
Abstract
Endothelial dysfunction and inflammation enhance vulnerability to hypertensive brain damage. To explore the participation of Angiotensin II (Ang II) in the mechanism of vulnerability to cerebral ischemia during hypertension, we examined the expression of inflammatory factors and the heat shock protein (HSP) response in cerebral microvessels from spontaneously hypertensive rats and their normotensive controls, Wistar Kyoto rats. We treated animals with vehicle or the Ang II AT1 receptor antagonist candesartan, 0.3 mg/kg/day, via subcutaneously implanted osmotic minipumps for 4 weeks. Spontaneously hypertensive rats expressed higher Angiotensin II AT1 receptor protein and mRNA than normotensive controls. Candesartan decreased the macrophage infiltration and reversed the enhanced tumor necrosis factor-α and interleukin-1β mRNA and nuclear factor-κB in microvessels in hypertensive rats. The transcription of many HSP family genes, including HSP60, HSP70 and HSP90, and heat shock factor-1 was higher in hypertensive rats and was downregulated by AT1 receptor blockade. Our results suggest a proinflammatory action of Ang II through AT1 receptor stimulation in cerebral microvessels during hypertension, and very potent antiinflammatory effects of the Ang II AT1 receptor antagonist. These compounds might be considered as potential therapeutic agents against ischemic and inflammatory diseases of the brain.
Introduction
Endothelial dysfunction and vascular inflammation enhance vulnerability to hypertensive brain damage and are major risk factors for brain ischemia and stroke in spontaneously hypertensive rats (SHR) (Amenta et al, 2003) and in humans (Di Napoli and Papa, 2003; Lawes et al, 2004). Angiotensin II (Ang II) is a proinflammatory agent involved in vascular alterations and endothelial cell injury in hypertension by activation of the nuclear factor (NF)-κB, cytokines and adhesion molecules (Pueyo et al, 2000; Pastore et al, 1999; Ruiz-Ortega et al, 2000; Suzuki et al, 2001; Touyz, 2003).
Experimental and clinical studies revealed that Ang II AT1 receptor antagonists reverse the inflammatory reaction in kidney and peripheral arteries (Mervaala et al, 1999; Navalkar et al, 2001; Hilgers et al, 2001; Dandona et al, 2003) and reversed the increased expression of intracellular adhesion molecule-1 (ICAM-1) and macrophage infiltration in microvessels from SHR (Ando et al, 2004). Long-term blockade of Ang II AT1 receptors in SHR reversed the pathological cerebrovascular remodeling, normalized the expression of endothelial and inducible nitric oxide synthase (eNOS and iNOS) and protected the animals against hypertensioninduced ischemic neuronal injury and stroke (Ito et al, 2002; Yamakawa et al, 2003).
To clarify the mechanisms of protection against ischemic injury by treatment with AT1 receptor antagonists, we focused on the heat shock protein (HSP) response in microvessels from SHR. Heat shock proteins, highly conserved throughout evolution, play essential roles as molecular chaperones facilitating the folding, intercellular transport, assembly and disassembly of cellular proteins. A wide variety of stressful stimuli, such as hypertension, inflammation, increased oxidative stress and ischemia, increase the intercellular synthesis of HSPs (Xu et al, 1996; Wallin et al, 2002; Pockley, 2003). Heat shock proteins may prevent damage and participate in cellular repair during injury (Christians et al, 2003), but they can also stimulate proinflammatory cytokine production (Asea et al, 2000). In the peripheral vasculature, hypertension increases expression of HSP70 and activates the heat shock factor-1 (HSF-1), the primary transcription factor regulating expression on many HSPs (Xu et al, 1996). HSP70 restriction fragment length polymorphism is associated with the development of hypertension (Hamet et al, 1992). Angiotensin II, through AT1 receptor activation, induces HSPs expression in peripheral tissues (Ishizaka et al, 2002).
We wished to gain a better understanding of the molecular mechanisms leading to the participation of Ang II in vulnerability to brain ischemia, and the potential therapeutic value of AT1 receptor antagonists. To this end, we studied AT1 receptors, NF-κB, additional inflammatory markers and HSP expression in brain microvessels from SHR and their normotensive controls from Wistar Kyoto (WKY) rats after prolonged treatment with an AT1 receptor antagonist.
Materials and methods
Animals and Treatment
Male SHR and WKY rats, 12-week-old, weighing 270 to 450 g, were purchased from Taconic Farms, Germantown, NY, USA. The National Institute of Mental Health Animal Care and Use Committee has approved all procedures. Both SHR and WKY rats were divided into two groups as Candesartan and vehicle treatment. Osmotic minipumps (Durect) were implanted subcutaneously to deliver the AT1 antagonist candesartan (0.3 mg/kg/day) or vehicle for 28 days. After 4 weeks administration, the brain microvessels were isolated. Candesartan was a gift from Astra Zeneca, R & D, Mölndal, Sweden.
We measured systolic blood pressures by the tail-cuff method at the end of the treatment. Reported blood pressure measurements are the mean of four determinations in each individual animal.
Isolation of Brain Microvessels
We anesthetized the animals with 30 mg/kg pentobarbital, intraperitoneally, and perfused them through the heart with saline. We removed the brains and rinsed them in cold isotonic sucrose buffer (0.32 mol/L sucrose, 3 mmol/L HEPES, pH 7.4). We discarded the cerebellum, pia mater and choroids plexus, and homogenized the rest of the brain (1.1 ± 1.3 g) with a Dounce homogenizer and a tightly fitting pestle, in 3 volumes of sucrose buffer, twice, each followed by centrifugation at 4°C for 10 mins at 1000g. We resuspended the sediments in sucrose buffer and centrifuged twice for 30 secs at 100g, and pooled the supernatants and washed them twice. We resuspended the final pellets in 1 mL sucrose buffer followed by centrifugation at 14,000g, and stored the precipitates (around 60 to 80 μL) at −80°C until use (Yamakawa et al, 2003).
The purification of the microvessels was evaluated by a microscope (stained dried smear with hematoxylin) and gamma-glutamyl transpeptidase activity (Yamakawa et al, 2003), a marker enzyme localized to brain microvessels. Its activity was over 10 times higher in microvessels than in the whole brain, with no significant differences between SHR and WKY rats, vehicle-treated or candesartan-treated (results not shown).
