSD via an action on the inhibitory 5-HT1A receptor, we applied in a series of 6 rats first the 5-HT1A receptor antagonist NAN-190 (SIGMA, Germany, 0.1 mmol/L) to the brain surface alone and then together with yohimbine (1.75 mmol/L) topically to the brain surface. Each substance was applied for 20 mins, then a prick was set. In 3 of these experiments, a small KCl crystal (weighing approximately 0.05 mg) was placed onto the untreated cortex 15 mins after the pricks. The α2-adrenoceptor agonist clonidine (SIGMA, Germany; 0.56 mmol/L, dissolved in 165 mmol/L NaCl; Reichl and Walland, 1980) was tested in 7 rats. In an additional group of 6 rats, we applied the subtype specific α2D-adrenoceptor antagonist BRL 44408 (Tocris, Biotrend GmbH, Germany, 1 mmol/L) to the brain surface alone and elicited CSDs, and afterwards we administered BRL 44408 and clonidine (0.56 mmol/L) at the brain surface and repeated CSD elicitation. In four of these rats, we tested not only prick but also KCl-elicited CSD. In another six rats, the β-adrenoceptor antagonist propranolol (SIGMA, Germany, 250 μmol/L, 330 μmol/L, 0.5 mmol/L, and 1 mmol/L, dissolved in Tyrode; Wiedemann et al, 1996) was administered to the cortical surface.
To test whether local hemodynamic responses that might have caused the inhibition of CSD were caused by the adrenergic agonists or antagonists, in four additional rats the same surgical preparation and trephination was made but no electrodes were inserted. The mean arterial blood pressure was measured via the catheter in the left femoral artery (Blood Pressure Monitor BP-1 with transducer, WPI Instruments, Sarasota, FL, USA). The cortical regional perfusion was measured continuously with a flow meter (Type Laser Blood Flow Monitor MBF3D, Moor Instruments, Devone, Exminster, UK) with sensors of a diameter of approximately 2 mm at two different points of the gray matter. One was located in the area where the substances were applied, and the other one was located at the frontal cortical site where usually the CSD was elicited by pricking and where no compounds were applied. Intervals of 5 mins were evaluated before and during drug application. Changes in regional cortical perfusion during application were calculated as percentage of predrug values. After measuring a baseline of 20 mins, the substances were applied for 15 mins to the cortical surface in the order NE, yohimbine, and clonidine. After 15 mins, the drug was removed and the cortex was washed thoroughly with Tyrode. The next substance was applied 30 mins after. As a positive control, the NO donator sodium nitroprusside (Schwarz Pharma, Germany, 1.5 mg/kg) was applied topically to the brain surface. At the end of the experiment, the rat was killed by an overdose of the anesthetic.
Results
Cortical Spreading Depression Parameters in the Untreated Rat Cerebral Cortex
In the untreated cortex, a prick usually elicited a single CSD that moved slowly across the hemisphere and was picked-up from all recording electrodes. No differences were found regarding CSD time course and shape between the CSDs recorded from the frontal and caudal electrodes. The amplitudes of CSDs at these sites were 17.6+5.1 and 15.1+6.5 mV and the durations were 145.3+62.6 and 154.8+59.8 secs (mean+s.d., n = 21), respectively. The CSDs propagated with a velocity of 4.6+1.4 mm/min (mean+s.d., n = 21) from the frontal to the caudal electrode. Under baseline conditions, KCl induced 4 to 8 CSDs following each other at intervals of 4 to 10 mins.
Application of α-Adrenergic Agonists or Antagonists
The effect of NE at concentrations of 10−-4 mol/L or 10−-3 mol/L was tested in six rats. In five of six rats CSD were influenced: In three rats, we found an inhibition of CSD migration 15 to 30 mins after NE application. In these cases, a CSD could be elicited and recorded in the untreated frontal cortical area but the CSD did not propagate through the treated area to the caudal electrode. Cortical spreading depression recurred after a washout period of 15 or 60 mins in two of these three rats. In the other two rats, CSD initiation and migration was inhibited. Neither under the frontal nor under the caudal electrode could a CSD be evoked after NE application. This effect was transient and a washout period of 15 to 60 mins reconstituted CSD (Figure 2A). Different concentrations of NE were necessary to achieve these results: In two rats the application of 10−-4 mol/L NE was sufficient to inhibit CSD migration (one rat) or CSD initiation (one rat). In the other three rats, a concentration of 10−-3 mol/L NE was needed (Figure 2B).
