
Abstract
This study investigated the expression of deiodinases of thyroid hormones in the rat brain after transient occlusion of the middle cerebral artery. The activity of type 2 deiodinase (D2), which catalyzes the deiodination of thyroxine into the more active thyroid hormone 3,5,3′-triiodothyronine, was strongly increased by cerebral ischemia at 6 and 24 hours in the striatum and at 24 hours in the cerebral cortex. The activity of type 3 deiodinase, which catalyzes the inactivation of thyroid hormones, was not affected by ischemia. In situ hybridization showed, as soon as 6 hours, an upregulation of the expression of D2 mRNA in the ipsilateral striatum, which disappeared at 24 hours. In the ipsilateral cortex, the induction of D2 mRNA started at 6 hours, was increased at 24 hours and finally declined at 72 hours. These results were confirmed by reverse transcription-PCR for D2 mRNA in the striatum and cerebral cortex. The upregulation of D2 mRNA after ischemia was mainly localized in astrocytic cell bodies. These results show that D2 is rapidly induced in astrocytes after ischemic stroke. Future work will include the exploration of the role of the upregulation of this enzyme, responsible for local 3,5,3′-triiodothyronine production as a neuroprotective mechanism in the brain.
Introduction
The major role of thyroid hormones has been well established for central nervous system development (for a review, see Bernal, 2002). Severe hypothyroidism during the neonatal period leads to structural alterations, including hypomyelination and defects of cell migration and differentiation, with long-lasting, irreversible effects on behavior and performance. These irreversible effects on development are classically opposed to reversible changes promoted by thyroid hormones in brain function in the adult. However, some reports have suggested that thyroid hormones contribute to the reduction of neurologic deficits after injury. For example, thyroid hormones seem to be essential for peripheral nerve repair by acceleration of axonal regeneration and neuromuscular reinnervation (for a review, see Barakat-Walter, 1999). Thyroid hormones would also promote axonal growth in the spinal cord and brain of adult animals (Harvey and Srebnik, 1967; Fertig et al, 1971; Guth, 1974; Heinicke, 1977). Thyroid hormones rescue the axotomized sensory neurons in dorsal root ganglia (Schenker et al, 2003). The protective role of 3, 5, 3′-triiodothyronine (T3) against apoptosis has been shown in culture of cerebellar granule neurons (Muller et al, 1995). In vivo, thyroxine (T4) administration attenuates the hippocampal neuronal damage caused by ischemia in the rat (Rami and Krieglstein, 1992) and provides protection against global ischemia after cardiac arrest and resuscitation in the dog (D'Alecy, 1997). The cerebral T3, which is classically considered as the biologically active hormone, is essentially generated through local conversion of T4 into T3 by the cerebral deiodinase of type 2 (D2) (for a review, see Bianco et al, 2002). Cerebral T3 concentration is also dependent on the activity of another enzyme, the deiodinase of type 3 (D3), which catalyzes the deiodination of thyroid hormones into inactive metabolites (for a review, see Bianco et al, 2002). Interestingly, intravenous T3 infusion failed to improve, in contrast to T4 infusion, neurologic outcome in the dog after global ischemia (D'Alecy, 1997). This observation may indicate that, also after injury, cerebral T3 is produced by the deiodination of cerebral T4 catalyzed by D2 and does not derive from the circulation. It thus appears of interest to explore if cerebral ischemia affects D2 and D3 expression, which may contribute to the protective mechanism against brain insult. In this way, an upregulation of D2 mRNA has been shown near the contusion site by in situ hybridization 3 days after traumatic brain injury in the rat (Zou et al, 1998). In the present work, we followed D2 and D3 activities in the cortex and striatum as soon as 6 hours after transient cerebral ischemia in the rat. We also studied the expression of D2 mRNA by in situ hybridization and identified its cellular localization. Our results show that D2 is rapidly induced in astrocytes after brain ischemia.
Materials and methods
Animals
All experiments were performed with male Sprague–Dawley rats (Iffa Credo, l'Arbresle, France) weighing 300 to 350 g in accordance with the French decree A 94120 (1991) and associated guidelines of the EEC directive 86/609/EEC (1986).
Induction of Transient Focal Cerebral Ischemia
Artery occlusions were performed according to procedures described previously (Lerouet et al, 2002). Rats were anesthetized with chloral hydrate (400 mg/kg, i.p., Prolabo, Fontenay-sous-Bois, France) and allowed to breathe spontaneously. The left middle cerebral artery (MCA) was exposed through a temporal craniotomy and occluded with a microclip (zen type temporary clip, 13 × 0.4 mm, Ohwa Tsusho, Tokyo, Japan), and both the left and right common carotid arteries (CCAs) were concomitantly clamped. The microclip was placed on the MCA at a site proximal to the lenticulostriate arteries to produce cortical and striatal infarctions, and kept in place for 20mins. The clip was then removed and recirculation within the CCAs was allowed. Reperfusion in the three arteries was checked under a microscope. The same surgery was performed in sham-operated rats but the MCA and the CCAs were not occluded. The rat body temperature was monitored throughout surgery by a rectal probe, and maintained at 37°C to 38°C with a normothermic blanket control unit (Harvard apparatus, Edenbridge, Kent, UK). After surgery, rats were returned to their home cage and fed with mashed lab food.
RNA Preparation
The ipsilateral striatum and parietal cerebral cortex were aseptically collected and immediately frozen in liquid nitrogen and stored at −80°C until RNA extraction. Samples were ground into powder in a mortar precooled with liquid nitrogen. Then, total RNA was extracted using TRIzol Reagent (Life Technologies, NY, USA) according to the instructions of the manufacturers and quantified by 260 nm absorption measurement.