Microarray
All procedures were conducted by Affymetrix GeneChip Expression Analysis Technique (Affymetrix). Affymetrix rat genome U34A array contains 8799 probes. At least two independent experiments were performed, with each using pooled RNA samples from five rats. The results were analyzed with Affymetrix Microarray Suite 5.0 and Data Mining Tool 3.0 software. In pairwise comparisons, the transcripts showing increase/marginal increase (I/MI) or decrease/marginal decrease (D/MD) call were scored using the Wilcoxon signed-rank test. We chose the score above 75 with the present transcripts as a moderately stringent cutoff for significance. A further description of the methodology, according to the Minimum Information About a Microarray Experiment (MIAME) guideline is provided in the supplementary information (Supplementary MIAME).
Real-time Polymerase Chain Reaction
Real-time polymerase chain reaction (PCR) was performed in a 25 μL reaction mixture consisting of 1 x SYBR® Green PCR Master Mix (Applied Biosystems), 1 μL cDNA and 0.3 μmol/L of each primer for a specific target on a Smart Cycler (Cepheid). The specific primers are listed in Table 1. The amplification was performed at 95°C for 10 mins, followed by 40 cycles at 95°C for 15 secs and 56 to 60°C for 60 secs. To obtain a calibration curve, serial dilutions of rat cDNA were used. The individual targets for each sample were quantified by determining the cycle threshold (Ct) and by using a calibration curve. The relative amount of the target was normalized with the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Specific primers used for real-time PCR

Western Blotting
Samples containing approximately 20 μg protein were electrophoresed on 10% Novex Tris-Glycine Gel (Invitrogen) and then transferred to PVDF membranes. The membranes were probed with rabbit antihuman Ang II AT1 receptor polyclonal antibody (N10), mouse antihuman HSP60 monoclonal antibody (H1), mouse antihuman HSP90 monoclonal antibody (F18) (Santa Cruz), mouse antihuman HSP70 monoclonal antibody (SPA-810), rabbit antihuman HSF-1 polyclonal antibody (SPA-901) (Stressgen) and rabbit antihuman phospho-NF-κB p65 (Ser536) polyclonal antibody (Cell Signaling). The protein bands were visualized by the Western Blotting Chemiluminescence Luminol Reagent (Santa Cruz) or SuperSignal West Femto Maximum sensitivity substrate (Pierce). ScionImage software (Scion) was used to quantify the results.
Immunohistochemistry and Immunofluorescence
Immunohistochemistry was performed as described earlier (Ando et al, 2004). Brain sections were treated with the primary antibodies for 60 mins at room temperature. In addition to the antibodies mentioned above, rabbit antirat tumor necrosis factor-α (TNF-α) polyclonal antibody (Chemicon) and mouse antirat interleukin-1β (IL-1β) monoclonal antibody (Serotec) were also used. A mouse antirat monoclonal antibody reacting with the ED2-like antigen was applied to visualize perivascular macrophages (HIS36) (BD Biosciences). The antibodies were visualized with the DAKO Envision System (DAKO). Positive results were evaluated by at least two investigators and counted bilaterally in two randomly selected areas (1 × 1 mm2 in size) from the forebrain cortex by double-blind methods. Groups were of four animals, two to five sections per animal.
For immunofluorescence staining, the slides were incubated with unlabeled primary antibody at 4°C overnight. A mouse anti-AT1 receptor monoclonal antibody (6:1000 dilution, a gift from University of Bern) and a rabbit antiglucose transporter (GLUT-1) polyclonal antibody (1:100 dilution, Chemicon) were used to visualize the AT1 receptor and brain endothelial cells. After three 5-min washing in PBS, the slides were incubated for 1 h with fluorescein isothiocyanate (FITC) conjugated antirabbit IgG (1:100, Vector) and Texas red-conjugated antimouse IgG (1:100, Vector). Slides were observed under a Leica Confocal system (Leica), using an x 100 objective lens.
Statistical Analysis
Data were expressed as mean ± s.e.m. Statistical analysis was performed by two-way ANOVA followed by post hoc analysis using Newman-Keuls test for multiple comparison.
Results
Blood Pressure
The systolic blood pressures were higher in SHR (203 ± 3.6 mm Hg) compared with WKY controls (115 ± 2.7 mm Hg, P<0.001). Treatment with candesartan for 4 weeks lowered the systolic blood pressure in SHR (132 ± 3.3 mm Hg, P<0.001) and in WKY rats (103 ± 0.9 mm Hg, P<0.001).
Overexpression of AT1 Receptor in Brain Microvessels from Hypertensive Rats
Isolated brain microvessels consisted of arterioles, venules and capillaries with minimum transverse diameter ≤50 μm. Immunofluorescence and immunohistology showed that AT1 receptors were localized to the endothelium of brain microvessels either in isolated tissues or in brain sections (Figure 1). We also found AT1 receptor expression in smooth muscle cells of small arterioles less than 50 μm diameter in SHR (Figure 1D). We did not find AT1 receptor expression in smooth muscle cells in small arterioles of WKY rats, probably because of limitations in the sensitivity of our method.
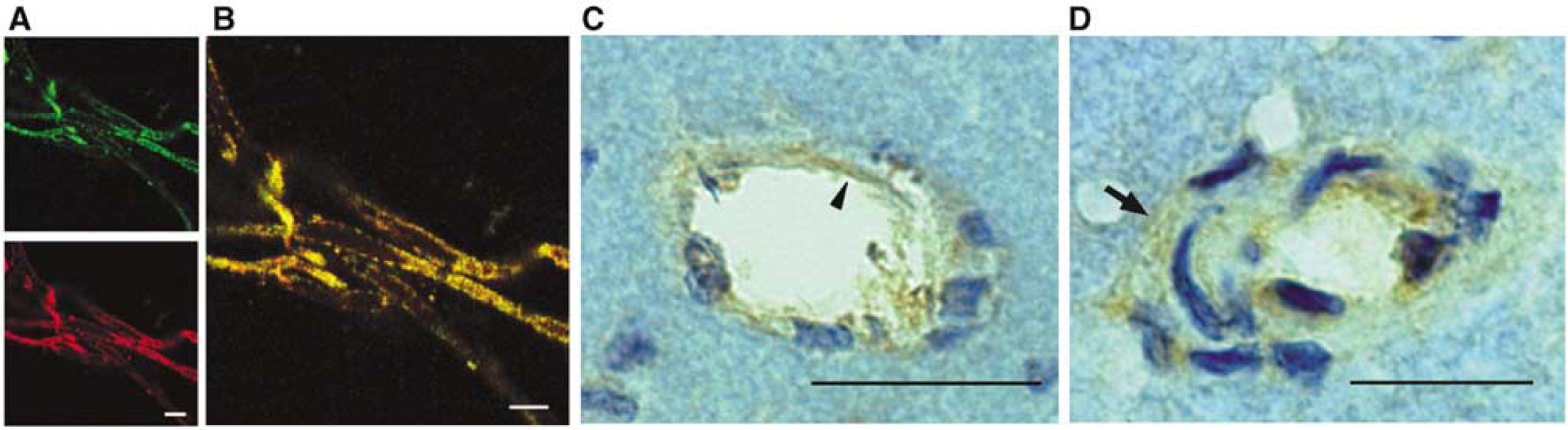
Expression of AT1 receptors in endothelium of brain microvessels. (
In our brain microvessel preparations, AT1 receptor mRNA and protein were increased in SHR when compared with WKY rats. There were no statistically significant changes in receptor mRNA or protein after treatment with the AT1 blocker (Figure 2).
