
Abstract
After exposure to asphyxia, infants may develop both prolonged, clinically evident seizures and shorter, clinically silent seizures; however, their effect on cerebral tissue oxygenation is unclear. We therefore examined the hypothesis that the increase in oxygen delivery during postasphyxial seizures might be insufficient to meet the needs of increased metabolism, thus causing a fall in tissue oxygenation, in unanesthetized near-term fetal sheep in utero (gestational age 125 ± 1 days). Fetuses were administered an infusion of the specific adenosine A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine, followed by 10 mins of asphyxia induced by complete umbilical cord occlusion. The fetuses then recovered for 3 days. Sixty-one episodes of electrophysiologically defined seizures were identified in five fetuses. Tissue PO2 (tPO2) did not change significantly during short seizures (<3.5 mins), 5.2 ± 0.2 versus baseline 5.6 ± 0.1 mm Hg (NS), but fell to 2.2 ± 0.2 mm Hg during seizures lasting more than 3.5 mins (P<0.001). During prolonged seizures, cortical blood flow did not begin to increase until tPO2 had begun to fall, and then rose more slowly than the increase in metabolism, with a widening of the brain to blood temperature gradient. In conclusion, in the immature brain, during prolonged, but not short seizures, there is a transient mismatch between cerebral blood flow and metabolism leading to significant cerebral deoxygenation.
Introduction
The presence of seizures in newborn infants with hypoxic-ischemic encephalopathy is known to be highly associated with adverse outcome (Caravale et al, 2003; Miller et al, 2004). However, there is considerable ongoing controversy over whether they simply reflect the presence of injury or themselves cause further injury (Holmes and Ben-Ari, 2001). Studies of induced seizures suggest that the immature brain is relatively resistant to injury (Fernandes et al, 1999; Haas et al, 2001; Lado et al, 2000; Pereira de Vasconcelos et al, 2002), but nevertheless, in some models severe status epilepticus can still be associated with marked neuronal loss, in a mixed apoptotic/necrotic pattern (Haas et al, 2001; Sankar et al, 1998; Thompson and Wasterlain 1997). During severe seizures, hypermetabolism, with local depletion of metabolites, and impaired cerebral perfusion leading to uncoupling of cerebral blood flow and cerebral metabolism, appear to be a prerequisite for injury (Fernandes et al, 1999; Fujikawa et al, 1988; Ingvar, 1986; Pereira de Vasconcelos et al, 2002). In the immature brain, there is also an independent relationship between increased seizure severity and severity of metabolic changes in the intervascular boundary zone measured by magnetic resonance spectroscopy (Miller et al, 2002). These data support the concept that altered cerebral metabolism during seizures in the newborn may lead to uncoupling of cerebral blood flow and metabolism.
Continuous electroencephalograph (EEG) monitoring of neonates after perinatal asphyxia has recently shown, however, that shorter discrete electrographic seizures, often without clinical correlates, are relatively common. The significance of these relatively short seizures is unclear, although their presence, in some studies, is associated with adverse outcome (McBride et al, 2000). There is little information on the relationship between the onset of seizures and changes in CBF, tissue oxygenation, and metabolism in the immature brain, in part because of the methodologies used, most of which do not allow for continuous and simultaneous correlation between these variables. In the adult rat, short seizures have been associated with reduced cerebral oxygenation (Kreisman et al, 1983) and there is some supportive, indirect evidence for this in the piglet (Park et al, 1987). In older children and adults, there is evidence of both increased and decreased cerebral oxygenation depending on seizure type (Adelson et al, 1999; Haginoya et al, 2002; Sokol et al, 2000). In the neonate, short electrographic seizures appear to be associated with an increase in cerebral blood flow velocity (Boylan et al, 1999) and in regional flow (Borch et al, 1998); however, it is unknown whether the increase in oxygen delivery is sufficient to meet the needs of increased metabolism.
We recently showed that inhibition of the adenosine A1 receptor during brief but profound asphyxia in the near-term fetal sheep led to enhanced hippocampal and striatal injury, which was accompanied by a phase of delayed discrete seizure events (Hunter et al, 2003a). The current study used continuous measurements of local cerebral tissue oxygenation, blood flow, and metabolism to investigate whether postasphyxial seizures in the immature brain are associated with reduced tissue oxygenation and impaired coupling of cerebral blood flow and metabolism.