Semi-Quantitative Reverse Transcription-PCR
Reverse transcription (RT) was performed for 1 hour incubation, at 42°C, with 1 μg of total RNA as template for each sample in a 20 μL reaction volume of reverse transcriptase buffer containing 10 mmol/L DTT, 0.5 mmol/L dNTPS mix and 200 ng of random primers, 20 U RNase inhibitor and 200 U MMLV reverse transcriptase (Life Technologies, NY, USA). Then, an appropriate volume of the same RT was used in subsequent D2 and S26 ribosomal protein PCR experiments. The PCR reaction was performed in 50 μL Expand High Fidelity buffer containing 1.5 mmol/L MgCl2, 0.2 mmol/L dNTPs, 0.1 μCi [α-32P]-dCTP (Amersham Pharmacia Biotech, Buckinghamshire, UK), 0.4 μmol/L of sense and antisense primers, 2.6 U Expand High Fidelity PCR System (Roche, Mannheim, Germany). The oligonucleotide primers were derived from the coding regions of rat D2 cDNA (Croteau et al, 1996) and of rat S26 ribosomal protein cDNA, which were expected to give 590 and 261 base-pair products, respectively. Primers for D2 were: sense, 5′-TTC AAA GGC TAC CCC ATA AG-3′, and antisense, 5′-ACT CGG TCA TTC TGC TCA AG-3′. Primers for S26 were: sense, 5′-GTG CGT GCC CAA GGA TAA GG-3′; and antisense, 5′-ATG GGC TTT GGT GGA GGT CG-3′. The conditions of amplification for D2 were: 2 mins at 94°C, followed by 30 cycles of denaturation (94°C, 1 min), annealing (58°C, 1min) and extension (72°C, 1min), followed by a final extension at 72°C for 2 mins. The conditions of amplification for S26 were: 2 mins at 94°C, followed by 25 cycles of denaturation (94°C, 1 min), annealing (64°C, 1min) and extension (72°C, 1 min), followed by a final extension at 72°C for 2 mins. Amplification products were separated on a 0.8% agarose gel and then transferred to a positively charged nylon membrane (Roche, Mannheim, Germany), exposed to Kodak X-ray film. The quantity of PCR product formed, which was evaluated by the quantity of incorporated radiolabelled nucleotide using an Instant Imager (Packard Instrument Co., Meridian, CT), increased in a linear manner as a function of the cDNA amount obtained from RT, until 100 ng for D2, and S26 ribosomal protein PCR. Reactions without template cDNA or with RNA extracts without RT were run as negative controls.
D2 and D3 Activity Assays
Striatum and parietal cortex from ipsilateral and contralateral sides were homogenized with a glass homogenizer at 4°C in 450 μL of 80 mmol/L β-glycerophosphate buffer, pH 7.4, containing 15 mmol/L MgCl2, 20 mmol/L EGTA, protease inhibitors (1 mmol/L phenylmethylsulfonyl fluoride, 50 μg/mL aprotinin, 4 μg/mL leupeptin, 10 μg/mL antipain, 1 mmol/L trypsin inhibitor, 1 mmol/L benzamidin, 10 μg/mL pepstatin) and 1 mmol/L of the phosphatase inhibitor orthovanadate (Na3VO4). Aliquots of homogenates were immediately frozen and kept at −80°C. The protein concentration of tissue homogenates was determined by the method of Bradford (1976) using bovine serum albumin as the standard. For D2 activity assays, homogenates with the same amount of protein (40 μg) were incubated at 37°C for 60 mins, in a final volume of 80 μL containing 20 mmol/L HEPES buffer (pH 7.4), 20 mmol/L dithiothreitol, 50 nmol/L T3 and 1 nmol/L [125I]T4 (Amersham Pharmacia Biotech, Buckinghamshire, UK). Reactions were stopped by adding 10 μL of NH4OH (10 mol/L) containing 10 μmol/L T3 and 10 μmol/L. The [125I]T3 produced was separated from [125I]T4 by descending paper chromatography (Courtin et al, 1986). Then, the radioactive products were counted for determination of D2 activity expressed as fmol T3/min mg proteins. Deiodination was linear regarding to both protein concentration and incubation time. For D3 activity assays, homogenates with the same amount of protein (40 μg) were incubated at 37°C for 60 mins, in a final volume of 80 μL containing 20 mmol/L HEPES buffer (pH 7.4), 20 mmol/L dithiothreitol and 5 nmol/L [125I]T3 (Amersham Pharmacia Biotech, Buckinghamshire, UK). Reactions were stopped by adding 10 μL of NH4OH (10 mol/L) containing 10 μmol/L T3 and 10 μmol/L T4. The [125I]3,3′T2 produced was separated from [125I]T3 by descending paper chromatography (Courtin et al, 1986). Then the radioactive products were counted for determination of D3 activity expressed as fmol 3,3′T2/min mg proteins. Deiodination was linear regarding both protein concentration and incubation time.
In Situ Hybridization Histochemistry and Immunohistochemistry
Tissue processing
Anesthesia was induced by pentobarbitone (60 mg kg, IP) and the animals were perfused transcardially with 1% paraformaldehyde in PBS. Brains were dissected out and postfixed in the same solution for 1 hour at 4°C and cryoprotected in the 1% paraformaldehyde solution containing 15% sucrose changed after 1 and 5 hours. They were frozen after a day in dry ice and 14-mm coronal sections were obtained in a cryostat. After drying at room temperature, slices were stored at −70°C.