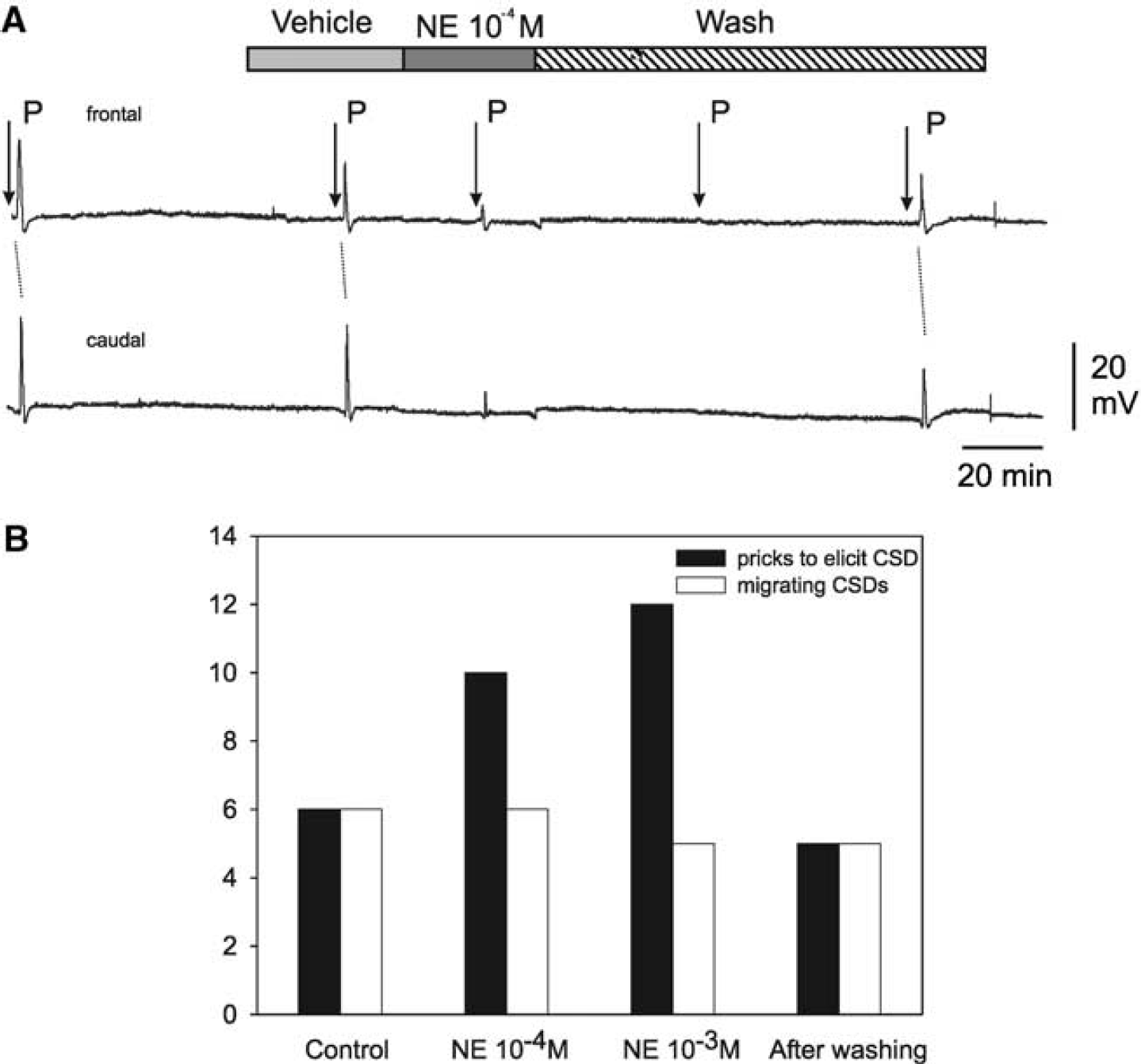
Effects of norepinephrine (NE) on cortical spreading depression (CSD). (
The α2-adrenergic antagonist yohimbine inhibited CSD (Figure 3B). In seven out of 11 rats, yohimbine inhibited CSD initiation, in three rats yohimbine had no effects on CSD. Cortical spreading depression were restored after 15 to 45 mins washout in four rats. In the other three rats, CSD were not restored even after 90 mins of washout. In one rat only CSD migration was blocked reversibly and restored after a washout for 60 mins. As after NE, the effect of yohimbine occurred within 10 to 50 mins after application.
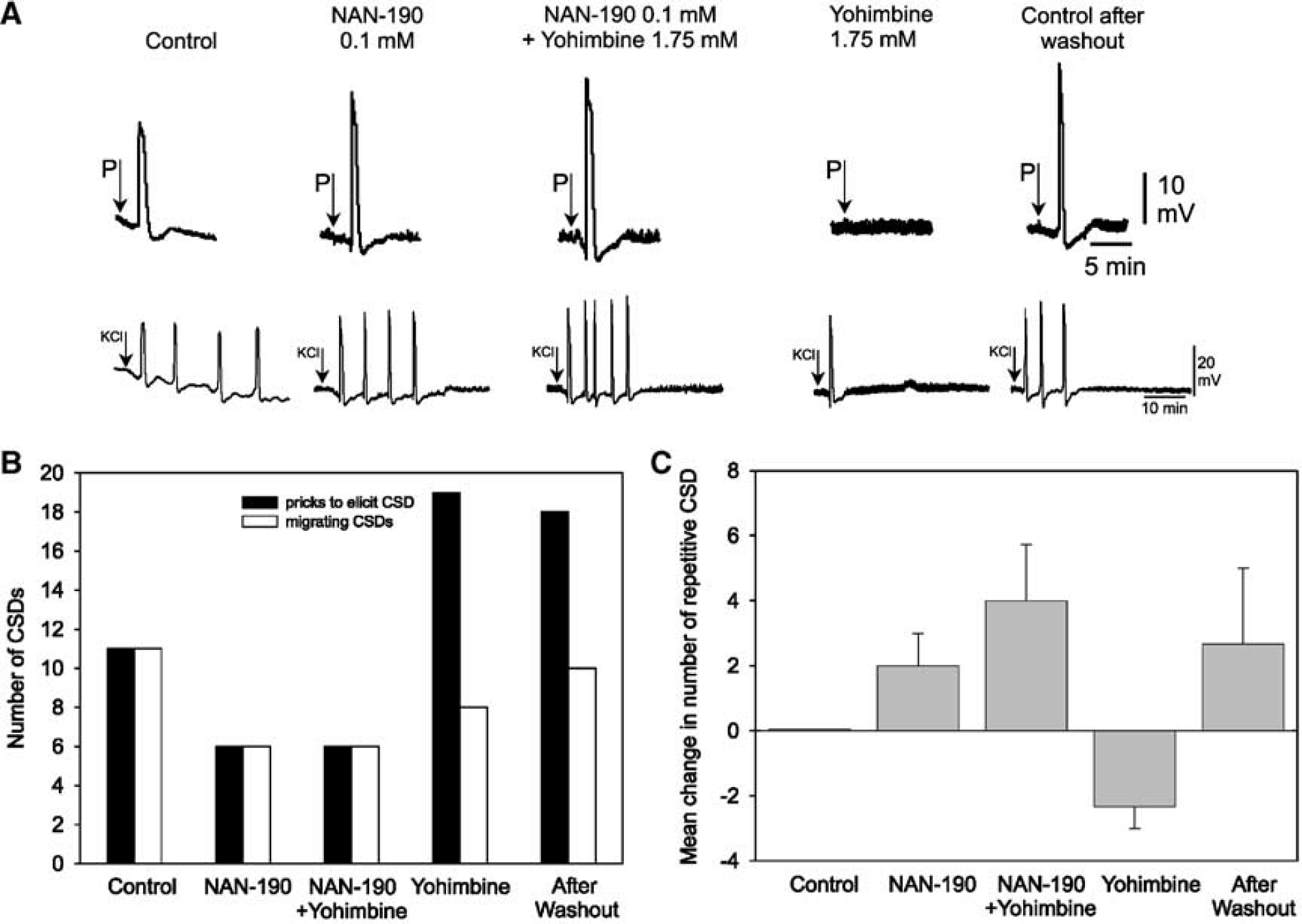
Effects of the 5-HT1A receptor antagonist NAN-190 and yohimbine on cortical spreading depression (CSD). (
Yohimbine is known to bind to α2-adrenoceptors as well as to 5-HT1A receptors. In another set of experiments we tested the influence of 5-HT1A-receptors on the yohimbine-induced inhibition of CSD (specimen see Figure 3A). In none of the animals, the application of NAN-190 alone to the cortical surface influenced prick-induced CSD (Figure 3B). When yohimbine was applied to the brain surface in the presence of the 5-HT1A-receptor antagonist NAN-190, its inhibitory action on prickinduced CSD was completely abolished (six of six rats). The effect of NAN-190 could be removed by washing with Tyrode for 45 to 60 mins, and then yohimbine again blocked CSD. The number of self-regenerating CSD after KCl was slightly increased by NAN-190 and by NAN-190 added to yohimbine. After washing away the mixture, yohimbine reversibly reduced the number of generating CSD (Figure 3C).
The role of α2-adrenoceptors was further tested by the application of the α2-adrenergic agonist clonidine (Figure 4). Clonidine produced a similar inhibition of prick-induced CSD as NE in six out of the seven rats. In three of the rats CSD migration was inhibited, in the other three rats CSD initiation was blocked. In a washout period of 45 to 120 mins, CSD was restored in 3 of the 6 rats (Figure 4B). In another set of experiments, it was tested whether the effect of clonidine depended on an action at the α2D-adrenoceptor subtype (specimen see Figure 4A). The application of the α2D-adrenoceptor antagonist BRL 44408 inhibited prick-induced CSD in two of the six rats tested (Figure 4B) and reduced the number of self-regenerating CSD after KCl in four of four rats tested (Figure 4C). When BRL 44408 was administered in the presence of clonidine, an inhibition of prick-induced CSD was still seen in two of the six rats. The number of self-regenerating CSD after KCl equally declined after BRL44408 plus clonidine and clonidine alone. These effects were only partially reversible by washing the brain with Tyrode for 45 to 60 mins. However, after that period in all rats attempts to elicit CSD were successful (Figure 4B and 4C).