Materials and methods
Animals and Surgical Procedures
Twelve Romney/Suffolk fetal sheep were instrumented at 118 to 119 days of gestation (term = 147 days) as previously described (Hunter et al, 2003a). All procedures were approved by the Animal Ethics Committee of the University of Auckland. Food but not water was withdrawn 18 h before surgery. Ewes were administered 5 mL of streptopen (procaine penicillin (250,000 IU) and dihydrostreptomycin (250 mg/mL), Pitman-Moore, Wellington, New Zealand) intramuscularly for prophylaxis 30 mins before the start of surgery. Anesthesia was induced by intravenous injection of Saffan (Alphaxalone and Alphadolone; 3 mg/kg, Schering-Plough Animal Health Ltd, Wellington, New Zealand) and general anesthesia maintained using 2% to 3% halothane in O2. The depth of anesthesia and maternal respiration were constantly monitored by trained anesthetic staff. Under anesthesia, a 20-gauge intravenous catheter was placed in a maternal front leg vein, and the ewes were placed on a constant infusion saline drip to maintain maternal fluid balance.
Using sterile techniques, through a mid-line maternal abdominal incision, the uterus was opened and the top half of the fetus exteriorized. Polyvinyl catheters were placed in each axillary artery, the right axillary vein, and the amniotic sac. Electrocardiograph (ECG) electrodes were sewn across the chest to record fetal heart rate (FHR) (AS633-3SSF; Cooner Wire Co., Chatsworth, CA, USA). Two pairs of EEG electrodes (AS633-5SSF; Cooner Wire Co.) were placed on the dura over the parasagittal parietal cortex (5 and 10 mm anterior to bregma and 5 mm lateral) and secured with cyanoacrylate glue. A reference electrode was sewn over the occiput. To record cortical impedance, a third pair of electrodes (AS633-3SSF; Cooner Wire Co.) was placed over the dura 5 mm lateral to the EEG electrodes (Gunn et al, 1997). A polyvinyl catheter was placed in the sagittal sinus approximately midway between the points where the coronal and lamboid sutures intersect the sagittal suture to measure oxygen content. This catheter was advanced 0.5 to 1.0 cm, such that the tip lay at or near the confluens sinum. An inflatable silicone occluder was placed around the umbilical cord of all fetuses (In Vivo Metric, Healdsburg, CA, USA).
A four-part composite probe (diameter ~400 μm) containing emitting and receiving laser Doppler channels, a PO2 electrode, and thermocouple was placed in the right parietal cortex approximately 5 mm lateral to the midline and 5 mm posterior to the coronal suture, to a depth of 5 mm below the dura (Oxford Optronix Inc., Oxford, UK) (Hunter et al, 2003a). An ultrasonic flow probe (3S, Transonic Systems Inc., Ithaca, NY, USA) was placed on the right carotid artery near the angle of the jaw. A thermocouple (IT-18 thermometer, Physitemp, Clifton, NJ, USA) was inserted into the lingual artery towards the carotid artery to measure arterial blood temperature, with a resolution of 0.01°C.
On completion of these procedures, all fetal leads were exteriorized through the maternal flank, the fetus returned to the uterus, the uterus and abdominal incisions repaired, and a maternal long saphenous vein catheterized to provide access for postoperative care and euthanasia. Antibiotics (80 mg gentamicin, Rousell, Auckland, New Zealand) were administered into the amniotic sac before the closure of the uterus. Postoperatively, all sheep were housed together in separate metabolic cages with free access to water and food. They were kept in a temperature-controlled room (16°C ± 1°C, humidity 50% ± 10%), in a 12-h day/night cycle. During postoperative recovery, antibiotics were administered daily for 5 days intravenously to the ewe (600 mg benzylpencillin sodium (crystapen) and 80 mg gentamicin). Fetal catheters were maintained patent by continuous infusion of heparinized isotonic saline (20 U/mL at 0.2 mL/h), and the maternal catheter maintained by daily flushing with heparinized saline. Experiments were started after 3 days of postoperative recovery, and fetal arterial blood was taken daily from the brachial artery for pH, blood gas, glucose, and lactate analysis to verify fetal health as previously reported (Hunter et al, 2003a), and microbial surveillance at postmortem showed no evidence of fetal infection.
Recordings
Fetal arterial blood pressure (mean arterial blood pressure; MAP) corrected for amniotic fluid pressure (Novatrans II, MX860; Medex Inc., Hilliard, OH, USA), carotid arterial blood flow (CaBF; T208 Ultrasonic Flowmeter; Transonic Systems Inc.), cortical blood flow (laser Doppler flowmetry, Oxyflow; Oxford Optronics Inc.), cortical oxygen tension (Oxylite; Oxford Optronics Inc.), cortical temperature (Oxylite; Oxford Optronics Inc.), lingual arterial temperature and EEG and impedance were recorded continuously at 512 Hz from 24 h before until 3 days after asphyxia. Data were stored to disk by custom software for offline analysis (Labview for Windows, National Instruments Ltd, Austin, TX, USA).