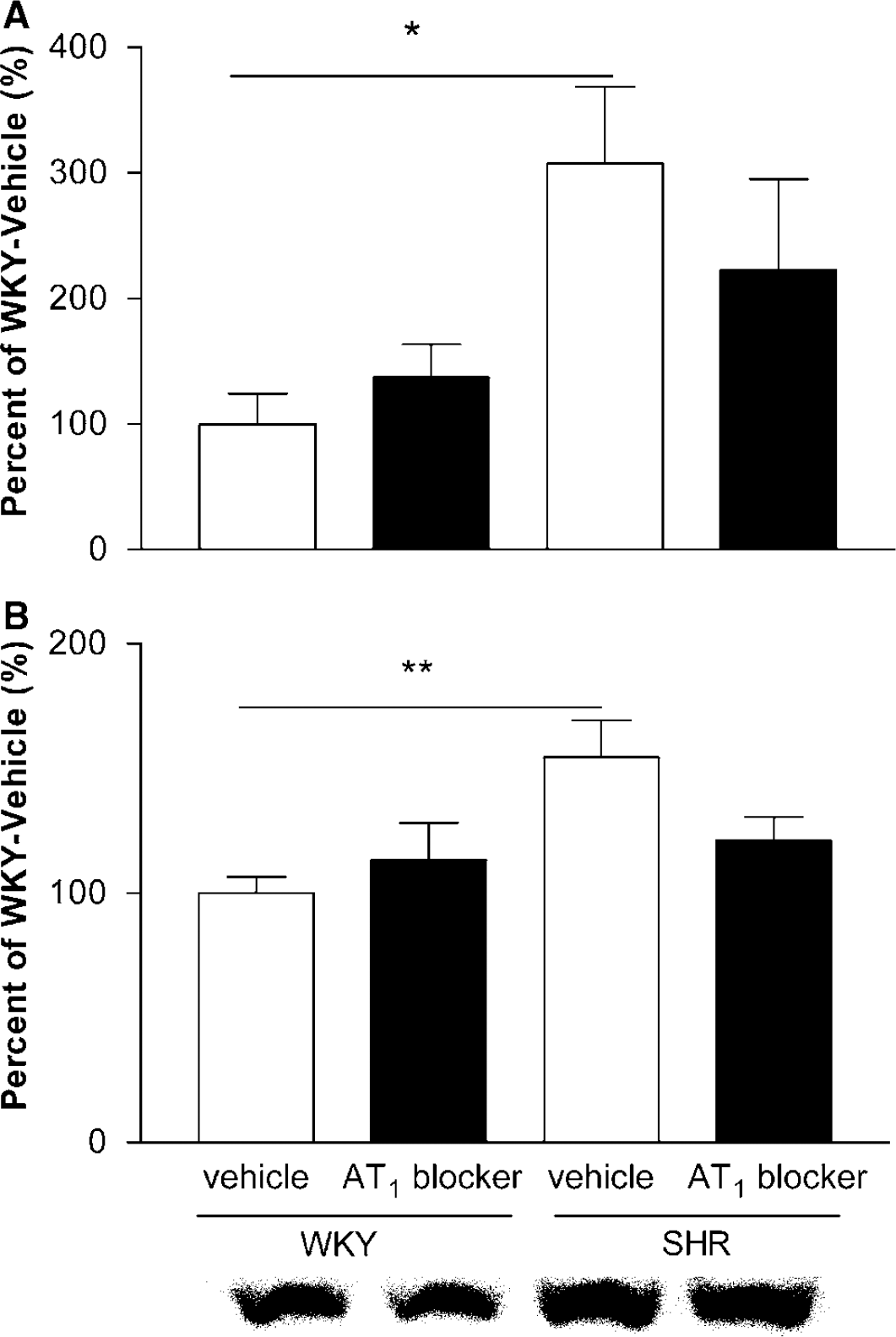
Expression of AT1 receptors in brain microvessels from SHR and WKY rats. The expression of AT1 receptor mRNA (
AT1 Receptor Blockade Decreases Perivascular Macrophage Infiltration in Hypertensive Rats
In WKY rats, a few perivascular resident macrophages were visualized by ED2-positive staining as flattened and elongated cells closely applied to and never separated from the microvascular wall (7.3 ± 2.0/mm2). The number of ED2-positive perivascular macrophages was increased in SHR when compared with WKY rats. In addition to perivascular resident macrophages, in SHR we observed many rounded infiltrating macrophages detached from the vessel wall (14.5 ± 2.1/mm2, P<0.05). AT1 receptor blockade decreased the number of infiltrating macrophages in SHR (8.7 ± 0.9/mm2, P<0.05). Data refer to Ando et al (2004). The representative pictures were shown in Figure 3.
