
Abstract
The tumor suppressor gene p53 plays an important role in the regulation of apoptosis through transcriptional activation of cell cycle control. Degradation of p53 hinders its role in apoptosis regulation. Recent studies have shown that MDM2-mediated ubiquitylation and the ubiquitin–proteasome system are critical regulating systems of p53 ubiquitylation. However, the mechanism regulating p53-mediated neuronal apoptosis after cerebral ischemia remains unknown. We examined the MDM2 pathway and the ubiquitin–proteasome system using a transient focal cerebral ischemia (tFCI) model and analyzed the interaction between p53 regulation and superoxide using copper/zinc superoxide dismutase (SOD1) transgenic mice after tFCI. p53 degradation and ubiquitylation were detected after tFCI. The accumulation of ubiquitylated p53 was inhibited and p53 degradation was facilitated by SOD1. Nuclear translocation and MDM2/Akt interaction were detected after tFCI and were inhibited by phosphatidylinositol 3-kinase inhibition and promoted by SOD1. Cytosolic translocation of the p53/MDM2 complex was detected after tFCI and was promoted by SOD1. Moreover, accumulation of multiubiquitin chains and direct oxidative injury to a proteasome were detected and inhibited by SOD1 after tFCI. These results suggest that SOD1 promotes the MDM2 pathway and the ubiquitin–proteasome system after tFCI and that production of reactive oxygen species after tFCI prevents p53 degradation by inhibiting both systems.
Introduction
Reactive oxygen species (ROS) have been implicated in neuronal cell death after a variety of toxic stimuli, and inhibition of them is an important therapeutic strategy for neuronal protection (Chan, 1994, 2001; Bossy-Wetzel et al, 2004). Superoxide anions are significantly produced in neuronal cells exposed to reperfusion injury during the early period after stroke and cause critical damage to these neuronal cells (Chan, 1994). Electron flow in isolated brain mitochondria produces superoxide anions, which are scavenged by superoxide dismutase. Copper/zinc-superoxide dismutase (SOD1), a cytosolic antioxidant isoenzyme, is highly protective against reperfusion injury including apoptotic neuronal cell death after stroke (Kinouchi et al, 1991; Chan, 1996; Fujimura et al, 1999b). Overexpression of SOD1 can also alter the balance between the apoptotic cell death and survival pathways, but superoxide is thought to be involved in a variety of apoptotic signaling pathways after stroke (Noshita et al, 2002; Saito et al, 2003b).
The tumor suppressor gene p53 encodes a sequence-specific transcription factor that controls the expression of genes whose products mediate either cell cycle arrest or apoptosis (Oren, 1999; Ogawara et al, 2002; Kawai et al, 2003). Recently, p53 was also found to be a target of MDM2 ubiquitylation and the ubiquitin–proteasome system in response to apoptotic stimuli (Mayo and Donner, 2001; Ogawara et al, 2002; Kawai et al, 2003). MDM2 inhibits p53 by abolishing its transcription regulatory activity in the nucleus (Momand et al, 1992; Chen et al, 1995; Haupt et al, 1997; Kubbutat et al, 1997; Kawai et al, 2003; Meulmeester et al, 2003). MDM2 also targets p53 for degradation by acting as an E3 ubiquitin–protein ligase in the cytoplasm (Haupt et al, 1997; Kubbutat et al, 1997). The entry of MDM2 into the nucleus is regulated by Akt phosphorylation (Mayo and Donner, 2001).
The ubiquitin–proteasome system has been studied for its control over the ordered degradation of proteins involved in apoptosis and for its regulation of cell survival by p53 degradation (Scheffner et al, 1995; Maki et al, 1996; Dimmeler et al, 1999; Yang and Yu, 2003). The ubiquitylated substrate proteins are recognized and degraded by the 26S proteasome (Schwartz and Ciechanover, 1999; Yang and Yu, 2003). Ubiquitylation is first activated by an E1-activating enzyme, then transferred to an E2 ubiquitin-conjugating enzyme, and finally ligated to the substrate by an E3 ubiquitin-protein ligase (Schwartz and Ciechanover, 1999; Yang and Yu, 2003). The 26S proteasome is a eukaryotic ATP-dependent protease complex composed of a core protease (the 20S subunit) and a pair of PA700 regulatory particles (Coux et al, 1996; Asai et al, 2002).
Many reports show that p53 functions as a proapoptotic factor and that it upregulates in apoptotic lesions after an in vivo stroke (Chopp et al, 1992; Li et al, 1994; McGahan et al, 1998; Watanabe et al, 1999; Maeda et al, 2001). However, its precise mechanisms remain unknown in reperfusion injury, which is associated with apoptosis in vivo. In this study, we examined the mechanism of p53 degradation and its relationship to oxidative stress after transient focal cerebral ischemia (tFCI).
Materials and methods
SOD1 Transgenic Mice
Heterozygous SOD1 transgenic (Tg) mice of the SOD1 TGHS/SF-218-3 strain with a CD-1 background, carrying human SOD1 genes with a threefold increase in SOD1, were derived from the founder stock described previously (Yang et al, 1994). They were further bred with CD-1 wild-type mice to generate heterozygous mice. The SOD1 Tg mice were identified by quantitative demonstration of SOD1 using nondenaturing gel electrophoresis, followed by nitroblue tetrazolium staining. There were no differences in the phenotypes, including the anatomy of the circle of Willis, between the SOD1 Tg mice and their wild-type littermates. There was no difference in the regional cerebral blood flow before or after tFCI between the SOD1 Tg and wild-type mice.