The EEG signal was low-pass filtered with a sixth-order low-pass Butterworth filter, with a cutoff frequency of 50 Hz. A power spectrum was then calculated from this 256 Hz sampled signal. For clarity of data display, the EEG intensity was log transformed (dB, 20 x log(intensity)) (Williams et al, 1991). Additionally, the raw EEG signal was processed through a digital FIR low-pass filter with a cutoff frequency of 30 Hz and stored at a sampling rate of 64 Hz for analysis of seizures. The impedance signal was also extracted as previously described (Williams et al, 1991). The impedance of a tissue rises concomitantly as cells depolarize and fluid shifts from the extracellular to the intracellular space, and thus impedance is a measure of cytotoxic edema. CaBF was measured as an index of changes in global CBF (Bishai et al, 2003; Gratton et al, 1996). Laser Doppler was used to measure local cortical blood flow (Bishai et al, 2003).
Brain Heat Production Using the Fick Principle
Changes in the heat production of the brain were calculated as an index of local cerebral metabolism using the Fick principle (Hunter et al, 2003b). The difference between the temperature of arterial blood (Tblood) supplying the brain and the brain tissue itself (Tbrain) gives the temperature increase resulting from brain metabolism. Multiplying this difference by blood flow provides the relative heat production in the local region of the brain in which the temperature sensor is placed; heat production was expressed as percent change from baseline.
Experimental Protocol
As previously reported, fetuses were randomized to receive either vehicle or 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) infused into the right axillary vein at a rate of 3.6 mg/min for 10 mins, and then at 0.75 mg/min for 60 mins (Hunter et al, 2003a). After infusion for 60 mins, asphyxia was induced by complete occlusion of the umbilical cord for 10 mins, confirmed by characteristic fetal cardiovascular and pH and blood gas changes (Hunter et al, 2003a). The infusion of DPCPX was discontinued at the end of the occlusion period. Three days after the asphyxial insult, the sheep were killed with sodium pentobarbitone (9 g intravenous to the ewe; Pentobarb 300, Chemstock International, Christchurch, New Zealand).
Data Analysis
Seizures were identified visually on the raw EEG record and defined as the concurrent appearance of sudden, repetitive, evolving stereotyped waveforms in the EEG signal lasting more than 10 secs and of an amplitude greater than 20 μV (Scher et al, 1993). Only episodes for which stable tPO2 recordings were available were analyzed. The effects of seizures on time sequence data were evaluated by repeated measures one-way ANOVA (SPSS v10, SPSS Inc., Chicago, IL, USA). The baseline period was taken as the average of the 25-min period immediately preceding the onset of each seizure. When an effect of time was found, data were compared with baseline using Dunnett's post-test. Regression analysis was used to examine the relationship between selected variables. Statistical significance was accepted at P<0.05. Data are mean ± standard deviation.
Results
A total of 61 episodes of postasphyxial seizure activity with good quality tPO2 recordings were identified from five fetuses, one treated with vehicle and four with DCPCX. Seizures began between 6 and 8 h after the asphyxial episode, and resolved within 24 h in each case. Fetuses exhibited between 5 and 13 episodes each. We have previously reported changes during the acute asphyxia period, and neuronal loss in four of these fetuses (Hunter et al, 2003a). The mean gestational age of the fetuses in this study was 125 ± 1 days and fetal weight at autopsy was 3.35 ± 0.21 kg.
A significant fall in tPO2 was seen only in longer seizures (>3.5 mins, Figure 1), with a sigmoidal relationship between the duration of seizures and the nadir of tPO2 (r2 = 0.79, P<0.001, Figure 2). In contrast, there was no correlation between the increase in EEG amplitude during seizures and nadir of tPO2 (r2 = 0.04, Figure 2). The mean preseizure baseline tPO2 was 5.6 ± 0.1 mm Hg. In events lasting less than 3.5 mins, tPO2 fell to a nadir of 5.2 ± 0.2 mm Hg (NS versus baseline values), while during events lasting for more than 3.5 mins tPO2 fell to 2.2 ± 0.2 mm Hg (P<0.001). The tPO2 did not fall further even in seizures lasting for more 10 mins (Figure 2). Using this retrospective classification of long and short events, one fetus showed predominantly short events, one showed predominantly long events, while three (including the vehicle-treated fetus) showed similar numbers of long and short events.