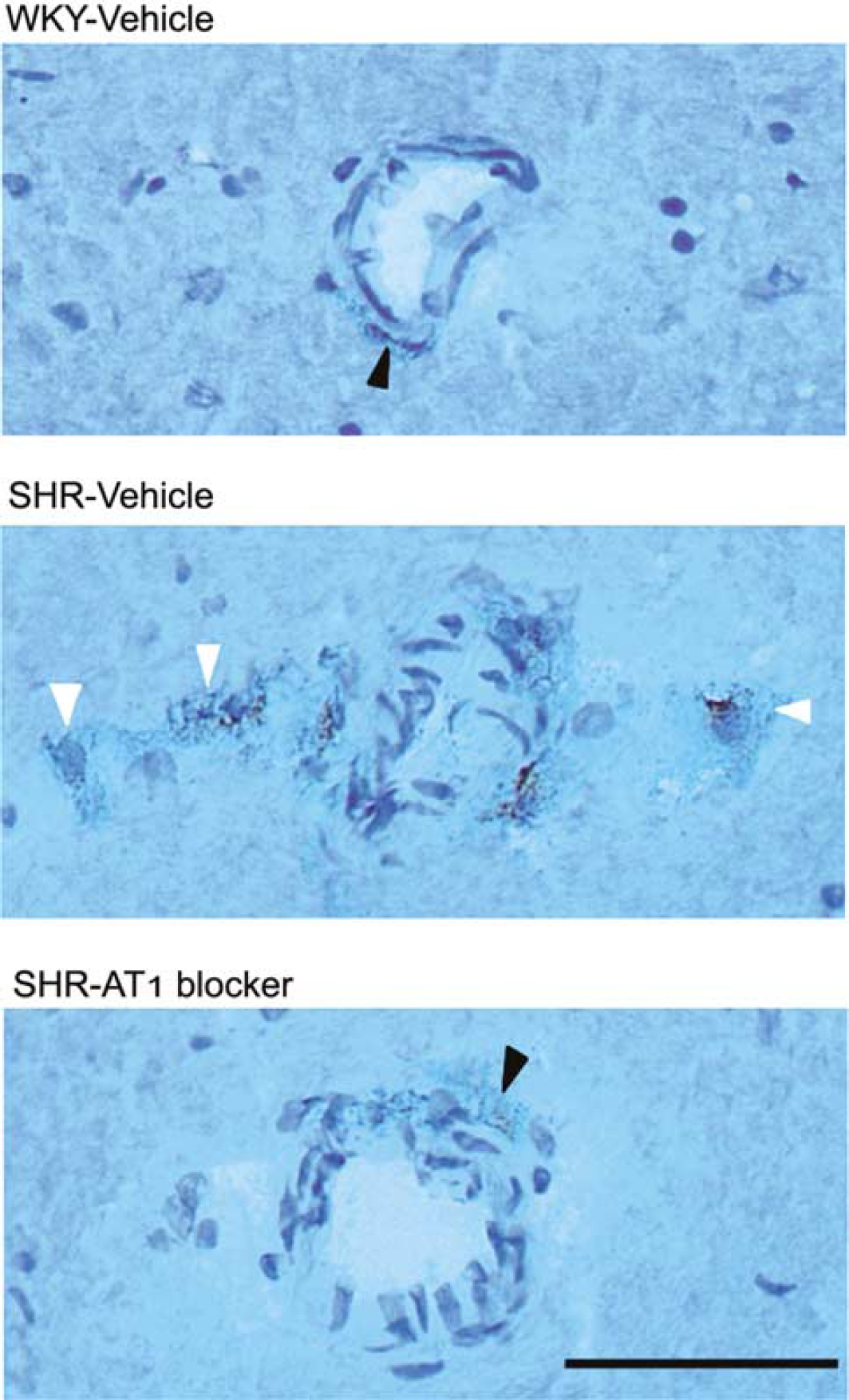
Effect of AT1 receptor blockade on perivascular macrophage infiltration in brain microvessels. Macrophages were identified with an antibody against ED2. Black arrowheads point to flattened resident macrophages, closely applied to the microvessel wall. White arrowheads point to round, infiltrating macrophages detached from the vessel wall. Infiltrating macrophages were rare in microvessels from WKY rats, numerous in microvessels from SHR and reduced in SHR after treatment with the AT1 receptor antagonist. Bar = 20μm.
AT1 Receptor Blockade Inhibits the Expression of Inflammatory Factors and Heat Shock Proteins
We examined the transcription factor NF-κB p50 subunit precursor p105 and the proinflammatory cytokines TNF-α and IL-1β mRNA by real-time PCR. The expressions of NF-κB p105, TNF-α and IL-1β mRNA were higher in SHR than in WKY rats, and they were normalized by AT1 receptor blockade (Figure 4). The expression of phospho-NF-κB p65 subunit was also higher in SHR and reversed by AT1 blocker treatment (Figure 8). Phospho-NF-κB p65 subunit, TNF-α and IL-1β were localized by immunohistochemistry on the endothelium of cerebral microvessels (Figures 7 and 8).
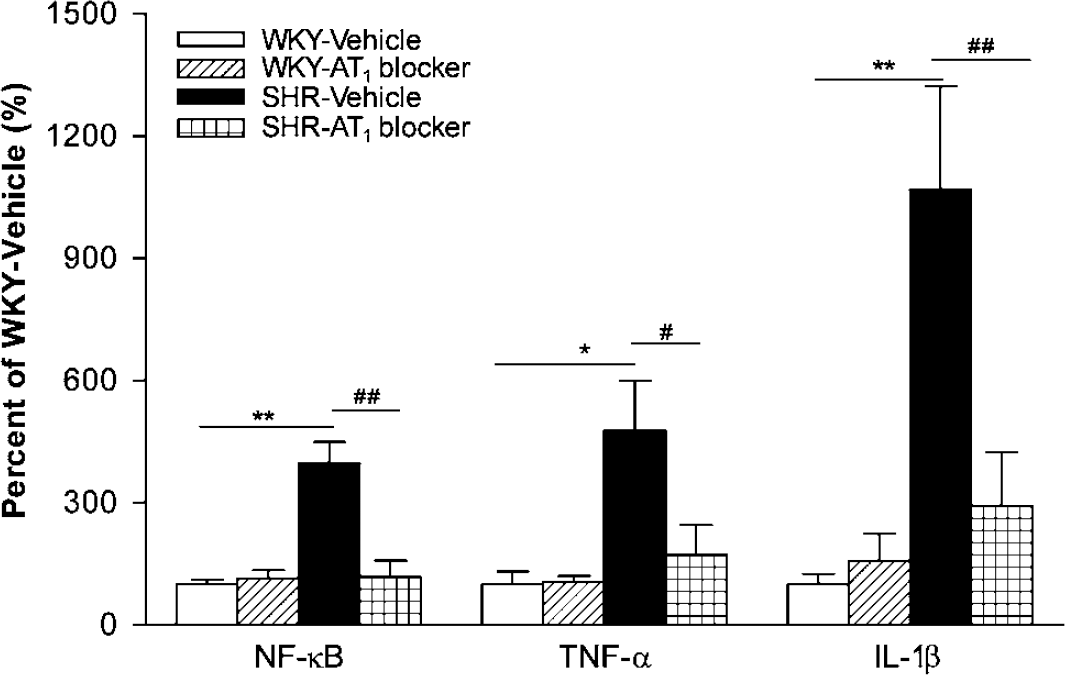
Effect of AT1 receptor blockade in expression of inflammatory factors in brain microvessels. The expression of NF-κB, TNF-α and IL-lβ mRNA in brain microvessels from SHR was higher than that in WKY rats, and was normalized after treatment with an AT receptor blocker (n = 4 to 6 per group). *P<0.05, **P<0.01 versus WKY-Vehicle; #P≪0.05, ##P<0.01 versus SHR-Vehicle.