Focal Cerebral Ischemia
Adult male mice (3 months old, 35 to 40 g) were subjected to tFCI by intraluminal middle cerebral artery (MCA) blockade with a nylon suture as described previously (Yang et al, 1994). The mice were anesthetized with 1.5% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. The rectal temperature was controlled at 37°C with a homeothermic blanket. Cannulation of a femoral artery allowed for the monitoring of blood pressure and arterial blood gases, with samples for analysis being taken immediately after cannulation, 10 mins after occlusion, and 10 minus after reperfusion. Blood gas was analyzed with a pH/blood gas analyzer (Chiron Diagnostics, Ltd, Essex, UK). After a midline skin incision, the left external carotid artery was exposed, and its branches were electrocoagulated. An 11-mm 5-0 surgical monofilament nylon suture, blunted at the end, was introduced into the left internal carotid artery through the external carotid artery stump. After 60 minus of MCA occlusion, blood flow was restored by the withdrawal of the nylon suture.
Drug Treatment
To examine the role of phosphatidylinositol 3-kinase (PI3-K) in the MDM2 signaling pathway, we administered a PI3-K inhibitor, LY294002, after tFCI. LY294002 (2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one) was purchased from Cell Signaling Technology (Beverly, MA, USA) and dissolved in dimethyl sulfoxide and phosphate-buffered saline (PBS) (LY294002, 50 nmol in 25% dimethyl sulfoxide in PBS; vehicle, 25% dimethyl sulfoxide in PBS), as described previously (Noshita et al, 2001). The scalp was incised on the midline and the skull was exposed. This drug and the vehicle were injected intracerebroventricularly (2 μl, bregma; 1.0 mm lateral, 0.2 mm posterior, 3.1 mm deep). The drug and the vehicle were injected 1 hour before MCA occlusion.
Immunofluorescent Double-Labeling Staining
To evaluate colocalization of MDM2 or p53 and neuron-specific nuclear protein (NeuN), we performed double immunofluorescent staining. The sections fixed by 4% paraformaldehyde were immunostained with an anti-MDM2 antibody (Chemicon International, Temecula, CA, USA) or an anti-p53 antibody (Cell Signaling Technology), with biotinylated goat anti-rabbit immunoglobulin G (IgG; Vector Laboratories, Burlingame, CA, USA) followed by fluorescein avidin DCS (Vector Laboratories). The sections were then incubated with a blocking solution and reacted with an anti-NeuN antibody (Chemicon International) as described above, followed by a Texas Red-conjugated donkey anti-mouse IgG antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) at a dilution of 1:400. The sections were placed on slides, which were then covered with VECTASHIELD mounting medium with 4′,6 diamidino-2-phenylindole (DAPI; Vector Laboratories). Fluorescence of fluorescein was observed at an excitation (Ex) of 495 nm and emission (Em) of >515 nm, and fluorescence of Texas Red was observed at an Ex of 510 nm and Em of >580 nm. Fluorescence of DAPI was observed at an Ex of 360 nm and Em of >460 nm.
Western Blot Analysis
Protein extraction of the cytosolic fraction was performed as described previously with some modification (Fujimura et al, 1999a). Samples were obtained from the cerebral cortex around the caudate putamen (ischemic core) on the ischemic sides where apoptotic neuronal cell death was observed after transient MCA occlusion, and from nonischemic controls (n=4 each) (Noshita et al, 2001). Fresh brain tissue was removed after 1, 2, 4, 8, and 24 hours of reperfusion (n=4 each) and homogenized by gently douncing 35 times in a glass tissue grinder (Wheaton, Millville, NJ, USA) in 7 volumes of cold suspension buffer (20 mmol/L Hepes-KOH, pH 7.5, 250 mmol/L sucrose, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, and 1 mmol/L EGTA plus 0.7% protease and phosphatase inhibitor cocktails (Sigma, St Louis, MO, USA)). The homogenate was centrifuged at 750 g for 10 minus at 4°C and then at 8000 g for 20 mins at 4°C. The 750 g pellets were used to obtain the crude nuclear fraction and this fraction was purified using a nuclear extraction kit (Sigma). The 8000 g pellets were used to obtain the mitochondrial fraction. The supernatant was further centrifuged at 100,000 g for 60 mins at 4°C, and was then used for the cytosolic analysis. After adding the same volume of Tris-glycine sodium dodecyl sulfate sample buffer (Invitrogen, Carlsbad, CA, USA) to the supernatant, we loaded equal amounts of the samples per lane. The primary antibodies were 1:800 dilution of rabbit polyclonal antibody against phosphorylated MDM2 (pMDM2) (Cell Signaling Technology), 1:1000 dilution of rabbit polyclonal antibody against p53 (Cell Signaling Technology), 1:1000 dilution of rabbit polyclonal antibody against TFIID (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and 1:10,000 dilution of anti-β-actin monoclonal antibody (Sigma). Western blots were performed with horseradish peroxidase-conjugated anti-rabbit IgG (Cell Signaling Technology) using enhanced chemiluminescence Western blotting detection reagents (Amersham International, Buckinghamshire, UK). The film was scanned with a GS-700 imaging densitometer (Bio-Rad Laboratories, Hercules, CA, USA) and the results were quantified using Multi-Analyst software (Bio-Rad).
Coimmunoprecipitation
Protein extraction of both the cytosolic and nuclear fractions was performed as described in the Western blotting method (Fujimura et al, 1999a). The procedure for precipitation was performed as described previously (Saito et al, 2003a, 2004). Samples were obtained from the MCA territory brain tissue on the ischemic side and from nonischemic controls (n=4 each). Fresh brain tissue was removed after 1, 2, 4, 8, and 24 hours of reperfusion (n=4 each). Protein concentrations were determined by the Bradford method (Bio-Rad). In all, 200 μg of protein from the cytosolic fraction and 100 μg from the nuclear fraction were used for coimmunoprecipitation. Whole brain extract was included as a positive control. The protein sample was incubated with 50% slurry of protein G-Sepharose (Amersham Pharmacia Biotech, Uppsala, Sweden) for 1 hour at 4°C, and this mixed sample was centrifuged at 12,000 g for 1 minute. The supernatant was incubated with 2 μg of polyclonal mouse anti-pMDM2 antibody (Cell Signaling Technology) and 15 μl of protein G-Sepharose (50% slurry) for 1 hour 4°C. The negative control was prepared with protein G-Sepharose without an antibody. The 14,000 g pellets were washed three times and used as the samples bound to each antibody. After adding the same volume of Tris-glycine sodium dodecyl sulfate sample buffer (Invitrogen) to the samples, we boiled these samples to remove the sepharose beads. After centrifugation at 14,000 g for 1 minute, the supernatant was immunoblotted with a 1:1000 dilution of an antiphosphorylated Akt antibody (Cell Signaling Technology) or a 1:800 dilution of the anti-p53 antibody (Cell Signaling Technology) as described in the Western blotting method.