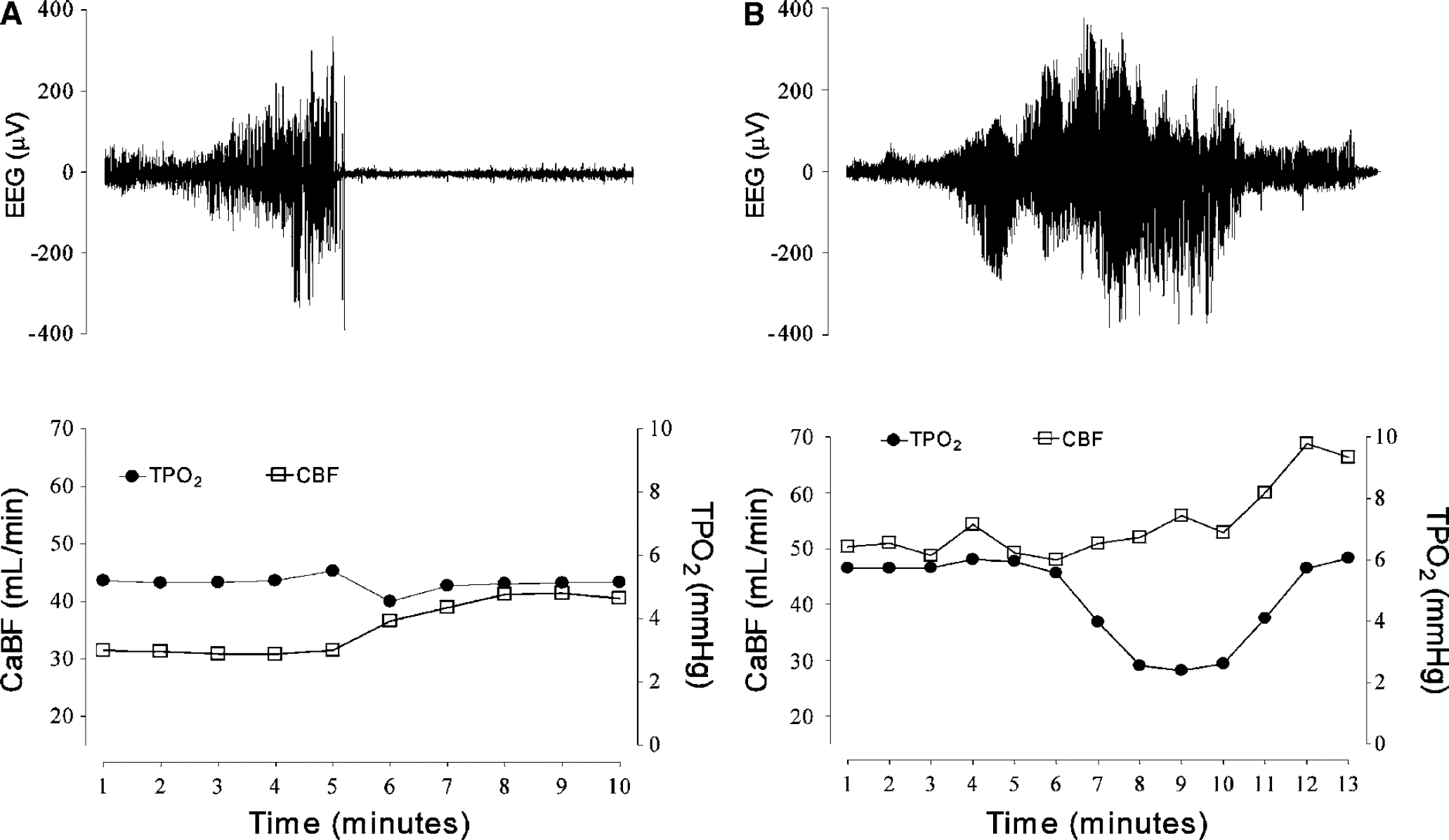
Examples from individual fetuses of short (panel
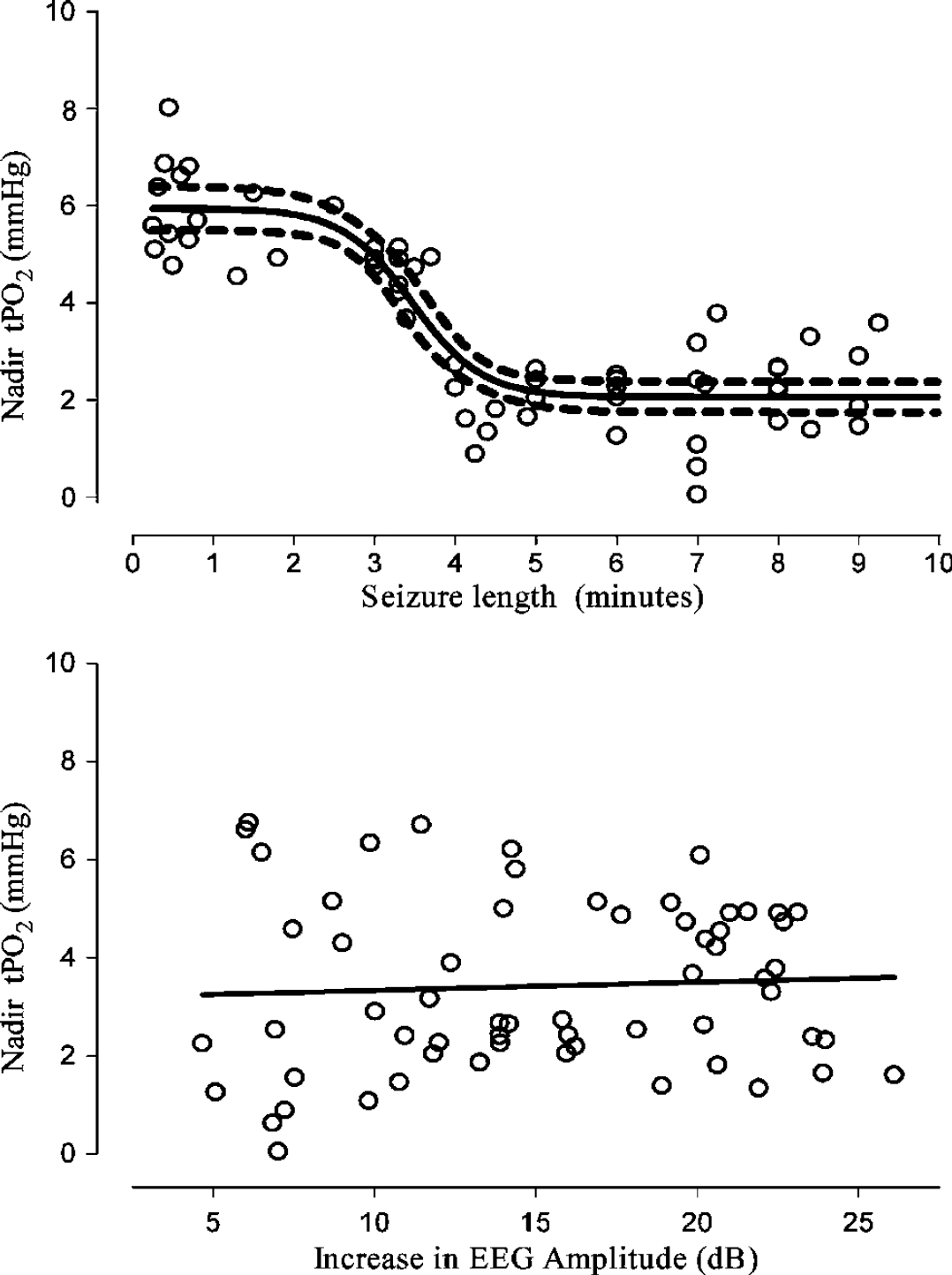
Relationship between the nadir of tissue oxygen levels (tPO2) during seizures and seizure duration (top panel) and the increase in EEG amplitude (in dB, bottom panel). There was no relationship between tPO2 changes and the increase in EEG amplitude, but there was a relationship with seizure duration. Tissue hypoxia was seen only in seizures lasting for more than 3.5 mins.
The time sequence of changes in mean EEG intensity (power), cortical impedance, CaBF, change in brain temperature, tissue oxygenation, and heat production before, during, and after the onset of 28 seizures that lasted for more than 3.5 mins are shown in Figure 3. The mean duration of these seizure events was 6.5 mins. EEG intensity increased immediately after the onset of the seizures, peaked at 6 mins, and then fell below baseline values from 3 mins after the end of the event. There was a very small but significant increase in cerebral impedance during seizures (peak impedance: 101.6% ± 1.2%, P<0.05). Delta temperature (the difference between cortical and arterial blood temperatures) was significantly increased at 4 and 5 mins after the onset of seizures. After the end of the seizure, delta temperature was significantly reduced from 2 to 10 mins, and then returned to baseline values.