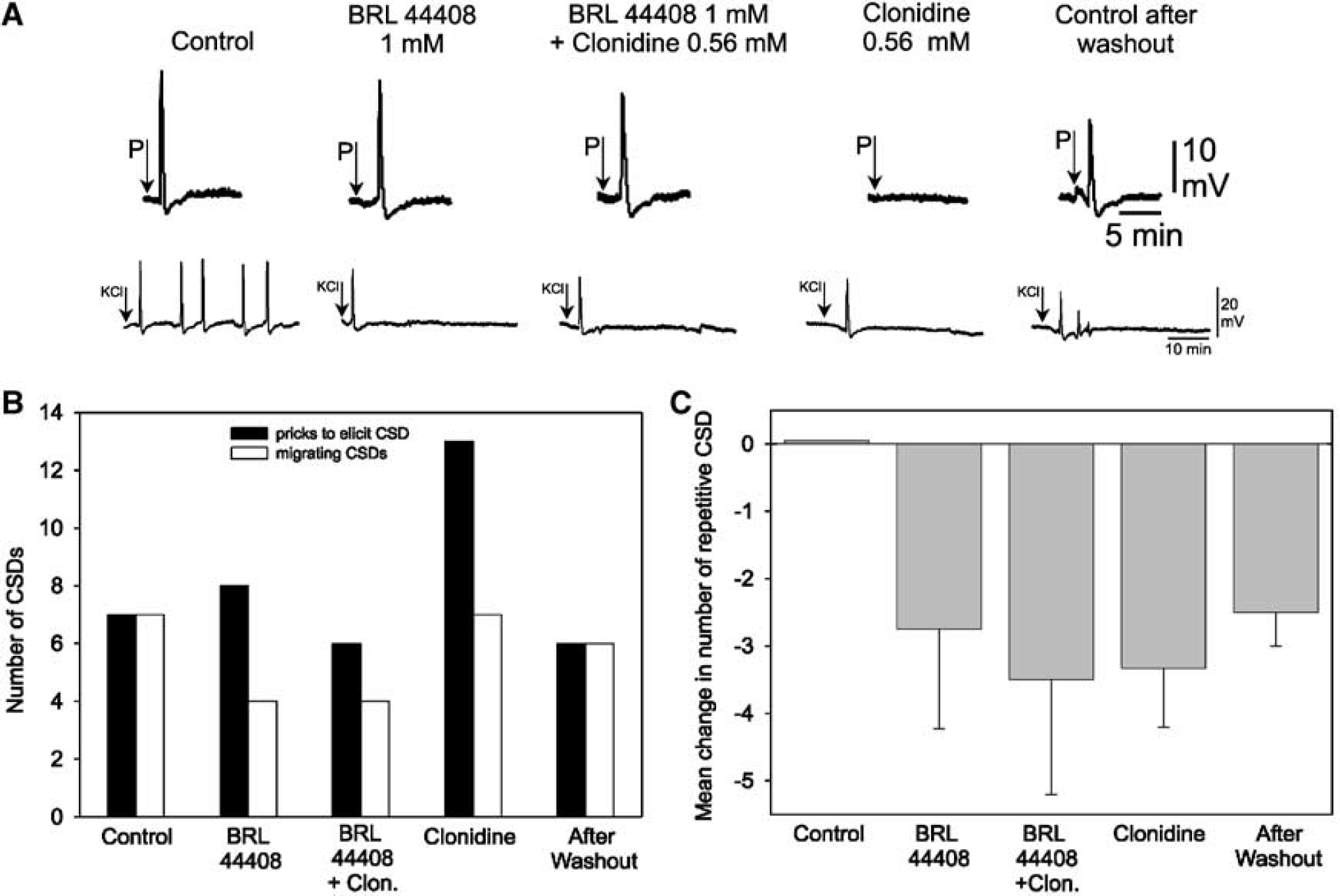
Effects of the α2D-adrenoceptor antagonist BRL 44408 and of the α2-adrenoceptor agonist clonidine on cortical spreading depression (CSD). (
Application of β-Adrenergic Antagonists
The effect of the beta-adrenergic antagonist propranolol on CSD was tested in six rats. Propranolol did not inhibit CSD initiation and had no influence on CSD under the frontal electrodes. But propranolol slowed or inhibited CSD migration in a dose-dependent manner: after 250 μmol/L CSD still traveled across the cortex in five out of six rats, after 330 μmol/L in four out of the six rats, after 500 μmol/L only in two out of six rats tested. A dose of 1 mmol/L blocked CSD migration in all rats (Figure 5). The migration time of those CSD that were still elicited increased from 137.4+16.4 secs (controls) to 241.8+77.2 secs (500 μmol/L). The other characteristics of CSD (duration, amplitude, etc.) remained unchanged.