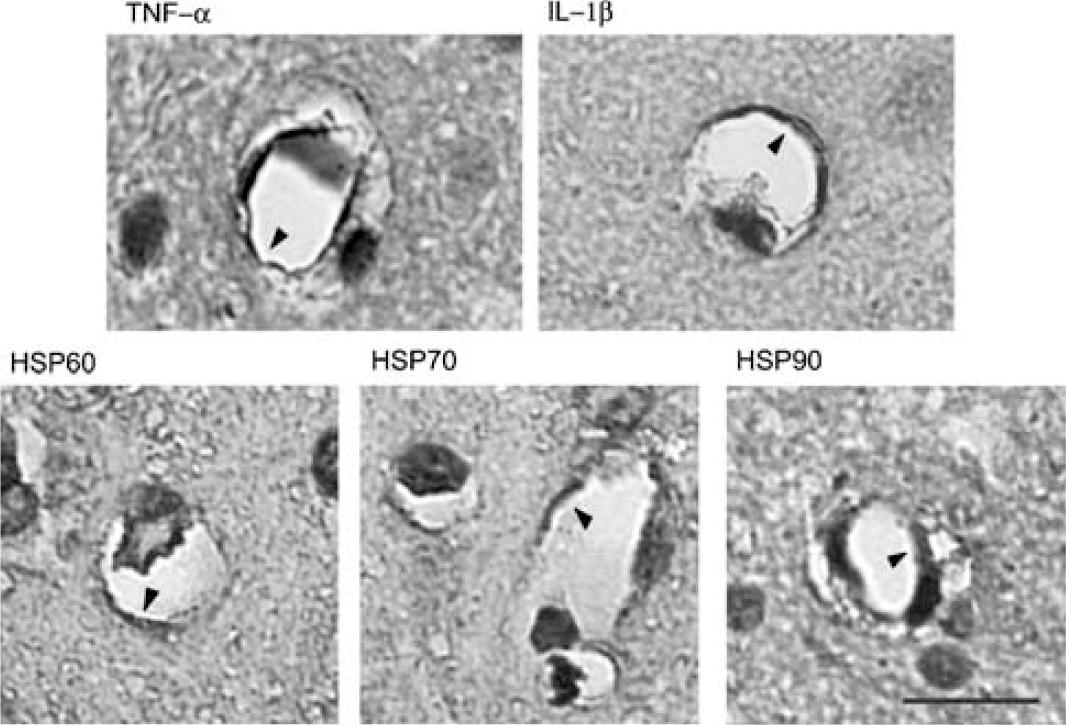
Immunocytochemical localization of TNF-α, IL-1β and HSPs in cerebral microvessels. Arrowheads point to the localization of TNF-α, IL-1β, HSP60, HSP70 and HSP90 in the endothelium of cerebral microvessels from SHR. Bar = 10 μm.
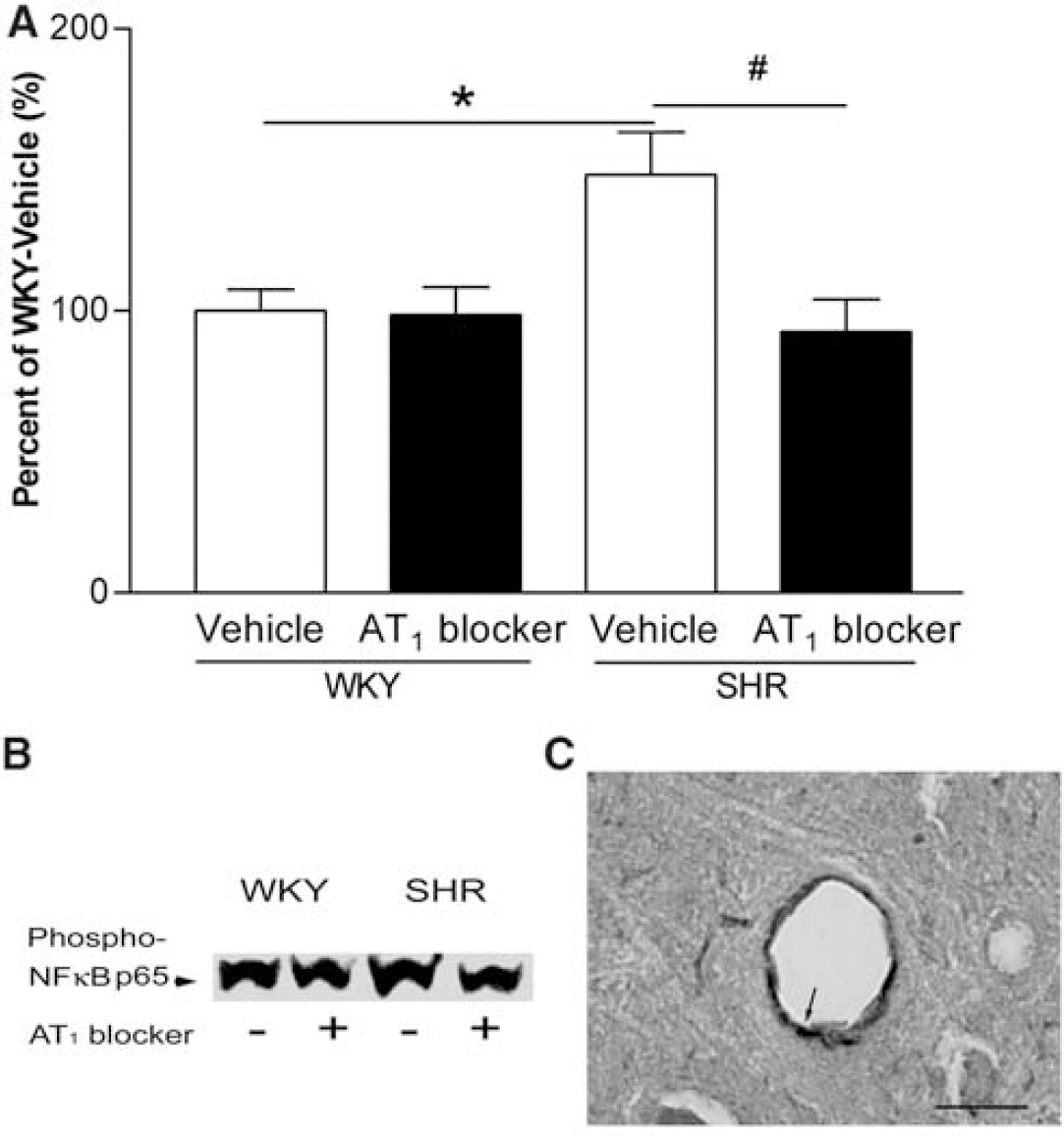
Effect of AT1 receptor blockade on expression of phosphorylated NF-κB p65 subunit in brain microvessels. (
Using the Affymetrix Rat Genome U34A arrays, we found that transcription of many HSP family genes was higher in microvessels from SHR when compared with WKY rats. Treatment with the AT1 antagonist decreased transcription of some of these genes in SHR, in WKY rats or in both strains (Table 2). Using real-time PCR and Western blotting, we confirmed the increased expression of HSP60, HSP70, HSP90 mRNA and protein in SHR and their normalization after AT1 receptor blockade. The expression of HSF-1 protein and mRNA was also increased in SHR, and normalized by AT1 receptor blockade (Figures 5 and 6). HSP60, HSP70 and HSP90 were localized to the microvessel endothelium (Figure 7) and revealed increased expression in SHR when compared with WKY rats by immunohistochemistry (results not shown) (Figure 8).