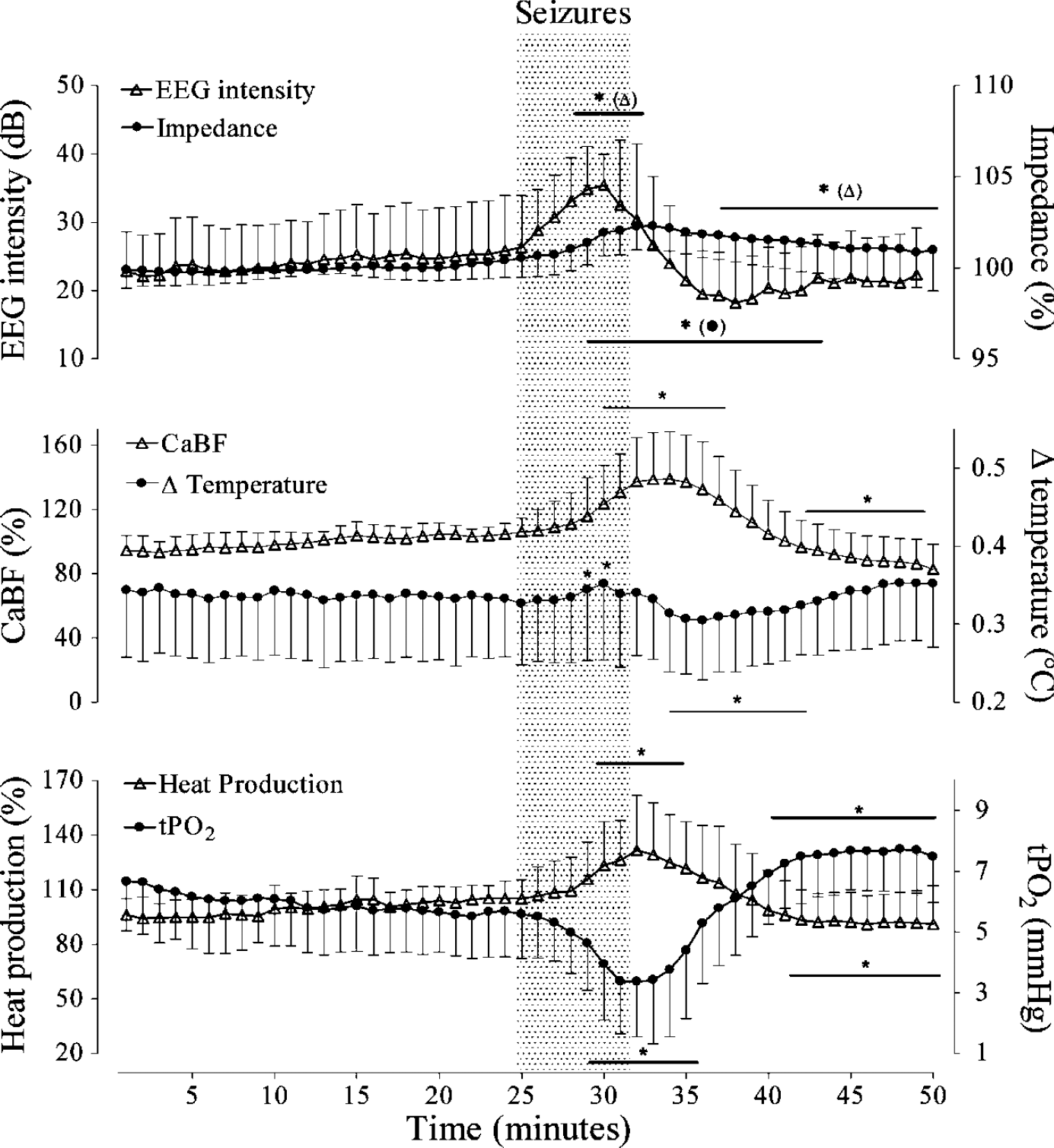
Time sequence of changes in mean EEG intensity (power), cortical impedance, CaBF, heat production, delta temperature, and tissue oxygenation (tPO2) before, during, and after the onset of 28 prolonged seizures (>3.5 mins, mean duration 6.5 mins). Data are mean ± s.d. *P<0.05 versus baseline.
Carotid arterial blood flow was significantly increased from 4 mins after the onset of the seizures until 6 mins after the end, reaching a peak of 35.7% ± 5.2% above baseline values (P<0.01). From 10 mins after the end of the seizures, CaBF was significantly suppressed compared to baseline. Tissue PO2 began to fall from 3 mins after the start of the seizures, and remained reduced until 3 mins after the end of the seizures (P<0.001). From 7 mins after the end of the seizures tPO2 increased above baseline levels (P<0.01). There was a close relationship between the timing of the fall in tPO2 and the onset of the increase in heat production, which was significant from 4 mins after the start of the events, with a maximum increase of 32.3% ± 6.1% (P<0.01). After the end of seizures, heat production fell, reaching control values after 4 mins and becoming significantly suppressed from 9 mins.