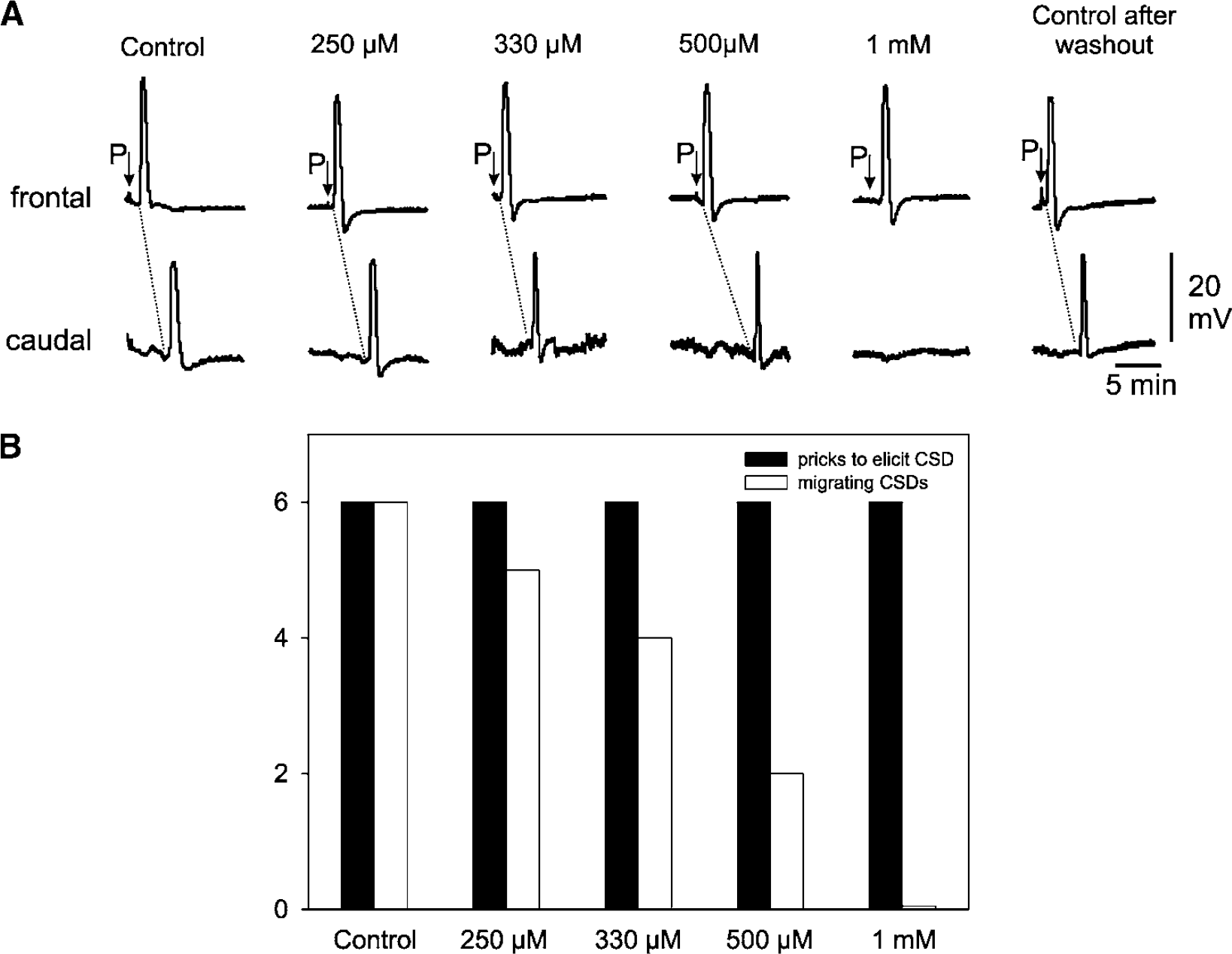
Effect of propranolol on cortical spreading depression (CSD). Propranolol applied to the cortical surface (n = 6 rats) diminished dose-dependently the number of CSD that traveled in the cortical gray matter. (
Blood Flow Measurements
Neither the topical application to the cortical surface of the adrenergic agonists nor of any of the antagonists decreased regional cerebral blood flow (rCBF) in the treated area and in the remote cortical area. No relevant change in systemic blood pressure was observed (Figure 6A). The rCBF was not influenced by NE, yohimbine, and clonidine in the treated and remote areas. When sodium nitroprusside was applied, rCBF increased in the treated area. In one of the three rats tested, there was an increase in rCBF in the remote area, which probably was caused by a spread of nitroprusside to the untreated area (Figure 6B).
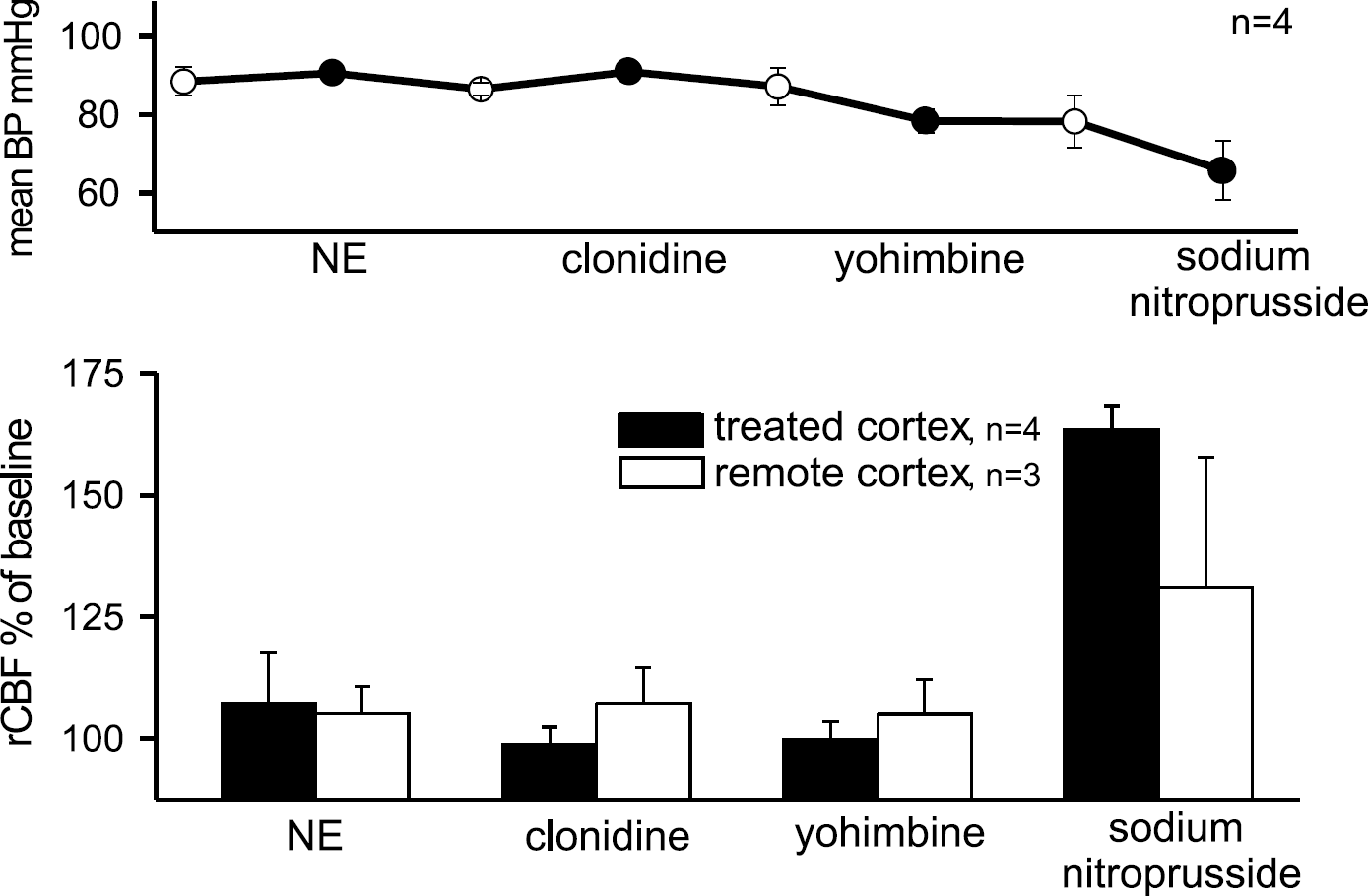
Effects of adrenergic agonists and antagonists on mean arterial blood pressure and regional cerebral blood flow (rCBF). (
Discussion
In the present study, we have shown for the first time that the migration of a CSD wave in the adult gray matter can be inhibited by topical application of either norepinephrine, the α2-adrenergic agonist clonidine, and the α2-adrenergic antagonist yohimbine as well as by topical application of the β-receptor antagonist propranolol. These findings may explain the neuroprotective effects of adrenergic agonists and antagonists in animal experiments after ischemic stroke. The latter could improve the outcome after an ischemic insult. Our results suggest that both α2- and β-receptors control the propagation of CSD.
Norepinephrine and adrenoceptor agonists/antagonists caused inhibition of CSD migration. In some of the animals, we saw not only an inhibition of CSD migration but also an inhibition of prick-induced CSD elicitation. Because the distance between the site of pricking and the recording electrodes in the remote cortex was very short (around 2 mm) we cannot exclude that small amounts of test substances expanded by capillary forces between cortical surface and dura to the remote cortex. In this case, an inhibition of CSD propagation would appear as an inhibition of CSD elicitation so that it becomes impossible to decide if the propagation or the elicitation of CSD had been blocked.