In situ hybridization for the detection of D2 mRNA was performed on tissue sections. Specific D2 sense and antisense riboprobes were synthetized in the presence of [35S]UTP and [35S]CTP using a 371 bp DNA template spanning nucleotides 537 to 907 from the rat D2 cDNA sequence (Croteau et al, 1996). This area contains little sequence homology with the rat type 1 deiodinase and D3 mRNAs. After alkaline hydrolysis at 60°C in lysis buffer (40 mmol/L NaHCO3, 60 mmol/L Na2CO3, pH 10.2) stopped after 20 mins by acidic buffer (NaAc 0.1 mol/L, 5% acetic acid, pH 4.5), the riboprobes were purified by quick spin G50 column (Roche Diagnostics GmbH, Mannheim, Germany) precipitated with yeast tRNA in ethanol–LiCl 8 mol/L, and suspended in a Tris-EDTA buffer containing 50 mmol/L DTT.
The 14-μm tissue sections from each animal were postfixed in 1% paraformaldehyde (5 minutes) and washed in two successive 4 × SSC baths (1 × SSC is 0.15 mol/L NaCl and 0.015 mol/L Na citrate). Thereafter, the sections were washed in a 0.1 mol/L triethanolamine buffer (pH 8.0) for 3 minutes, acetylated with 0.25% acetic anhydride in 0.1 mol/L triethanolamine buffer (pH 8.0) for 10 minutes and washed in four successive baths containing 70% to 100% ethanol for 1 minute each before drying. Hybridization was performed at 50°C for 16 hours in a hybridization buffer containing 40% formamide, 8% dextran sulfate, 0.8 × Denhard's solution, 3.2 × SSC, 10 mmol/L Na2HPO4, 400 μg/mL salmon sperm DNA, 800 μg/mL yeast tRNA, 100 mmol/L DTT, SDS 0.1%, and thiosulfate 0.1% with 35S-labelled riboprobes at 2 × 107cpm/mL. Usually, 100 μL of buffer was added on each coronal section and coverslipped with hybrislip (VWR, Strasbourg, France). Hybrislip and excess probe were removed in two baths (2 × SSC containing 1 mmol/L DTT) of 5 mins at 4°C, then a bath in the same buffer for 30 mins was performed at room temperature, followed by incubation with RNAse A (20 μg/mL) in 10 mmol/L Tris–HCl, pH 7.5, 0.5 mol/L NaCl, and 1 mmol/L DTT at 37°C for 10 mins. After washing in two successive baths (2 × SSC containing 1 mmol/L DTT) at room temperature, stringency washes were performed in 50% formamide 2 × SSC containing 1 mmol/L DTT at 60°C for 1 hour, then in 2 × SSC containing 1 mmol/L DTT at 60°C for 15 mins, then in 0.1 × SSC containing 1 mmol/L DTT at 60°C, and then in 0.1 × SSC containing 1 mmol/L DTT at room temperature.
To analyze the cell type that expresses D2 mRNA, a combination of in situ hybridization histochemistry and immunohistochemistry was performed on the same tissue section using a double-labelling technique inspired by that used by Guadano-Ferraz et al, 1997. Briefly, after hybridization and washes, the sections were incubated in 1% horse serum with the monoclonal antibody against Glial Filament Acidic Protein (GFAP) mouse antibody (1/1000; Sigma Chemical Co., St Louis, MO) overnight at 4°C, and then with a biotinylated secondary antimouse IgG (H+L) rat absorbed affinity purified (1/200; Vector Laboratories, Burlingame, CA, USA) in 1% horse serum for 2 hours at room temperature, followed by avidin–biotin–peroxidase complex (ABC) from Elite kit (Vector laboratories, Burlingame, CA). Peroxidase was then visualized with diaminobenzidine (0.05%) and H2O2 (0.03%). Sections were air-dried and exposed to Biomax MR-1 film (Kodak) for 1 or 2 weeks. For cellular resolution, the sections were dipped in Ilfold photographic emulsion K5 (Ilford scientific product, Mobberley, UK), exposed for 3 weeks to the cold, developed, fixed, dehydrated and coverslipped. When only in situ hybridization was performed, the sections were counterstained with toluidine blue. Optical observations were made with a Zeiss microcope (Carl Zeiss, Oberkochen, Germany).
Quantitative analysis
After film scanning, the autoradiographic signals were quantified using the NIH image program, version XSm in a Macintosh computer (Apple computer Inc., Cupertino, CA). Data are expressed as the optical density obtained from different brain regions after subtracting the film background. The value for each brain region of individual animals was recorded as the average of four measurements taken from four vicinus sections.
Results
Measurement of deiodinase activities shows an important increase in D2 activity (three-fold) in ipsilateral striatum as soon as 6 hours after transient focal cerebral ischemia (Figure 1A; left panel). D2 activity remained elevated at 24 hours. In the parieto-frontal cortex, D2 activity was strongly increased (six-fold) at 24 hours after ischemia (Figure 1B; left panel). No D2 upregulation was observed in the contralateral striatum and cortex of ischemic animals or in the ipsilateral striatum and cortex of sham-operated animals (Figures 1A and 1B; left panel). D3 activity was not affected by ischemia either in the striatum or parieto-frontal cortex (Figures 1A and 1B; right panel).