Multiubiquitin Chain Sandwich Enzyme-Linked Immunosorbent Assay
For quantification of multiple ubiquitin chains, we performed a sandwich enzyme-linked immunosorbent assay (ELISA) using a monoclonal antibody, FK2 (AFFINITI, Mamhead, Exeter, UK), which specifically recognizes multiple ubiquitylated proteins (Tamura et al, 1991; Takada et al, 1995; Ide et al, 1999). The procedure was performed as reported previously (Ide et al, 1999). Samples were obtained from the entire MCA territory on the ischemic side and from nonischemic controls (n=4 each). The cytosolic tissue samples were prepared as described in the Western blotting method. A cytosolic volume containing 100 μg of protein was used for the assay. Samples and controls were incubated on microtiter plates coated with the FK2 antibody via the avidin–biotin reaction, and, after subsequent washing, they were incubated in the FK2 antibody and peroxidase-conjugated streptavidin with the use of biotinylated IgG (Vector Laboratories). Detection of color development was performed with 3,3′,5,5′-tetramethylbenzidine (0.15 mg/ml in 1 mmol/l sodium acetate, pH 5.5 with 0.0025% of H2O2 (Sigma)) and the absorbance was observed at 450 nm.
Proteasome Activity Assay
For quantification of 20S proteasome activity, we used a commercial detection kit (Chemicon International) and followed the manufacturer's protocol. Tissue samples were prepared as described in the Western blotting method, with some modification. Tissue was lysed in Tris-HCl lysed buffer, and centrifuged at 13,000 g for 10 minus at 4°C. The supernatants were assayed for their ability to degrade the substrate. Activity was detected as that of fluorophore 7-amino-4-methylcoumarin (AMC) after cleavage from the labeled substrate LLVY-AMC (Tsukahara et al, 1988; Asai et al, 2002). The free AMC fluorescence was observed using 380- and 460-nm filter sets in a fluorometer.
Oxidative Injury Detection Assay
Following the manufacturer's protocol, we used a commercial detection assay (OxyBlot oxidation detection kit; Chemicon International) to examine oxidative injury directly to the proteins. Samples were obtained from the entire MCA territory on the ischemic side and from nonischemic controls (n=4 each). The cytosolic tissue samples were prepared as described in the Western blotting method. A cytosolic volume containing 50 μg of protein was used for the assay. The samples were reacted with 2,4-dinitrophenylhydrazine (DNP) for 20 minus at room temperature. DNP specifically reacted with the oxidized residue of the protein samples, and the DNP-binding sites were detected using an anti-DNP antibody by Western blot or coimmunoprecipitation as described above (Levine et al, 1994).
Quantification and Statistical Analysis
The data are expressed as mean±s.d. Comparisons among multiple groups were performed with a one-way analysis of variance with appropriate post hoc tests (SigmaStat software; Jandel Corporation, San Rafael, CA, USA). Comparisons between two groups were achieved with the Student's t-test. Significance was accepted with P<0.05.
Results
Physiological Data and Cerebral Infarction
Physiological data showed no significant differences in body temperature, MABP, or arterial blood gas analysis between the groups. The preischemic physiological values were as follows (in mmol/L Hg): 36.4±0.3°C body temperature, 83±3.1 MABP, 7.3±0.1 pH, 164.9±19 PaO2, 30±10 PaCO2 (values are mean±s.d; n=4). There was no deviation from these values over the period of assessment. An ischemic lesion of the core of the caudate putamen was visible as a pale, slightly stained area in the ischemic hemisphere as early as 1 hour after reperfusion and extended to the entire MCA territory at 4 hours by cresyl violet staining (data not shown). The time-dependent increase in infarction in the mouse brains with the intraluminal suture blockade is consistent with previous reports that used the same FCI model in mice (Yang et al, 1994).
Neuronal p53 Translocated from the Nucleus to the Cytoplasm after Transient Focal Cerebral Ischemia
Double immunofluorescent staining for p53 and NeuN showed that p53 expression colocalized with neurons in the ischemic cortex 24 hours after reperfusion (n=4; Figure 1A). Western blot analysis showed that p53 was detected as a 53-kDa band (n=4; Figure 1B). Nuclear p53 gradually increased 1 and 2 hours after stroke, and thereafter it began to decrease at 4 hours and was significantly decreased at 24 hours (Figure 1B). In contrast, expression of cytosolic p53 was barely detected in the sham-operated samples, but was significantly increased 4 and 8 hours after tFCI (Figure 1B). Thereafter, cytosolic p53 began to decrease (Figure 1B). These results suggest that p53 was induced in the nucleus during the early period of reperfusion injury and that it subsequently translocated from the nucleus to the cytoplasm after tFCI.