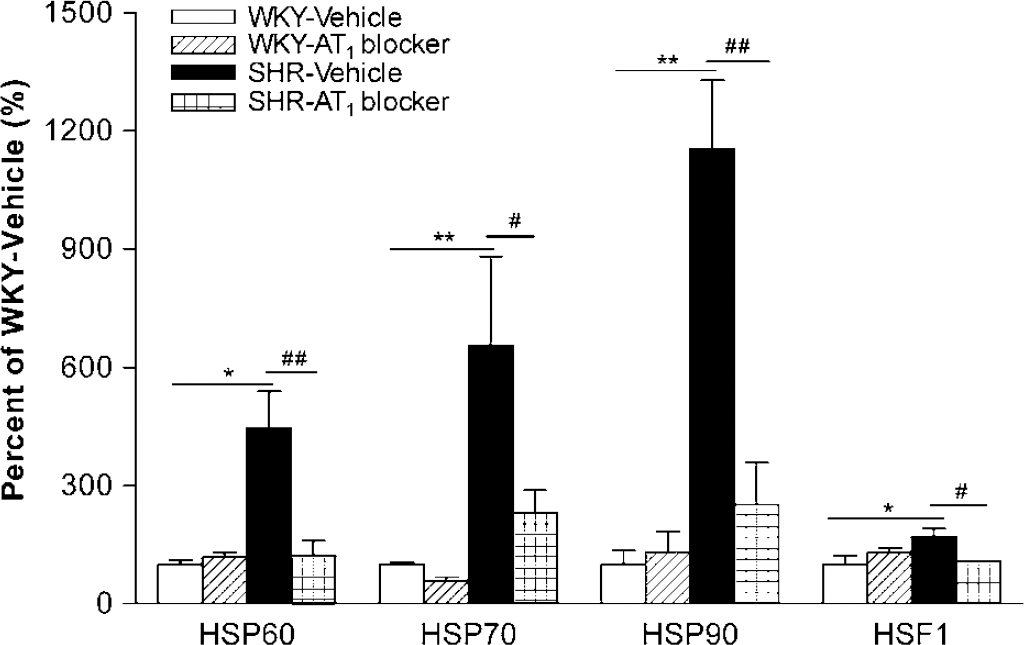
Effect of AT1 receptor blockade on HSPs and HSF-1 mRNA in cerebral microvessels. The expression of HSP60, HSP70, HSP90 and HSF-1 mRNA in brain microvessels from SHR was higher than that in WKY rats, and was normalized after AT1 receptor blockade (n = 4 to 6 per group). *P<0.05, **P<0.01 versus WKY-Vehicle; #P<0.05, ##P<0.01 versus SHR-Vehicle.
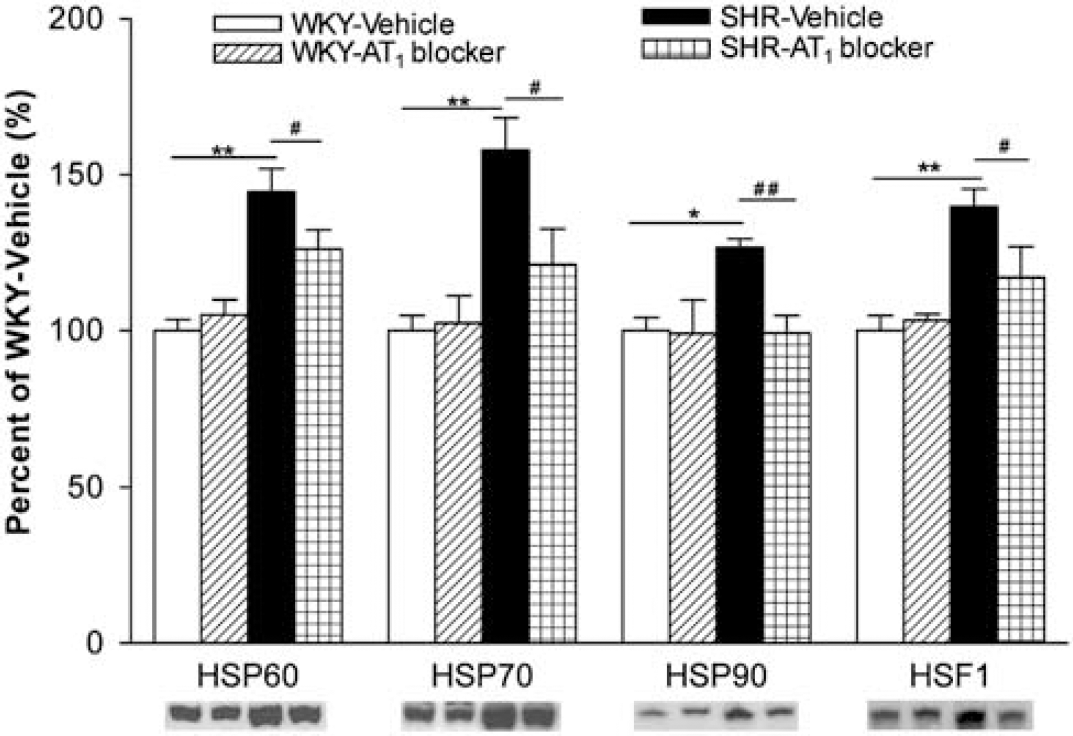
Effect of AT1 receptor blockade on HSPs and HSF-1 protein in cerebral microvessels. Expression of HSP60, HSP70, HSP90 and HSF-1 in brain microvessels from SHR was higher than that in WKY rats, and was normalized after treatment with the AT1 receptor antagonist (n = 6 to 8 per group). *P<0.05, **P<0.01 versus WKY-Vehicle; #P<0.05, ##P<0.01 versus SHR-Vehicle.