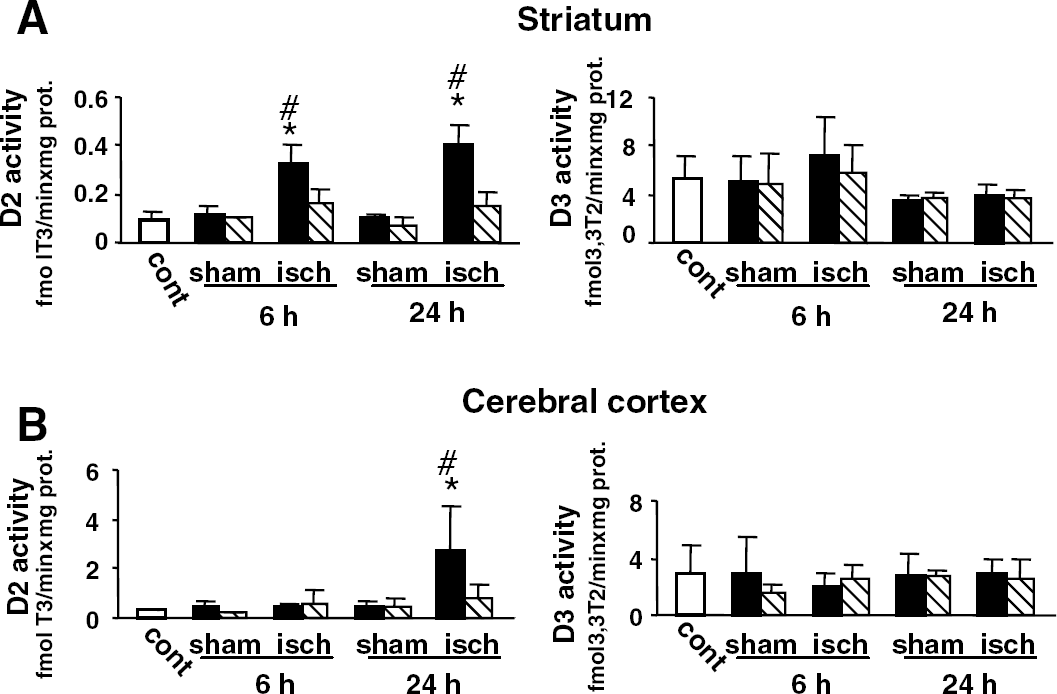
Effects of transient focal cerebral ischemia on the activities of D2 and D3 in the striatum and the cerebral cortex. D2 activities (left) and D3 activities (right) were measured as described in Materials and methods in homogenates of the striatum (
To determine the localization of D2 mRNA expression, in situ hybridization was performed on anterior coronal sections with a riboprobe similar to the one used to characterize D2 mRNA expression in the brain by in situ hybridization for the first time (Guadano-Ferraz et al, 1997). As shown in Figure 2, the results for uninjured control rats agree closely with those previously reported (Guadano-Ferraz et al, 1997), with D2 mRNA expression in the dorsal portion of the lateral septal nucleus, the caudate-putamen nucleus (striatum) and the cerebral cortex with main localization in layers I to IV (Figure 2F). Sham operation did not alter the D2 mRNA expression at 6 or 24 hours (Figures 2D and 2E). There was a very strong upregulation of D2 mRNA in the ipsilateral striatum at 6 hours after ischemia (Figure 2A), which disappeared at 24 and 72 hours (Figures 2B and 2C). The signal was also stronger in layers II to IV and VI of the ipsilateral fronto-parietal cortex at 6 hours after ischemia. The upregulation of D2 mRNA was amplified at 24 hours in layers II to IV and VI of the fronto-parietal cortex, whereas a frontal area showed a definitive signal loss in all layers after 24 hours (Figure 2B). This loss of D2 mRNA expression was thereafter extended to all the ischemic parietal cortex at 72 hours (Figure 2C). Finally, D2 mRNA upregulation was limited to the juxta-lesional frontal cortex or to the periphery of the lateral ventricle after 72 hours. To quantify changes in D2 mRNA contents, data from the autoradiograms were quantified for the different experimental groups as explained in Materials and methods, and shown in Figures 2H, 2I and 2J. No significant variations were observed in the contralateral area after ischemia or in the ipsilateral or contralateral side after sham operation compared with non-operated rats. Compared with control or sham-operated rats, the signal was potently upregulated at 6 hours after ischemia in the head of caudate nucleus of the ipsilateral striatum (+300%), but was definitively lost after 24 hours (Figure 2H). D2 mRNA expression was also slightly increased (+50%) at 6 hours after ischemia in the motor area of the ipsilateral frontal cortex (layers V to VI) and lost at 24 hours (Figure 2I). Compared with sham-operated rats, the signal was not significantly increased at 6 hours after ischemia in the parietal cortex (layers II to IV), but was increased at 24 hours (+50%) (Figure 2J). Reverse transcription-PCR analysis of D2 mRNA expression in the ipsilateral side (Figure 3) confirmed the in situ hybridization. D2 mRNA was transiently increased in the ipsilateral striatum 6 hours after ischemia. D2 mRNA was also increased in the ipsilateral parietal cortex as soon as 6 hours after ischemia and remained increased at 24 hours. Thus, D2 mRNA (Figures 2 and 3) appeared increased, as D2 activity (Figure 1), in the striatum as soon as 6 hours, whereas in the cerebral cortex D2 mRNA (Figures 2 and 3) began to increase as soon as 6 hours, and this before the upregulation of D2 activity.