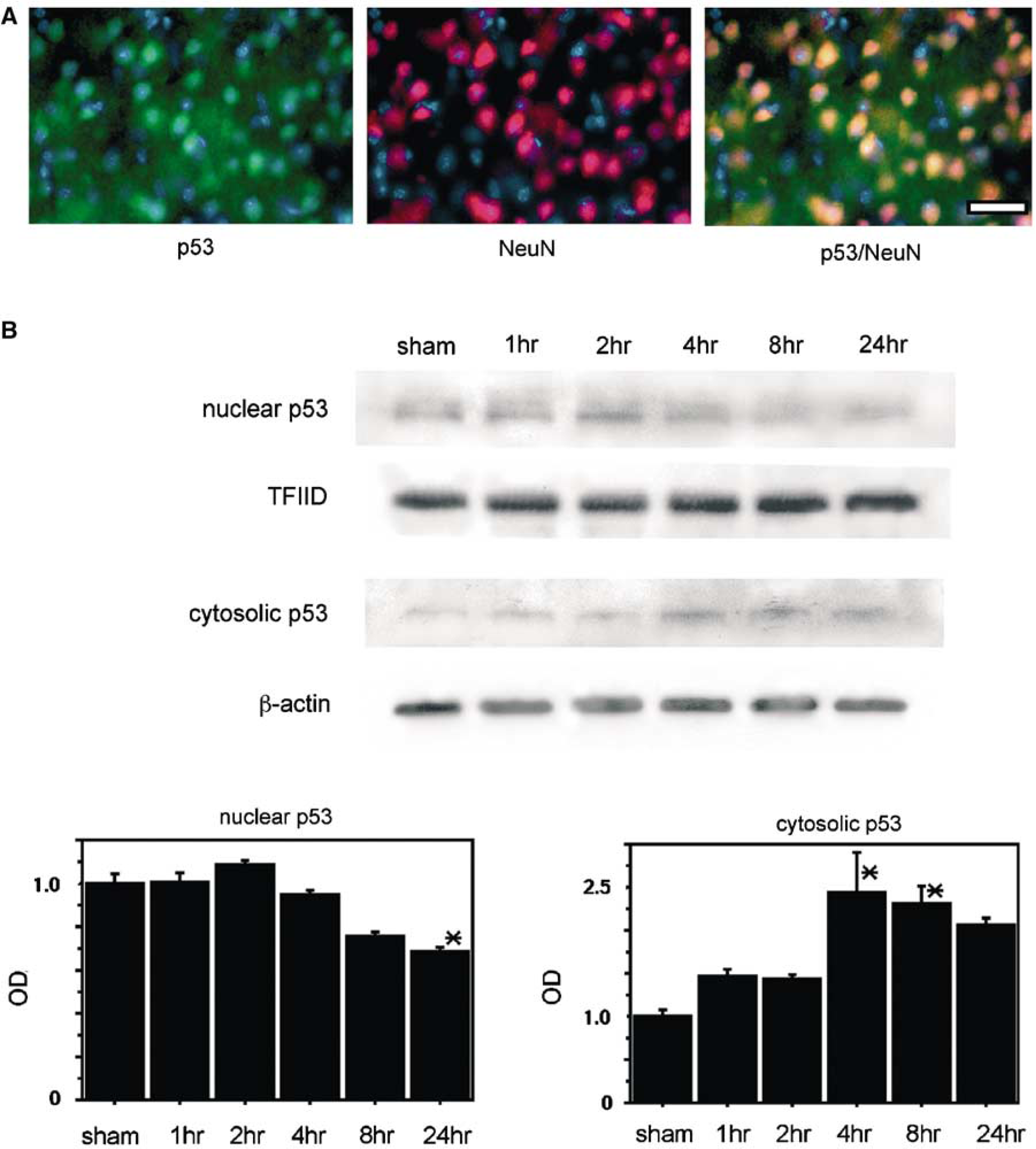
(
Accumulation of Ubiquitylated p53 after Transient Focal Cerebral Ischemia
We examined the ubiquitylated proteins among the whole cytosolic proteins after tFCI using the multiubiquitin chain sandwich ELISA. Accumulation of multiubiquitin gradually increased and reached a peak 8 hours after tFCI (n=4; Figure 2A), which was in accord with a previous report (Ide et al, 1999). To examine p53 ubiquitylation, we analyzed multiubiquitin expression precipitated by a p53 antibody in the cytosolic samples. Coimmunoprecipitation revealed that ubiquitylated p53 accumulated in the cytoplasm after tFCI (n=4; Figure 2B) and remarkably increased 4 and 8 hours after tFCI (Figure 2B). These results suggest that p53 ubiquitylation accumulated during the early period of reperfusion injury after tFCI but that it rapidly degraded.
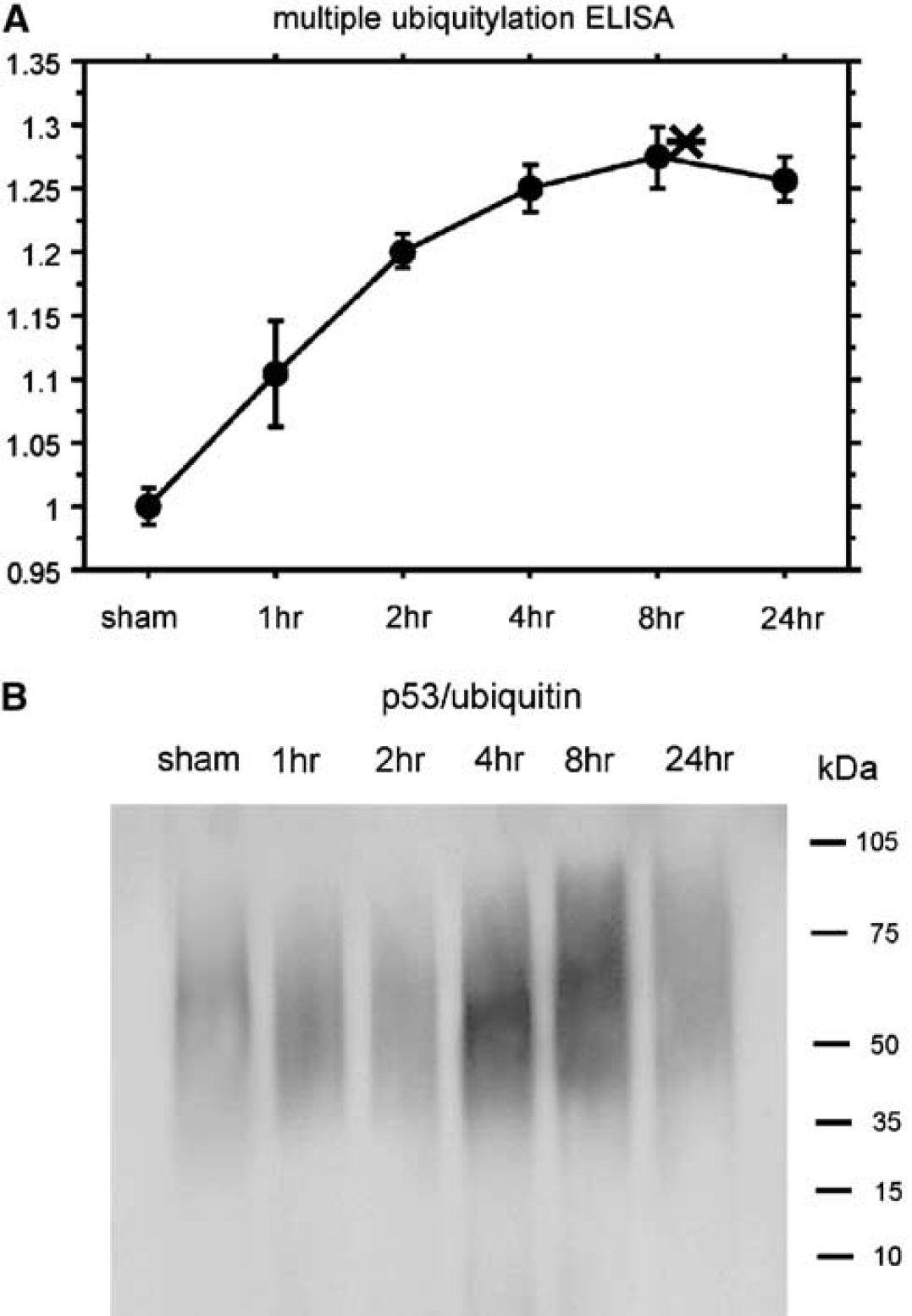
(
Oxidative Stress and Proteasome Activity after Transient Focal Cerebral Ischemia
We analyzed 20S protease activity, which is the core active site of 26S proteasome (DeMartino and Slaughter, 1999). Proteasome activity began to decrease 1 hour after tFCI and significantly decreased at 4 hours (n=4; Figure 3A), after which it gradually recovered (Figure 3A). This result lends support to a previous report (Asai et al, 2002). Using coimmunoprecipitation, we analyzed direct oxidative injury to the proteasome precipitated by the anti-DNP antibody, which specifically detected the