Heat shock protein genes in SHR and WKY rats with and without treatment with the Angiotensin II AT1 receptor antagonist
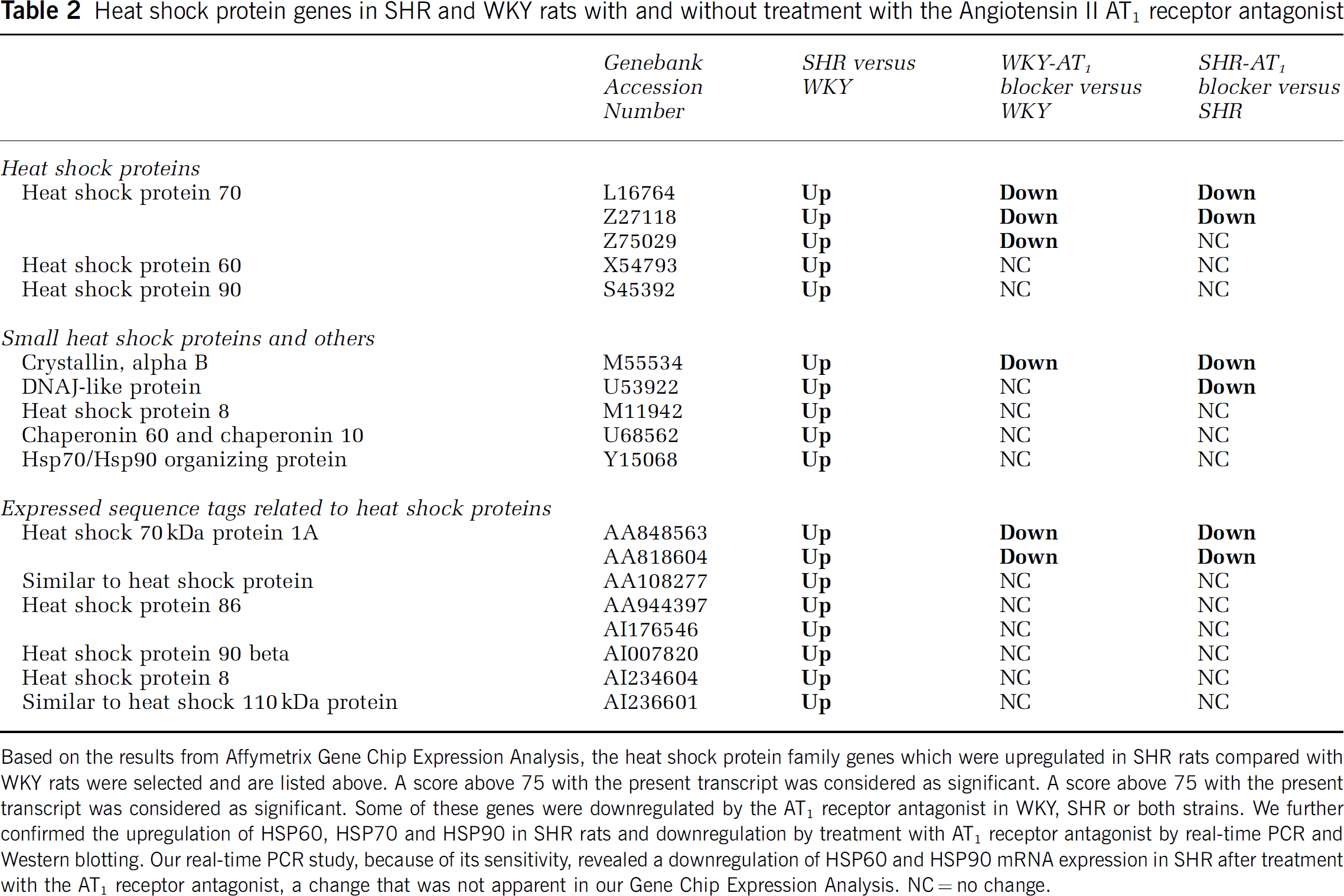
Based on the results from Affymetrix Gene Chip Expression Analysis, the heat shock protein family genes which were upregulated in SHR rats compared with WKY rats were selected and are listed above. A score above 75 with the present transcript was considered as significant. A score above 75 with the present transcript was considered as significant. Some of these genes were downregulated by the AT1 receptor antagonist in WKY, SHR or both strains. We further confirmed the upregulation of HSP60, HSP70 and HSP90 in SHR rats and downregulation by treatment with AT1 receptor antagonist by real-time PCR and Western blotting. Our real-time PCR study, because of its sensitivity, revealed a downregulation of HSP60 and HSP90 mRNA expression in SHR after treatment with the AT1 receptor antagonist, a change that was not apparent in our Gene Chip Expression Analysis. NC = no change.
Discussion
The protein and mRNA AT1 receptor expression was higher in SHR when compared with WKY rats. The AT1 receptors were localized in endothelial cells in both strains and in smooth muscle cells of arterioles in SHR in our brain microvessel preparation. This suggested enhanced cerebrovascular Ang II tone in genetic hypertension. AT1 receptors mediate Ang II internalization and transport, protecting the peptide from degradation and facilitating its intracellular effects (Rose and Audus, 1998, 1999). AT1 receptor blockade, by inhibiting Ang II effects and preventing its internalization, antagonizes the effects of Ang II in the cerebral vasculature. AT1 receptor mRNA and protein did not change in WKY rats, and only partially but not significantly decreased in SHR after treatment with the AT1 receptor antagonist. This indicated that blockade of AT1 receptors did not significantly affect receptor synthesis.