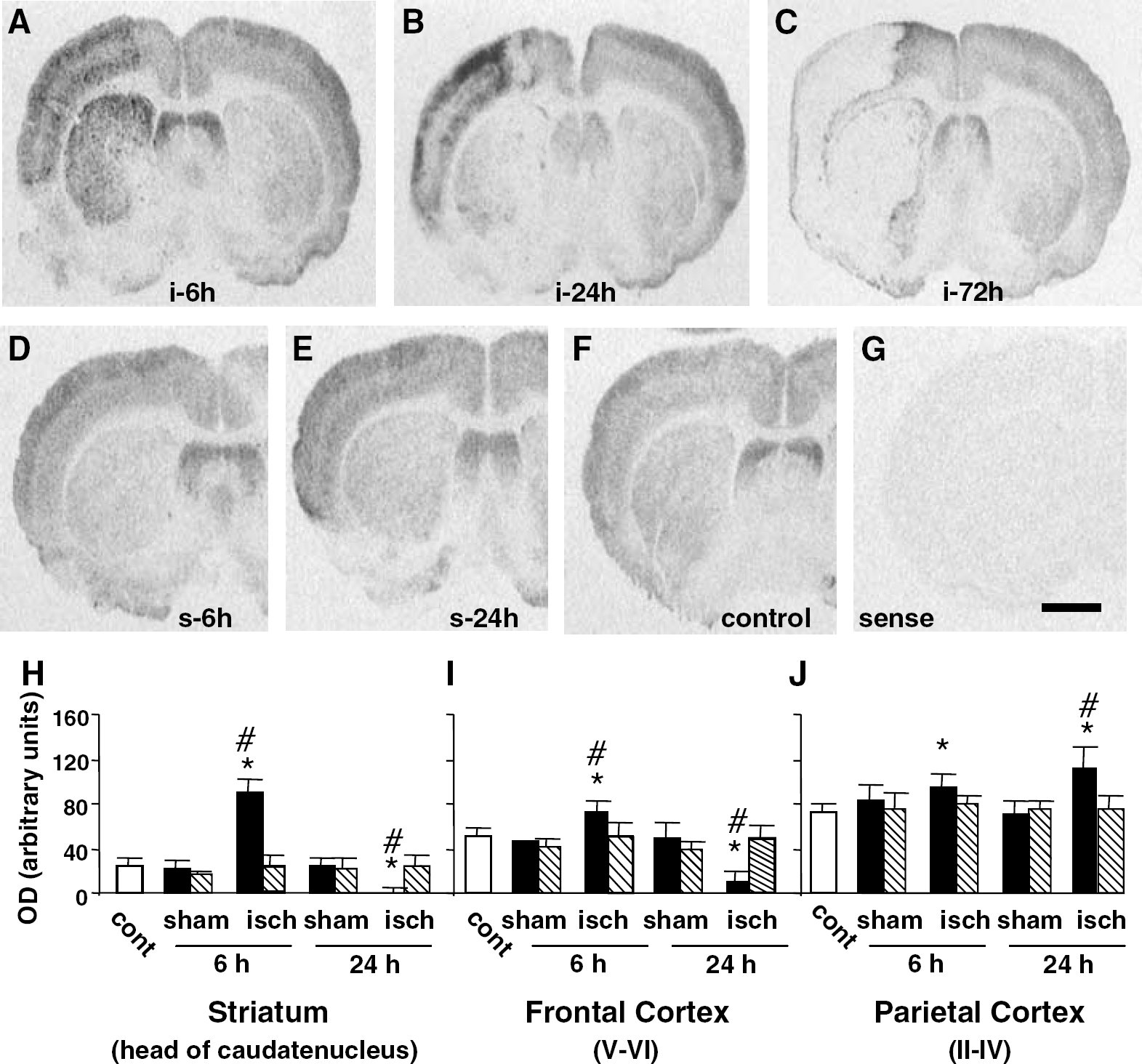
In situ hybridization for D2 mRNA on sections of rat brain after transient ischemia. Film autoradiograms (panels
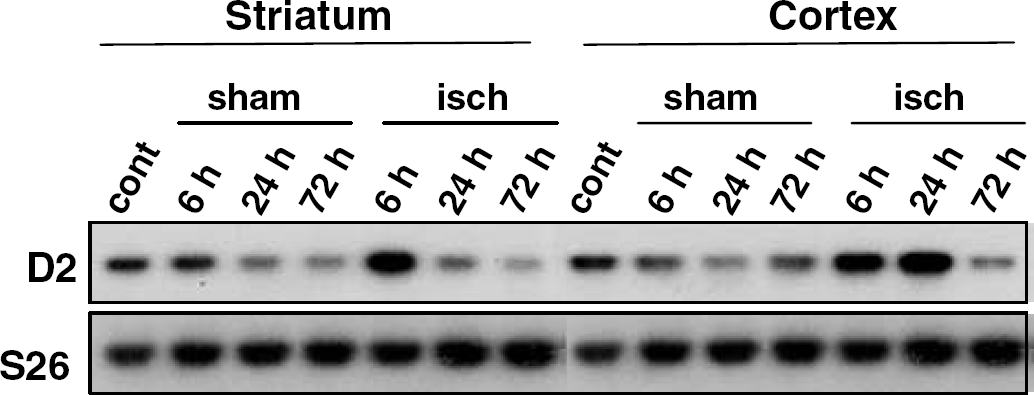
D2 mRNA expression in the ipsilateral cerebral cortex and striatum after ischemia. D2 mRNA levels were assessed by RT-PCR analysis in control (cont) rats, sham-operated (sham) and ischemic (isch) rats, at 6, 24, and 72 hours after surgery. S26 mRNA levels were also followed as internal controls.