site of DNP binding to the carbonyl residue of the oxidatively injured proteins after tFCI. Coimmunoprecipitation revealed that oxidative injury to the proteasome (proteasome/DNP) began to increase 1 hour after tFCI and significantly increased at 4 hours (n=4; Figure 3B). The peak time point of the decrease in proteasome activation was in accord with the peak point of direct oxidative injury to the proteasome after tFCI. These results suggest that oxidative injury may decrease proteasome activity after tFCI.
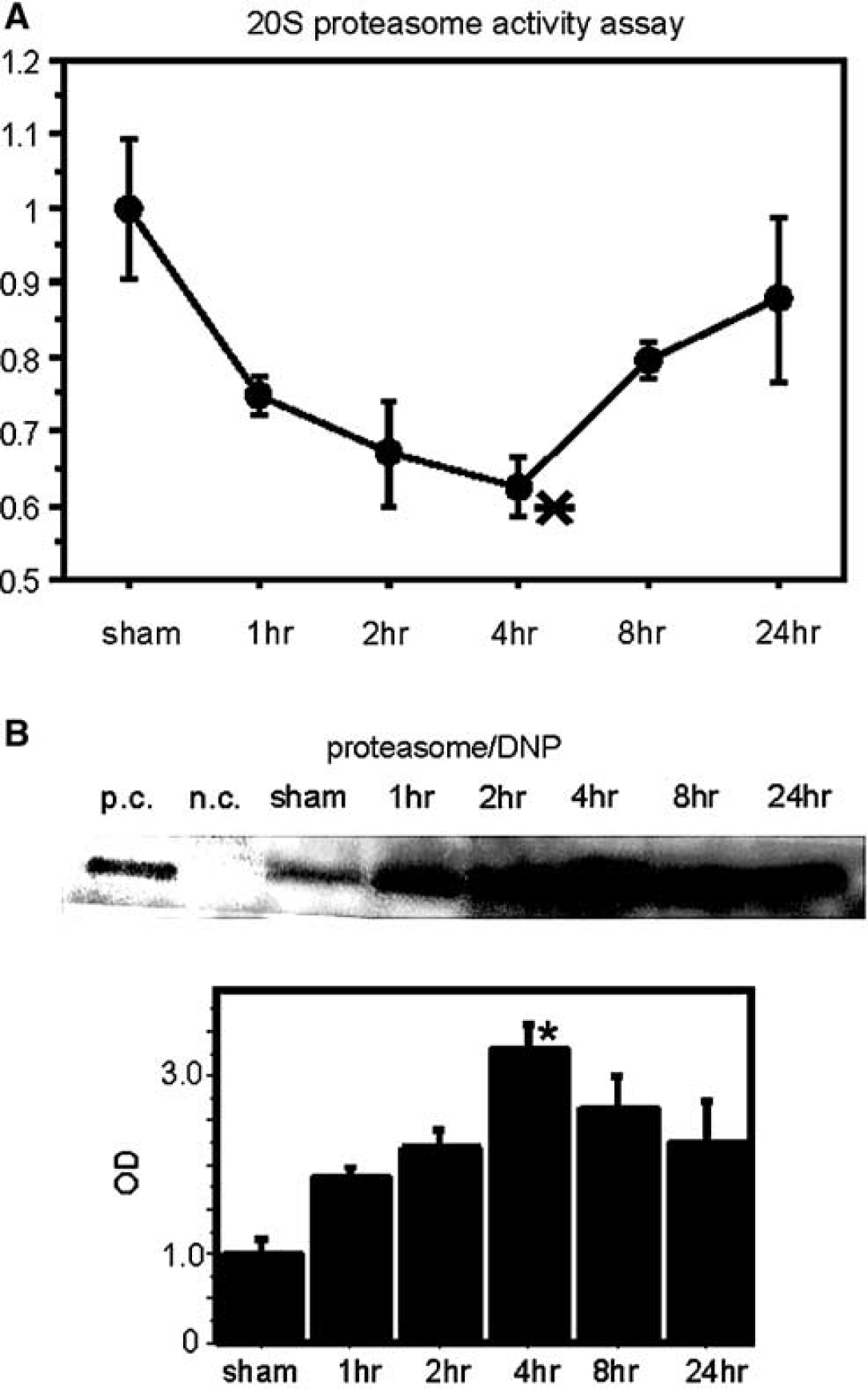
(
Neuronal MDM2 and Nuclear Translocation of pMDM2 after Transient Focal Cerebral Ischemia
We examined the MDM2 signaling pathway to analyze upstream p53 degradation after tFCI. Double immunofluorescent staining for MDM2 and NeuN showed that MDM2 protein expression colocalized with neurons in the ischemic cortex 24 hours after reperfusion (n=4; Figure 4A). We used the antibody which can detect both phosphorylated and nonphosphorylated MDM2, to examine neuronal expression by immunohistochemistry. Western blot analysis showed that cytosolic pMDM2 was detected as a 90-kDa band (n=4; Figure 4B) and that it transiently increased 1 and 2 hours after tFCI, after which it gradually decreased (Figure 4B). In contrast, nuclear pMDM2 gradually increased after tFCI and then significantly increased 24 hours after tFCI (Figure 4B). These results suggest that MDM2 translocates from the cytosolic space into the nucleus during the early period of reperfusion injury after tFCI.