We chose to treat the animals with the AT1 receptor antagonist candesartan for 4 weeks, a treatment previously shown to reverse cerebrovascular remodeling and experimental ischemic injury (Ito et al, 2002; Yamakawa et al, 2003; Nishimura et al, 2000) and to reverse the overexpression of ICAM-1 and the infiltration of macrophages in brain microvessels from SHR (Ando et al, 2004). It is well known that NF-κB family proteins play a pivotal role in inflammation and immune response (Dandona et al, 2003). The best understood heterodimer (p65/p50) is sequestered in the cytoplasm by its inhibitor proteins (IκBs) in most cells. In response to proinflammatory signals, IκB kinase (IKK) rapidly phosphorylates the IκBs. The phosphorylated IκBs are then rapidly degraded via the ubiquitin-proteasome pathway, thereby releasing NF-κB to enter the nucleus and regulate gene transcription. The p65 subunit is also phosphorylated by IKK, along with phosphorylation and degradation of IκBs in the cytokine-induced NF-κB activation pathway (Sakurai et al, 1999). We found a strong inflammatory reaction in brain microvessels from SHR, including a higher expression of the phospho-NF-κB p65 protein and NF-κB, TNF-α and IL-1β mRNA when compared with normotensive WKY rats. In addition, confirming our previous findings (Ando et al, 2004), there were large numbers of infiltrating macrophages from SHR, which were absent in normotensive animals. AT1 receptor antagonism decreased the expression of NF-κB, TNF-α and IL-1β and eliminated the macrophage infiltration in cerebral microvessels from SHR.
Our results support recent findings (Suzuki et al, 2001; Takemori et al, 2000; Ando et al, 2004), indicating that the strong inflammatory response via NF-κB pathway in cerebral microvessels of genetically hypertensive rats is dependent on Ang II stimulation, and can be suppressed by blockade of its AT1 receptors. Reversal of cerebrovascular inflammation by blockade of the Ang II system could be an important additional mechanism of protection against ischemic brain injury, as is the case in peripheral organs (Hilgers et al, 2001; Dandona et al, 2003). These antiinflammatory effects can explain some of the underlying mechanisms of the decrease in cardiovascular morbidity and mortality, which follow a 7-day course of candesartan when the treatment is administered during the first week after acute stroke (Schrader et al, 2003).
The administration of candesartan, as performed here, normalizes blood pressure in SHR and slightly lowers blood pressure in WKY. However, several lines of evidence indicate that the antiinflammatory effect of AT1 receptor antagonists is not related to the blood pressure decrease. We reported that prevention of cerebrovascular remodeling and protection against ischemia by AT1 antagonists are not associated with the decrease in blood pressure but dependent on the blockade of the Ang II system, since beta-adrenergic or calcium-blocking agents are not effective (Nishimura et al, 2000; Ito et al, 2002). Treatment of candesartan at a low dosage of 0.1 mg/kg reduced the stroke incidence and urinary protein excretion without affecting the blood pressure (Inada et al, 1997). In peripheral organs, the antiinflammatory effect of AT1 antagonists, in hypertensive rats and in patients, are independent of their effects on blood pressure (Tokuda et al, 2004; Dohi et al, 2003).
Because alterations in HSP expression are relevant to hypertension and inflammation, we studied the changes of HSP family genes using DNA microarray. The GeneChip we used provides a large coverage of the rat gene expression, which enables us to understand many signaling pathways and molecular mechanisms and to study different hypothesis-driven questions. We could answer specific questions: What is the expression of HSPs in microvessels from hypertensive rats? If altered, does AT1 receptor antagonism normalize the HSP response? What is the effect of treatment with the AT1 antagonist on inflammatory factors? Does it correlate with prevention of macrophage infiltration?
Our analysis revealed that many genes of the HSP family were upregulated in SHR. We found the expression of HSP60, HSP70 and HSP90 in the microvessel endothelium, and confirmed the gene upregulation and protein overexpression of HSP60, HSP70 and HSP90 in microvessels of SHR. The expression of HSF-1 and that of its mRNA were also increased in SHR. The overexpression of HSPs and HSF-1 protein and mRNA was normalized by treatment with the AT1 receptor antagonist. Our results are consistent with the observation that long-term administration of Ang II induces renal expression of HSPs by mechanisms unrelated to hypertension and dependent on AT1 activation (Ishizaka et al, 2002).
Heat shock proteins are intracellular signaling molecules inducing a range of proinflammatory responses including secretion of adhesion molecules and cytokines (Wallin et al, 2002). Members of the HSP family are able to stimulate cells of the innate immune system directly and act as dangersignaling molecules (Hilgers et al, 2001). Proinflammatory cytokines induce activation of HSF1-DNA binding followed by induction of HSP70 expression (Schett et al, 1998). The induction of HSPs previously primed by inflammation accelerates cell death by apoptosis (Buchman et al, 1993; Abello and Buchman, 1994). Heat shock protein 70 bound with high affinity to the plasma membrane elicited a rapid intercellular calcium flux, activated NF-κB and upregulated the expression of proinflammatory cytokines (Asea et al, 2000). In human endothelial cells, HSP60 triggered activation of NF-κB complexes, induced E-selectin, ICAM-1 and VCAM-1 expression similar to levels induced by Escherichia coli lipopolysaccharide (Kol et al, 1999). In this case, reversal of increased HSP and HSF-1 expression by AT1 receptor blockade might be considered an important mechanism to prevent inflammation, cell damage and apoptosis in brain microvessels, contributing to the end-organ protective effect of Ang II system blockade. Alternatively, HSPs can exert powerful protective effects against subsequent stress and inflammation (Pockley, 2003; Schett et al, 1998). Transgenic mice overexpressing rat HSP70 showed reduced infarction and neuronal injury after ischemia and seizures (Tsuchiya et al, 2003). The enhanced HSP expression in cerebral microvessels of SHR observed here could also be interpreted as a cytoprotective mechanism no longer needed after inflammation is reversed by AT1 receptor antagonism.
Our results indicate a tonic proinflammatory action of Ang II in cerebral microvessels during hypertension, through endothelial AT1 receptor stimulation, leading to increased expression of proinflammatory cytokines and enhanced HSP response. The AT1 receptor antagonist candesartan has very potent antiinflammatory properties in brain microvessels. In addition to their antihypertensive and antigrowth properties, the cerebrovascular antiinflammatory effects of Ang II AT1 receptor antagonists might be of major importance to protect the brain against neuronal damage due to ischemia. These compounds might be potential therapeutic agents against other inflammatory diseases of the brain.
Footnotes
Acknowledgements
The authors thank Dr AG Elkahloun (Microarray Unit, National Human Genome Research Institute) and Dr Zu-Xi Yu (National Heart, Lung and Blood Institute) for the technical support on microarray and confocal microscopy. Competing interests statement: The authors declare that they have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.