Because propagation of CSD is mainly driven by excitatory neurotransmitters, for example, glutamate (Somjen, 2001), we suppose that the inhibitory effect of NE is because of the reduced/inhibited release of glutamate. Inhibitory effects of NE on cortical and on hippocampal neuronal activity have been reported in the literature. The topical injection of 1 mmol/L NE solution into the hippocampus terminated focal epileptic activity; the same effect was seen after injection of 2 mmol/L isoproterenol (Ferraro et al, 1994). Norepinephrine inhibited discharges of most of the neurons in the middle cortical layers of cat somatosensory cortex (Warren and Dykes, 1996). Norepinephrine inhibited the spreading of paroxysmal epileptic activity to the limbic system (Ferry et al, 1997), and inhibited postsynaptic currents in cat substantia gelatinosa neurons (Kawasaki et al, 2003). However, excitatory effects of NE on neuronal activity have also been shown. Devilbiss and Waterhouse (2000) found both suppression and facilitation of glutamate-evoked discharges in cortical gray matter neurons. Arcos et al (2003) reported on increased firing rates in rat subthalamic nucleus neurons after application of NE and attributed this to effects via the excitatory α1-adrenoceptor. Whether this receptor that activates the adenylate cyclase (Ruffolo et al, 1995) plays a role in CSD is unknown.
The inhibition of CSD initiation/migration is probably because of an action of NE on presynaptic neuronal α2-adrenoceptors. These receptors inhibit via G-proteins the adenylate cyclase, which in turn inhibits calcium currents necessary for the release of glutamate (Chen et al, 1991; Ruffolo et al, 1995; Hein et al, 1999). The inhibitory effect of the α2-adrenoceptor agonist clonidine on CSD supports the idea that at least part of the NE effect is caused by an action at α2-adrenoceptors. Numerous studies have shown that clonidine inhibited presynaptic calcium currents that cause the release of neurotransmitters (Reichl and Walland, 1980; Davis et al, 1991; Trombley, 1992; Boehm and Huck, 1996; Boehm, 1999). Thus, inhibition of glutamate release could explain why α2-adrenoceptor agonists such as norepinephine or clonidine inhibit CSD. The α2-adrenoceptor agonist dexmedetomidine (Talke and Bickler, 1996) decreased the release of glutamate after hypoxia. Rilmenidine, another α2-adrenoceptor agonist, reduced the volume of focal ischemic infarction (Reis et al, 1994). Because CSD are thought to be important for the increase of the penumbra zone the inhibition of CSD by α2-adrenoceptor agonists is a feasible explanation of the observed neuroprotective effects.