The relationship between cortical blood flow and CaBF and MAP was studied in a subset of prolonged seizures (n = 13, Figure 4). Cortical blood flow closely paralleled the changes in CaBF, with a similar increase, which was maximal shortly after the end of the seizure (P<0.01), followed by a slightly slower fall compared with CaBF. During seizures, MAP was significantly elevated from 5 mins (P<0.01) and rapidly returned to baseline values after seizures.
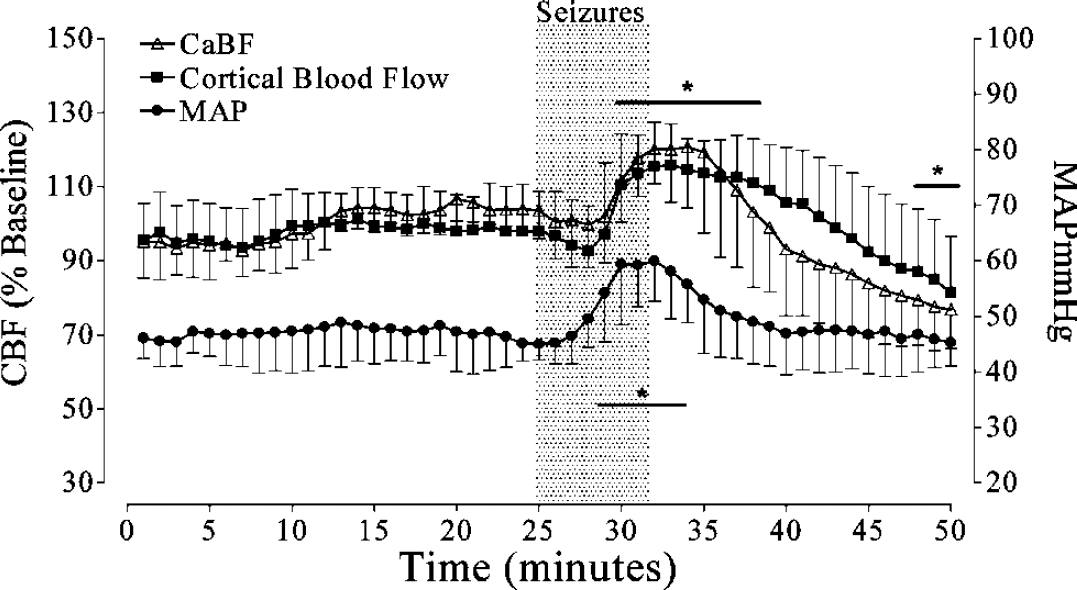
Time sequence of changes in cortical blood flow, in relation to changes in CaBF and MAP in a subset of seizures (n = 13). Note that the increase in cortical flow closely paralleled the changes in CaBF during seizures (P<0.01), followed by a slightly slower fall after the end of the seizure than CaBF. *P<0.05 versus baseline (significance not shown for CaBF; see Figure 3). Data are mean ± s.d.
Discussion
The present study shows for the first time that local cortical hypoxia occurs during postasphyxial events in the immature brain but only during longer seizures, lasting for more than a critical cutoff of 3.5 mins. This fall in cortical oxygenation was not because of a greater amplitude of the longer events, but rather because of a brief delay before CBF increased during seizures. Blood flow did not begin to rise until after the fall in tPO2 was established, suggesting that overt tissue hypoxia was the key mechanism triggering increased perfusion. Critically, when seizures continued after this point, CBF increased such that tPO2 levels stabilized with no further fall even in the longest seizures seen in the present study (10 mins plus).
The key mechanism of the delayed fall in tPO2 in the present study was the delayed rise in cerebral perfusion during longer events as measured by both caBF, an index of global flow (Bishai et al, 2003), and local cortical changes measured by laser Doppler (Lan et al, 2000). This observation is supported by the small but significant widening of the temperature gradient between arterial blood and brain at 4 and 5 mins after the start of events, denoting a greater increase in global cerebral metabolism than in CBF. Normal neural activation can, under some conditions, be associated with a lag of up to 5 secs before CBF rises, leading to a small, ‘initial dip’ in tissue oxygenation, followed by an increase (Thompson et al, 2004). Thus, the much larger delay in the present study is consistent with at least transient loss of the normal close coupling of cerebral flow and metabolism. The mechanism of this transient uncoupling is unknown. There is evidence that sympathetic activation limits hyperperfusion during seizures (Kurth et al, 1988); however, there are no detailed time course data on sympathetic activity in this setting.