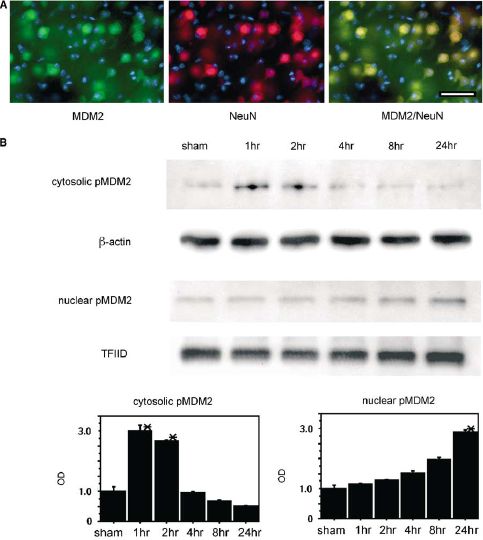
(
Direct Interaction of Phosphorylated Akt with pMDM2 and Nuclear Translocation of p53 and the MDM2 Complex after Transient Focal Cerebral Ischemia
Coimmunoprecipitation revealed that the complex of phosphorylated Akt (pAkt) and pMDM2 (pAkt/pMDM2) transiently increased 1 hour after tFCI (n=4; Figure 5A). Coimmunoprecipitation revealed that the complex of p53 and MDM2 (p53/MDM2) was detected in the nuclear fraction and the cytosolic fraction after tFCI (n=4; Figure 5B). Nuclear expression of p53/MDM2 transiently increased 1 hour after tFCI and thereafter it gradually decreased (Figure 5B). Cytosolic p53/MDM2 gradually increased in a time-dependent manner after tFCI (Figure 5B). These results suggest that after tFCI MDM2 reacts directly with pAkt before reacting with p53.
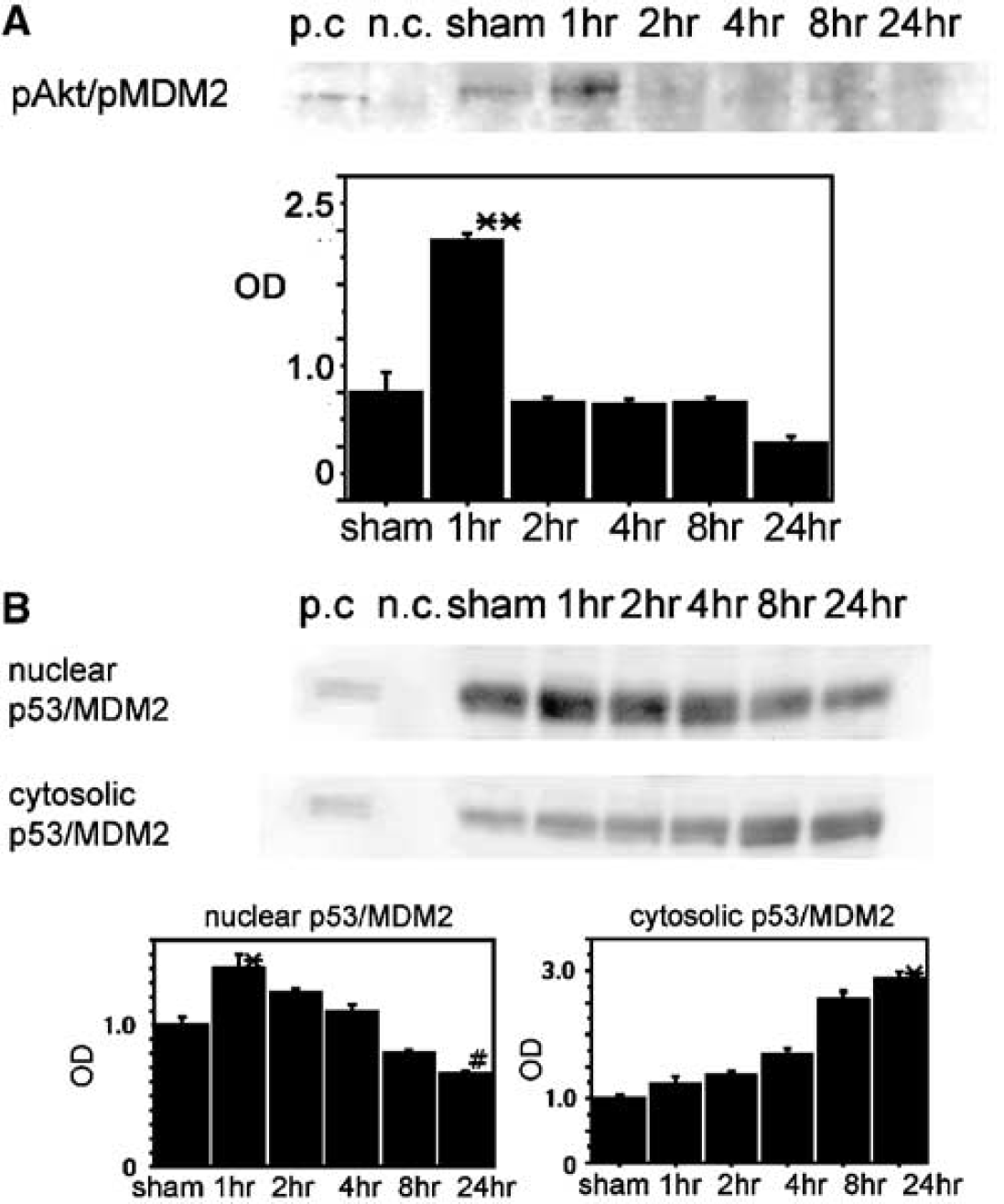
(
PI3-K Affects the Reaction of pMDM2 with pAkt and the Nuclear Translocation of pMDM2
Coimmunoprecipitation showed that the expression of pAkt/pMDM2 was inhibited in the LY294002-treated mice compared with the vehicle-treated mice 1 hour after tFCI (n=4; Figure 6A). Western blot analysis also revealed that expression of nuclear translocated pMDM2 was reduced in the LY294002-treated mice compared with the vehicle-treated mice 24 hours after tFCI (n=4; Figure 6B). These results suggest that PI3-K inhibition may prevent the reaction of pMDM2 with pAkt and the nuclear translocation of pMDM2 after reperfusion injury after tFCI.