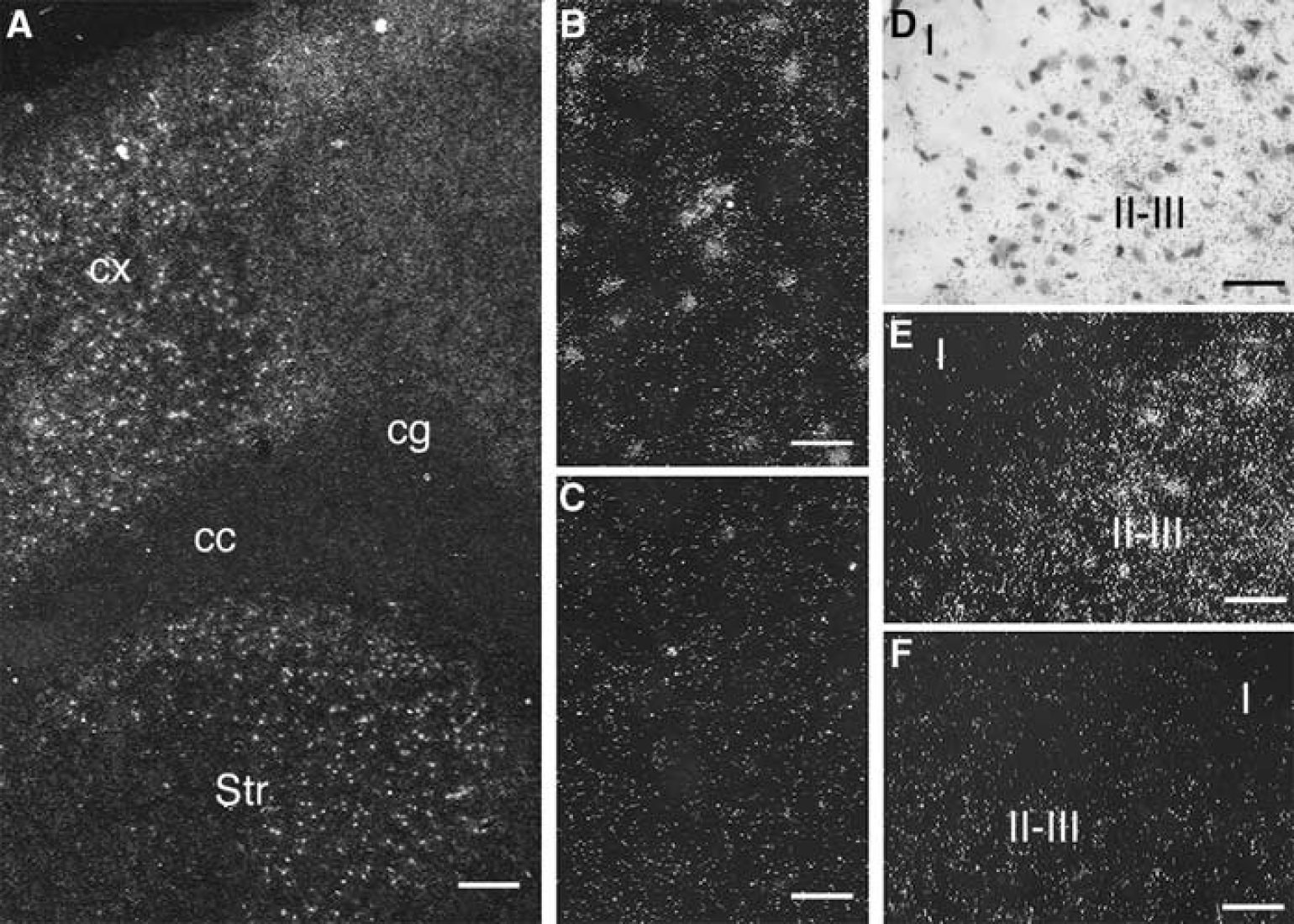
To more precisely localize the signal, we performed emulsion autoradiography and counterstaining of the sections. The dark-field image at low magnification (Figure 4A) showed the upregulation of D2 mRNA as clusters of silver grains in layers II to VI in the frontal cortex and head of caudate nucleus, 6 hours after ischemia. At higher magnification, these grain clusters appeared distinctly in the ipsilateral striatum area (Figure 4B), over the sparsely distributed basal signal of D2 mRNA expression as seen in the contralateral one (Figure 4C). Ischemia did not promote upregulation of D2 mRNA in the superficial layer I of the parietal cortex 24 hours after ischemia (bright-field Figure 4D and dark-field Figure 4E), compared with the contralateral side (dark-field Figure 4F). Ischemia promoted upregulation of D2 mRNA in layers II to III of the ipsilateral parietal cortex 24 hours after ischemia (Figures 4D to 4E). Increased concentrations of silver grains were not superposed on large neuronal nuclei, but were located over their surrounding areas with some high concentrations of grains over small nuclei (Figures 4D and 5H). No nuclear localization was present either in the striatum or cerebral cortex from control (Figures 5A and 5B) or sham-operated rats (not shown) as expected by the previous report of Guadano-Ferraz et al (1997). Silver grains were accumulated over small nuclei 6 hours after ischemia in the striatum (Figure 5E) and the parietal cortex (Figure 5F), and still present 24 hours after injury in the parietal cortex (Figure 5H). Outside the clusters, there is also an increased signal in the striatum and the parietal cortex 6 or 24 hours after ischemia (Figures 5E, 5F and 5H). To determine the cell type for D2 mRNA localization, we performed in situ hybridization for D2 mRNA and immunohistochemistry with an anti-GFAP antibody on the same sections. In the striatum and the cerebral cortex from control rats, some silver grains were clearly located in association with astrocytic processes (Figures 5C and 5D), as previously reported (Guadano-Ferraz et al, 1997). After ischemia, most of the grain clusters appeared mainly associated with the cell bodies expressing GFAP in the ipsilateral striatum at 6 hours (Figure 5G) and in the ipsilateral parietal cortex at 24 hours (Figures 5I to 5J). A labelling of cellular processes expressing GFAP was also observed (Figures 5G, 5I and 5J). Thus, after ischemia, upregulation of D2 mRNA in the striatum and cortex is localized in cells labelled by immunostaining of GFAP, that is, astrocytes.