The reason for the fall in tPO2 during prolonged seizures but not short events is likely to reflect the time required for seizures to propagate widely from their primary focus. In the present model, injury to the hippocampus and striatum was greater than in the cortex, and thus the primary focus of the seizures is likely to have been subcortical (Hunter et al, 2003a). Alternatively, this distribution may reflect the well-established vulnerability of the cornu amonis regions of the hippocampus to excitotoxic injury (Johnston et al, 2002), which might be enhanced by seizures (Tan et al, 1992). In studies of postnatal epilepsy, positron emission tomographic scanning shows highly localized changes in CBF (Borch et al, 1998), which would not necessarily be reflected in either measure of CBF used in the present study. Thus, the present data suggest that the time delay for global heat production to rise, and for tPO2 to fall after the onset of epileptiform activity, reflects the time for propagation of the seizure and thus for a general increase in cerebral metabolism.
After termination of seizure events in the present study, there was a period of marked suppression of EEG activity coupled with a persisting increase in perfusion, such that there was an overshoot increase in tPO2 levels. This phenomenon is highly suggestive of true luxury perfusion that we speculate may help to wash out anerobic metabolites. Again, the precise mechanism is unknown, but previous studies have strongly implicated adenosine in mediating seizure arrest and postictal refractoriness (During and Spencer, 1992). Adenosine is known to be released during seizures in the newborn piglet (Park et al, 1987), but more studies are required to dissect the roles of adenosine and other inhibitory neuromodulators in the immature brain.
Given that short seizures were not associated with tissue deoxygenation, it is improbable that they could be associated with further metabolic injury outside the primary focus. In contrast, the significance of tissue deoxygenation during longer seizures is unclear; however, cerebral metabolism could be maintained and the fall in tPO2 ultimately reached a stable nadir. In previous adult rat studies, the onset of injury was correlated with ultimate failure of metabolism (Ingvar, 1986). Thus, these data suggest that it is unlikely that these seizures were injurious. Supporting this conclusion, the increase in cytotoxic edema as measured by the rise in cerebral impedance during each seizure was only 1% to 2%, far less, for example, than during the primary period of umbilical cord occlusion (Hunter et al, 2003a), indicating minimal cortical depolarization. Nevertheless, it is important to note the possibility that further, secondary insults such as hypotension or hypocapnia that reduce cerebral perfusion (Vannucci et al, 1997) may have the potential in combination with tissue hypoxia to cause exacerbated injury during prolonged seizures.
Similar to the clinical situation, we have previously reported that the presence of seizures in the present model is highly related to greater underlying neuronal loss (Hunter et al, 2003a). However, the present study cannot directly assess the contribution of duration of seizures to injury, since all animals demonstrated a range of seizure durations. Currently, there is little previous direct evidence. While, in the 10-day-old rat, kainite-induced seizures after hypoxia-ischemia significantly exacerbated brain injury (Wirrell et al, 2001), this effect was primarily mediated by secondary hyperthermia (Yager et al, 2004). Further, although suppression of postischemic seizures in near-term fetal sheep with an N-methyl-
A potential limitation of the present study is that for most of the cases, the primary hypoxic injury was induced using a combination of adenosine blockade with asphyxia, which potentially could affect subsequent seizure activity, reduce the rate of increase in metabolism during events, or alter the relationship between increases in metabolism and flow. However, the highly specific adenosine A1 antagonist that was used did not affect cardiovascular or CBF changes during asphyxia (Hunter et al, 2003a), and because of its short half-life would have been cleared before the onset of seizures. Consistent with this, the vehicle-treated asphyxia fetus that developed seizures showed a similar relationship between duration of events and the onset of tissue oxygenation. Finally, it is important to recognize that the precise relationship between duration of seizures and the timing of deoxygenation is very likely to be affected by factors such as the stage of maturation of the brain, the cause and nature of the seizures (e.g., idiopathic versus hypoxia-ischemia), the presence of pyrexia, or induced hypothermia, and potentially by species (Park et al, 1987). This may underlie the variable results found with near-infrared spectroscopy studies of brain oxygenation in older children and adults. For example, in one report, purely electrographic seizures were accompanied by rapid reductions in oxygenation, whereas electroclinical seizures were characterized by an increase (Adelson et al, 1999). In contrast, others found no change during an electrographic seizure and increased oxygenation during complex partial seizures, but a fall in oxygenation during secondarily generalized seizures (Sokol et al, 2000).
In conclusion, these data show that short seizures are not associated with tissue deoxygenation, and thus are unlikely to be associated with further global metabolic injury. In contrast, during more prolonged seizures, there is a transient mismatch between cerebral blood flow and metabolism leading to significant tissue deoxygenation. Although oxygenation ultimately reaches a stable nadir, speculatively more severe tissue deoxygenation could occur in the presence of secondary insults such as hypotension or hypocapnia.