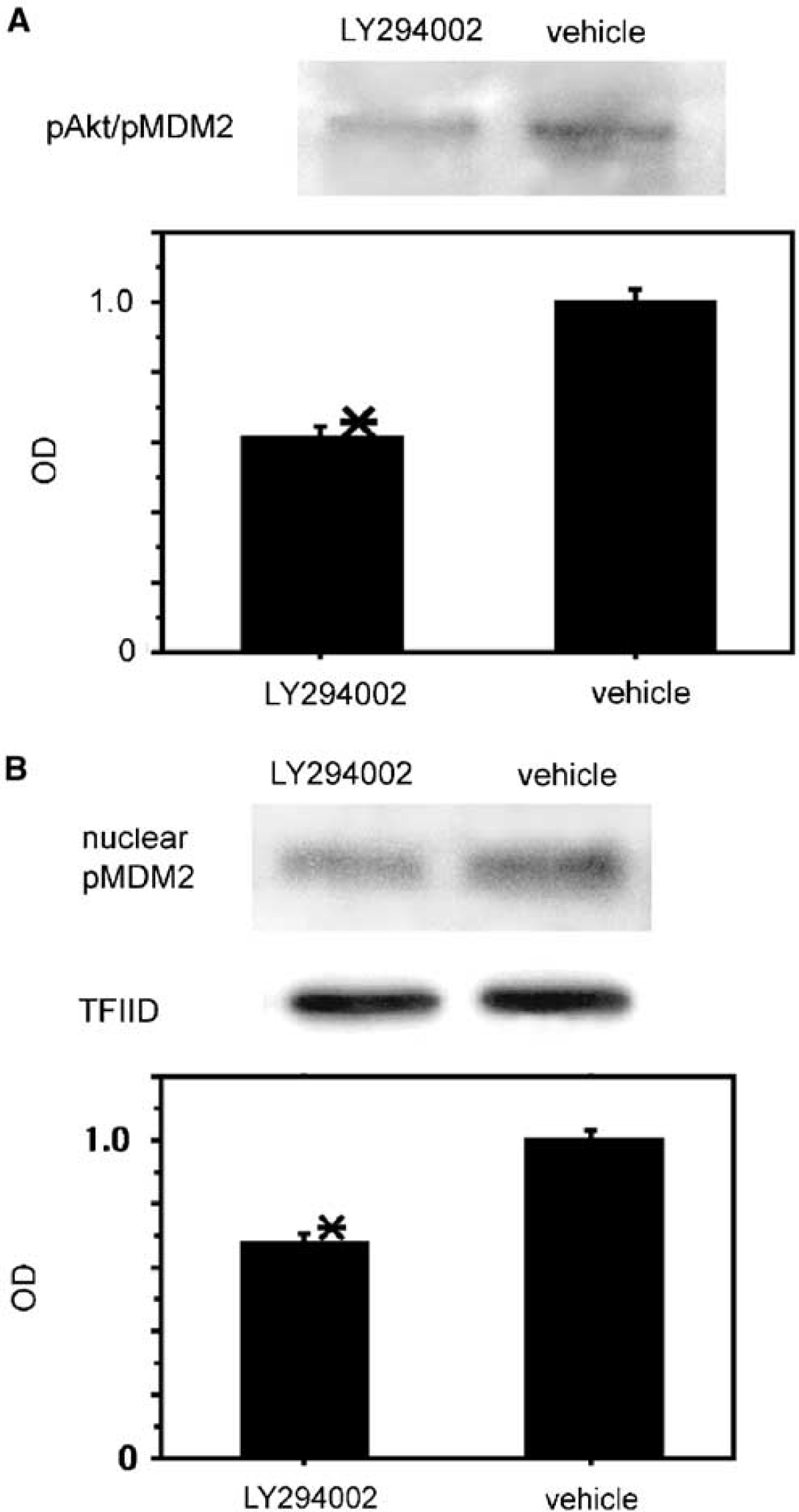
(
Overexpression of SOD1 Facilitated p53 Degradation and Reduced p53 Ubiquitylation after Transient Focal Cerebral Ischemia
Western blotting revealed that nuclear p53 significantly decreased 4 and 24 hours after tFCI in the SOD1 Tg mice compared with the wild-type mice (n=4; Figures 7A and 7B). Western blotting revealed that cytosolic p53 significantly increased 4 hours after tFCI, however, it significantly decreased 24 hours after tFCI in the SOD1 Tg mice compared with the wild-type mice (n=6; Figures 7A and 7B). To clarify the mechanism of this effective role of SOD1, we examined the relationship between superoxide and protein ubiquitylation, and the relationship between superoxide and both the MDM2 signaling pathway and the ubiquitin–proteasome system, which are involved in the regulation of protein ubiquitylation after tFCI. The sandwich ELISA showed that multiubiquitin accumulation was significantly reduced in the SOD1 Tg mice compared with the wild-type mice 8 hours after tFCI (n=4; Figure 7C). The cytosolic accumulation of p53/ubiquitin was remarkably reduced in the SOD1 Tg mice compared with the wild-type mice 8 hours after tFCI (n=4; Figure 7D). These results suggest that overexpression of SOD1 facilitates the cytosolic translocation of p53 and degradation of p53 after tFCI and that it prevents accumulation of multiple ubiquitylation and ubiquitylated p53 after tFCI.