Interestingly, the application of the α2-adrenoceptor antagonist yohimbine inhibited CSD as well. Because this substance produced an inhibition by itself, we could not test it against NE. Kovács and Hernádi (2003) have shown that both clonidine and yohimbine inhibited neuronal spiking activity, and yohimbine had similar effects as NE in rat prefrontal cortex in vivo. However, it is known that α2-adrenoceptor ligands such as yohimbine and to some extent clonidine exhibit also agonistic activity at 5-HT1A receptors (Newman-Tancredi et al, 1998). The latter were found in the whole rat brain, for example, at cortical pyramidal neurons of the rat cingulate cortex (Czyrak et al, 2003). 5-HT1A receptors inhibit neuronal activity (Hajos et al, 2003), probably via controlling the release of glutamate (Czyrak et al, 2003). The activation of 5-HT1A receptors attenuated amplitude and duration of CSD in cortical slices (Kruger et al, 1999). To test for the role of 5-HT1A receptors in yohimbine-induced inhibition, we first blocked the 5-HT1A receptors and found no change in prick-induced CSD. When we applied yohimbine in the presence of the blocker, CSD still migrated while we found a complete inhibition of prick-induced CSD by yohimbine alone. The number of self-propagating CSD elicited by KCl was higher during blockade of 5-HT1A receptors and during yohimbine applied in the presence of the 5-HT1A receptor blocker NAN-190 than under baseline conditions. We conclude, therefore, that the inhibition of CSD by the α2-adrenoceptor antagonist yohimbine is because of its agonistic action at the inhibitory 5-HT1A receptor. Thus, the agonist clonidine inhibited CSD by activation of α2-adrenoceptors while the action of yohimbine as an α2-antagonist is masked by its action at 5-HT1A receptors.
In rat cerebral cortex, the presynaptic α2-adrenoceptors are predominantly of the α2D subtype (Ho et al, 1998), which is analogous to the human α2A subtype (Norenberg et al, 1997; Docherty, 1998). The α2B- or α2C-subtypes do not play a significant role in rat cerebral cortex (Ho et al, 1998). To explore whether clonidine inhibits CSD via the cortical α2D-adrenoceptor subtype, we applied the subtype-specific α2D-adrenoceptor antagonist BRL 44408 that was shown to reverse the clonidine-induced inhibition of glutamate release (Li and Eisenach, 2001). Interestingly, BRL 44408 partially inhibited prick-induced CSD by itself and reduced the number of CSD after KCl application. It did not prevent the inhibitory effects of clonidine on CSD. BRL 44408, which is the only available α2D-antagonist, might have partial agonistic properties (Pauwels et al, 2000). Additionally, it has been shown that BRL 44408 has also affinity to imidazoline binding sites independent to its action on adrenoceptors (Callado et al, 1996). The activation of imidazoline receptors inhibits voltage-gated P/Q- and N-type calcium channels thereby decreasing the release of glutamate (Milhaud et al, 2002). Indeed, self-regenerating CSD after KCl were inhibited when P/Q- and N-type calcium channels were blocked (Richter et al, 2002).
Inhibition of CSD migration by propranolol was found in retinal spreading depression (Wiedemann et al, 1996). Similar as in the retina, CSD migration was inhibited after 500 or 1,000 μmol/L propranolol, and propagation velocity was slowed after 330 μmol/L. These effects could be because of a presynaptic role of the β-adrenoceptors. Nicholas et al (1993) have labeled β1- and β22-adrenoceptors in rat cerebral cortex, both on neurons and on astrocytes in adult rats (Sutin and Shao, 1992). In the amygdale cortex and in the cerebral cortex of the rat, β-adrenoceptors were found presynaptically and facilitated the release of glutamate via stimulation of the adenylate cyclase that activates P-/Q-type calcium channels (Hieble et al, 1995; Huang et al, 1996; Wang et al, 2002). Application of propranolol in that study abolished glutamate release. It is very likely, therefore, that the inhibition of CSD propagation by propranolol is because of reduction of glutamate release. Whether this inhibition of CSD is an explanation of the prophylactic efficacy of β-adrenoceptor antagonists in the treatment of human migraine deserves further studies since only a minority of migraineurs suffers from migraine with aura. If migraine is caused by cortical hyperexcitability (Welch, 2003), β-adrenoceptor antagonists could exert their prophylactic effects by decreasing neuronal excitability, thereby declining the frequency of migraine attacks.