In situ hybridization for D2 mRNA on sections of rat brain after transient ischemia—dark- and bright-field images showing the distribution of silver grains after coating the hybridized sections with photographic emulsion. The sections at the level of the septum were exposed for 5 weeks. The section in (
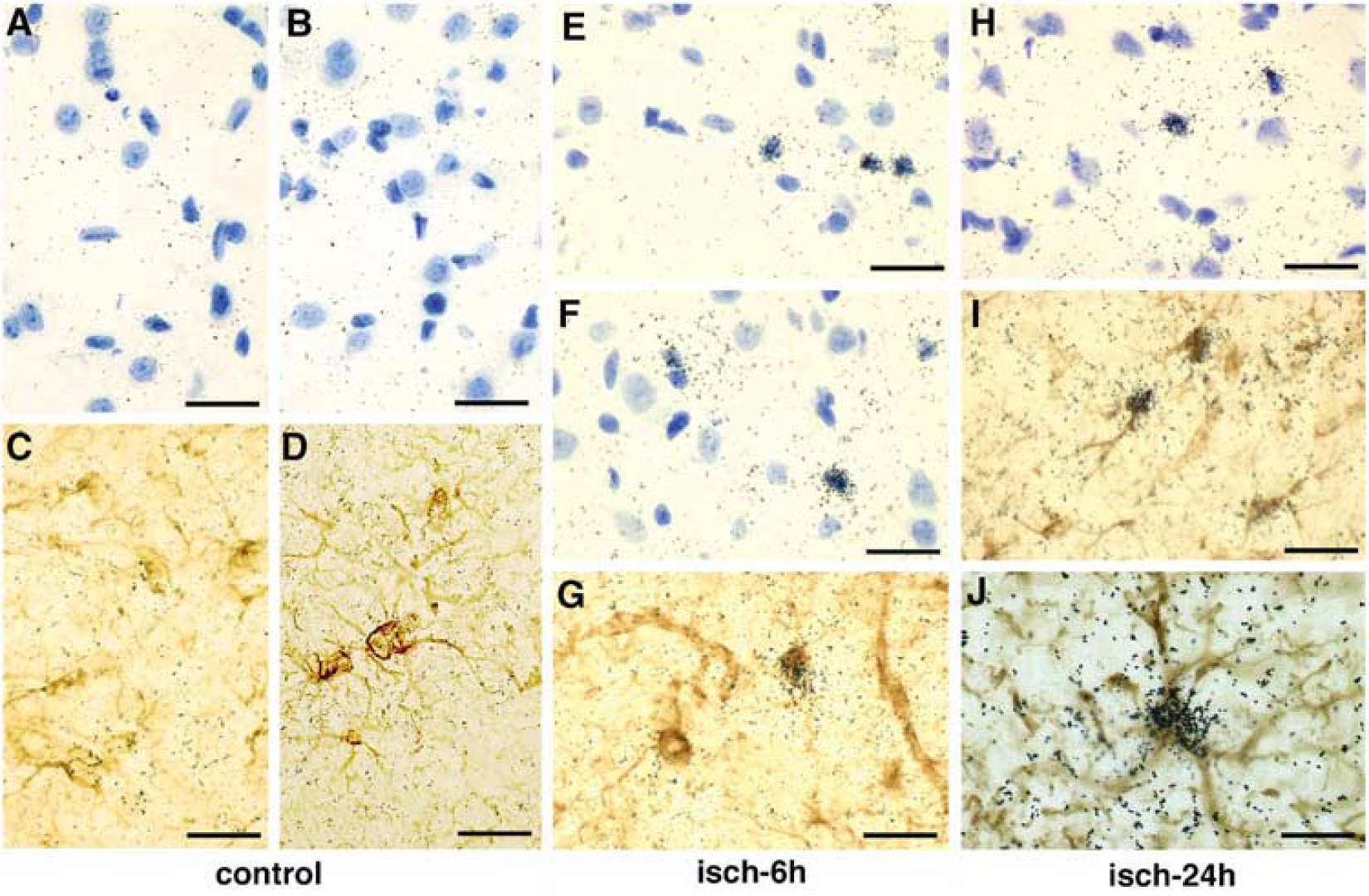
High-magnification photomicrographs taken from sections of the ischemic rat brain after in situ hybridization for D2 mRNA. The coronal sections at the level of the septum were obtained from control rat (
Discussion
This work represents the first demonstration of a rapid and potent upregulation of type 2 deiodinase after transient focal cerebral ischemia in the rat. D2 activity is strongly increased in the ipsilateral striatum by 6 hours, and at least until 24 hours after transient occlusion of the MCA. The expression of D2 mRNA followed by in situ hybridization or RT-PCR is also rapidly upregulated in the ipsilateral striatum. An important increase in D2 activity is also observed at 24 hours in the cerebral cortex, with an increase in D2 mRNA beginning at 6 hours and reinforced after 24 hours in the parietal cortex.