It is unlikely that the inhibitory effects of NE, clonidine, yohimbine, and propranolol on CSD were of vascular origin. There is a body of evidence in the literature that NE reduced the cortical rCBF via contracting the arterioles in the gray matter (Edvinsson et al, 1979; Iadecola et al, 1987; Takahashi et al, 2000) and propranolol reduced the oxygen consumption (Chi et al, 1999). Those effects were only seen, however, when the substances were administered intravenously and could evoke a systemic effect. We applied the substances topically to the cortical surface and did not see profound changes in blood pressure, rCBF or diameter of the vessels except a slight increase in rCBF after clonidine. There is no data in the literature that such an increase could inhibit CSD. In contrast, CSD is facilitated under ischemic conditions. The data of Wiedemann et al (1996) provide strong support for a neuronal action of the substances because in their retinal preparation SD was inhibited in the absence of blood vessels.
In summary, our data provide neurophysiological evidence that both α2- and β-adrenergic mechanisms control the propagation of CSD probably via the release of glutamate. This effect might depend on the regulation of presynaptic cAMP concentration, which is known to determine glutamate release from the terminals (Figure 7). This theory is further corroborated by the finding that CSD is also blocked by 5-HT1A receptors that also regulate cAMP level and glutamate release (Wang et al, 2002). The prophylactic effect of β-adrenoceptor antagonists in human migraine could therefore be based on decreased neuronal excitability and/or inhibition of CSD. Inhibition of CSD could also be an explanation for the neuroprotective effect of α2-adrenoceptor agonists in the penumbra zone after ischemic stroke. A future clinical use of those compounds, however, needs further investigations.
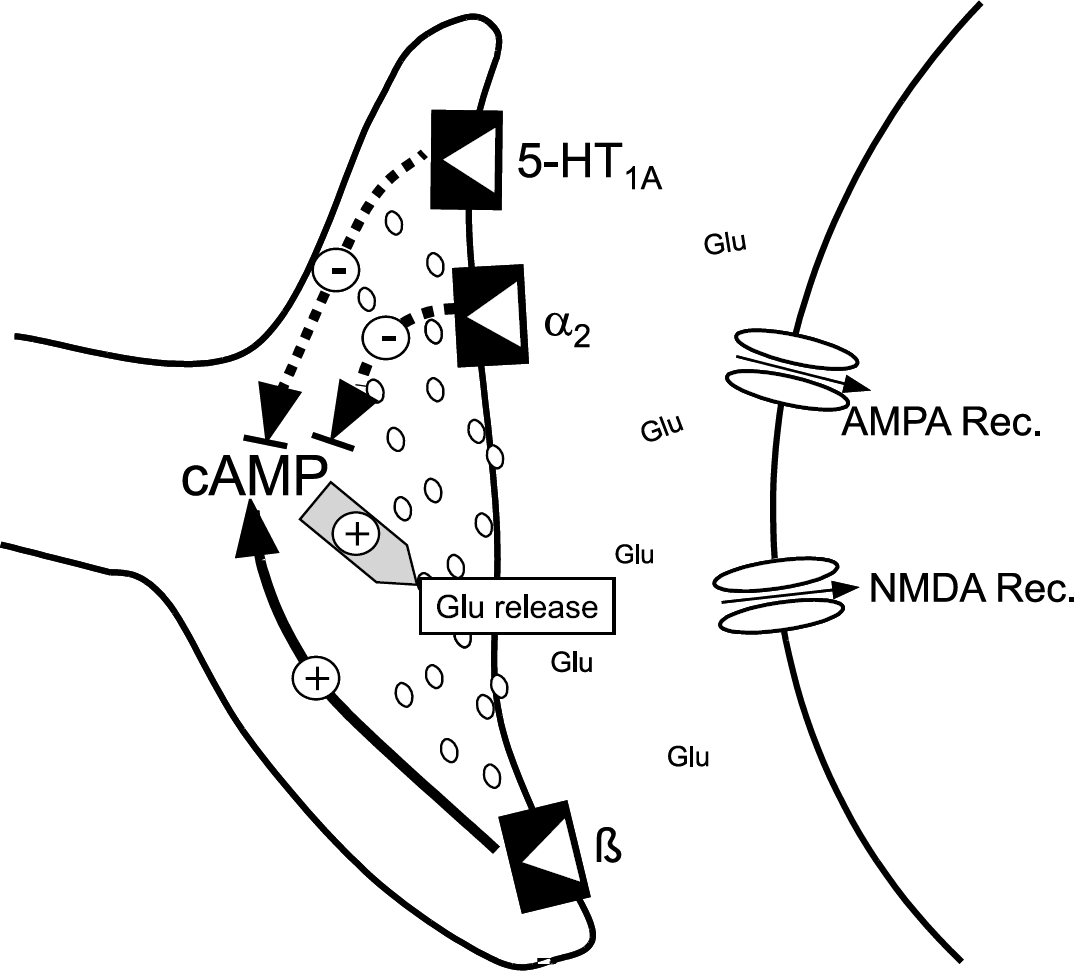
Model of adrenergic effects on cortical spreading depression (CSD). Presynaptically located α2-adrenoceptos and β-adrenoceptors influence the migration of CSD via controlling the cAMP-controlled synaptic release of glutamate. A similar mechanism presumably causes the inhibitory effect of yohimbine on CSD by its action at 5-HT1A-receptors. Solid lines show facilitatory effects, dotted lines inhibitory effects.
Footnotes
Acknowledgements
We thank Mrs Helga Müller for excellent technical assistance. We thank Dr Reinhard Bauer (Institute of Pathophysiology and Pathobiochemistry, Medical Faculty, Jena) for his help in the blood flow measurements.