The upregulation of D2 activity appears earlier in the striatum than in the parietal cortex after transient ischemia. This is probably correlated with the more reduced cerebral blood flow and the faster establishment of infarction in the striatum than in the fronto-parietal cortex (Lerouet et al, 2002, Kametsu et al, 2003).
Upregulation of D2 mRNA has been previously described in another type of brain injury, the traumatic brain injury (Zou et al, 1998). D2 mRNA detection was initially detected at 3 days in the ipsilateral cortex, near the contusion. It is possible that D2 mRNA appearance depends on the nature of brain injury. However, that the in situ hybridization performed with the oligonucleotide probe by Zou et al (1998) was unable to detect D2 mRNA expression in the earlier stage, because it is unable to detect any D2 mRNA expression in non-traumatized rats is not excluded. Our observations underline the rapid mechanism of D2 upregulation, which may confer a potential role to local T3 generation, as soon as a few hours after ischemia. T4 infusion partially protects against brain damage after global ischemia in the dog (D'Alecy, 1997). Repetitive administration of T4, before and immediately after ischemia, attenuates the hippocampal neuronal damage in the rat (Rami and Krieglstein, 1992). However, some interspecies differences exist since hypothyroidism has been reported to protect the brain against ischemia in gerbils (Shuaib et al, 1994).
In control rats, D2 mRNA is expressed primarily in some subpopulations of astrocytes, the protoplasmic astrocytes and tanycytes, with the majority of the signal being located in the cellular processes (Guadano-Ferraz et al, 1997). The upregulation of D2 mRNA is also mainly observed in GFAP-positive astrocytes as soon as 6 hours after ischemia in our model. The labelled astrocytes seem absent from layer I of the cerebral cortex and the corpus callosum until 24 hours, suggesting that astrocytes expressing D2 mRNA soon after ischemia are also primarily protoplasmic astrocytes. After 72 hours, this D2 mRNA expression is observed around the lesioned area in the corpus callosum and layer I of the cerebral cortex (not shown). A differential sensitivity of protoplasmic versus fibrous cortical astrocytes to permanent blood deprivation has been reported in mice, with very rapid loss of GFAP in protoplasmic astrocytes (Lukaszevicz et al, 2002). However, the early reperfusion after focal cerebral ischemia, as in our model, preserves the GFAP expression (Lee et al, 2003). These observations underline the peculiar sensitivity of protoplasmic astrocytes to blood deprivation and the importance of rapid reperfusion in preventing cell damage. Thus, protoplasmic cortical astrocytes, which are preserved in our model, may also contribute to the neuronal protection via D2 upregulation.
The molecular mechanisms responsible for the ischemia-induced increase in D2 remain to be clarified. Many factors have been described to increase D2 in cultured astrocytes by transcriptional and translational mechanisms: cyclic AMP, glucocorticoids, FGFs, phorbol esters, TSH, and selenite (Leonard, 1988; Courtin et al, 1988, 1989, 1990; Saunier et al, 1993; Pallud et al, 1997). T4 and reverse T3 promote the degradation of D2 by a posttranslational mechanism in astrocytes (Leonard et al, 1990). This inactivation requires selective conjugation of D2 to ubiquitin as observed in other cell systems (Steinsapir et al, 1998, 2000; Gereben et al, 2000). The rapid upregulation of D2 mRNA in ipsilateral striatum suggests a transcriptional mechanism. Recently, some transcriptional factors have been shown to be implicated in D2 promoter transactivation. Induction of D2 mRNA by cyclic AMP involves the binding of the transcription factor CREB to a canonical sequence cAMP Responsive Element (CRE) in its promoter sequence (Canettieri et al, 2000; Gereben et al, 2001; for a review, see Bianco et al, 2002). CREB phosphorylation has been proposed for mediating ischemic tolerance (Nakajima et al, 2002). However, CREB knockout mice exhibit the same neurologic score as the wild-type mice after ischemia (Hata et al, 1998). NFkappaB, which is induced in ischemia (Gabriel et al, 1999), and activates D2 promoter in HEK 293 cells (Fekete et al, 2004), is also a possible transcription factor implicated in D2 induction after ischemia. In the cerebral cortex, the delayed increase in D2 activity after the beginning of D2 mRNA accumulation might be due to posttranscriptional mechanisms. Otherwise, a posttranslational mechanism may contribute to the elevated D2 activity after 24 hours in consideration of the decline in D2 mRNA in the striatum. The ipsilateral signal of D2 mRNA and activity, associated with the absence of contralateral response after focal ischemia, allows the discarding of a role for variations in circulating concentrations of thyroid hormone (Silva et al, 1984; Obregon et al, 1991; Croteau et al, 1996; Guadano-Ferraz et al, 1999). Interestingly, a Von Hippel-Landau protein-interacting deubiquitinating enzyme, VDU1, may contribute to amplifying the increase in D2 activity (Curcio-Morelli et al, 2003). It would be interesting to explore the regulation of VDU1, a target for ubiquitin–proteasome degradation (Li et al, 2002), in the brain after transient ischemia.
The rapid upregulation of D2 after ischemia, and the expected local increase in T3 in presence of T4, may constitute a neuroprotective mechanism. Future work must establish the role of D2 by studying, for example, the impact of D2 knockout on the extension of injury after ischemia. It would be also interesting to identify the induction pathways to increase neuroprotection by increasing D2 induction.
Footnotes
Acknowledgements
We are grateful to Rachida Guennoun and Monique Gouezou for their technical advice. The authors thank Olivier Trassard for help with preparation of the figures. WW L was also a recipient of the Société de Secours des Amis des Sciences.