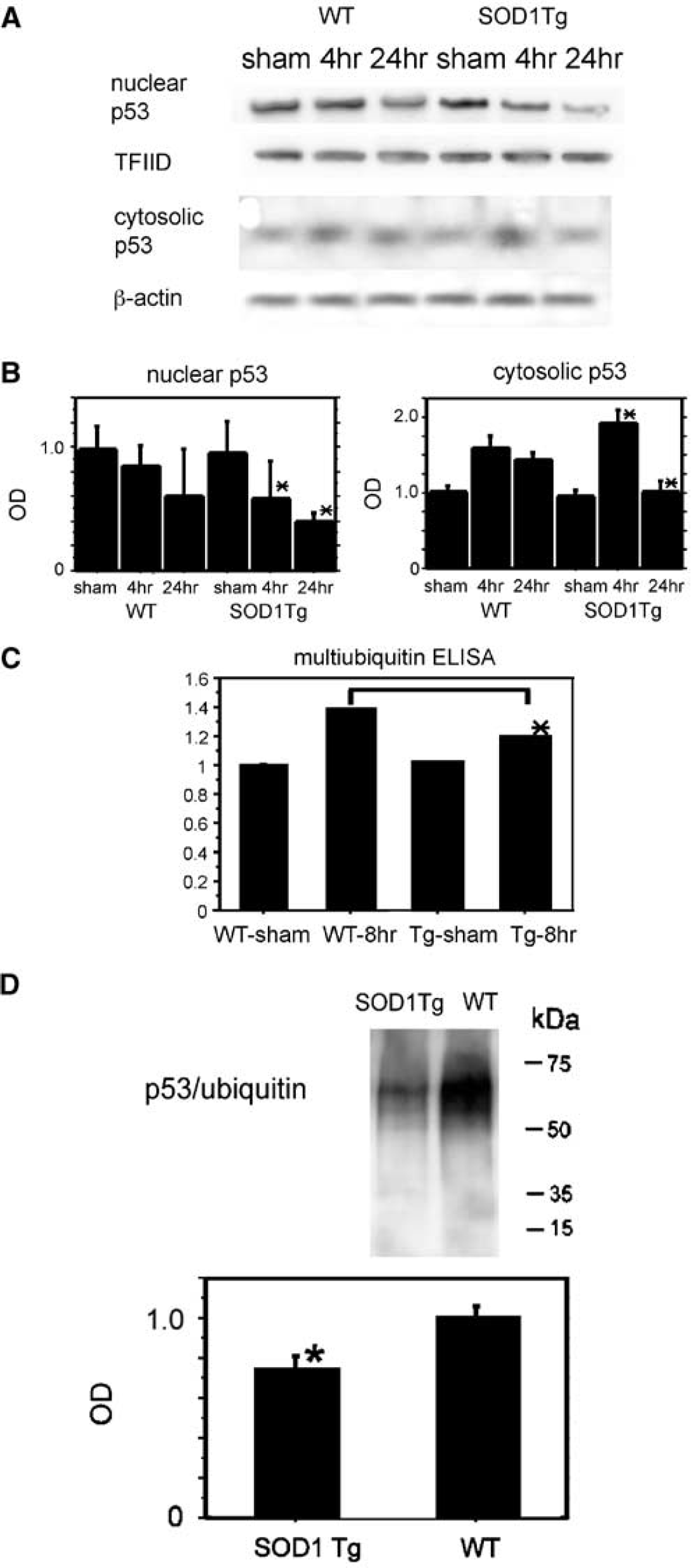
(
Overexpression of SOD1 Prevented Proteasome Inhibition and Direct Oxidative Injury to the Proteasome after Transient Focal Cerebral Ischemia
A proteasome activity assay revealed that inhibition of proteasome activity was reduced in the SOD1 Tg mice compared with the wild-type mice 4 hours after tFCI (n=4; Figure 8A). Coimmunoprecipitation showed that proteasome/DNP expression was significantly reduced in the SOD1 Tg mice compared with the wild-type mice 4 hours after tFCI (n=4; Figure 8B). These results suggest that oxygen radicals directly oxidize the proteasome after tFCI and that overexpression of SOD1 protects against oxidative damage by scavenging superoxides after tFCI.
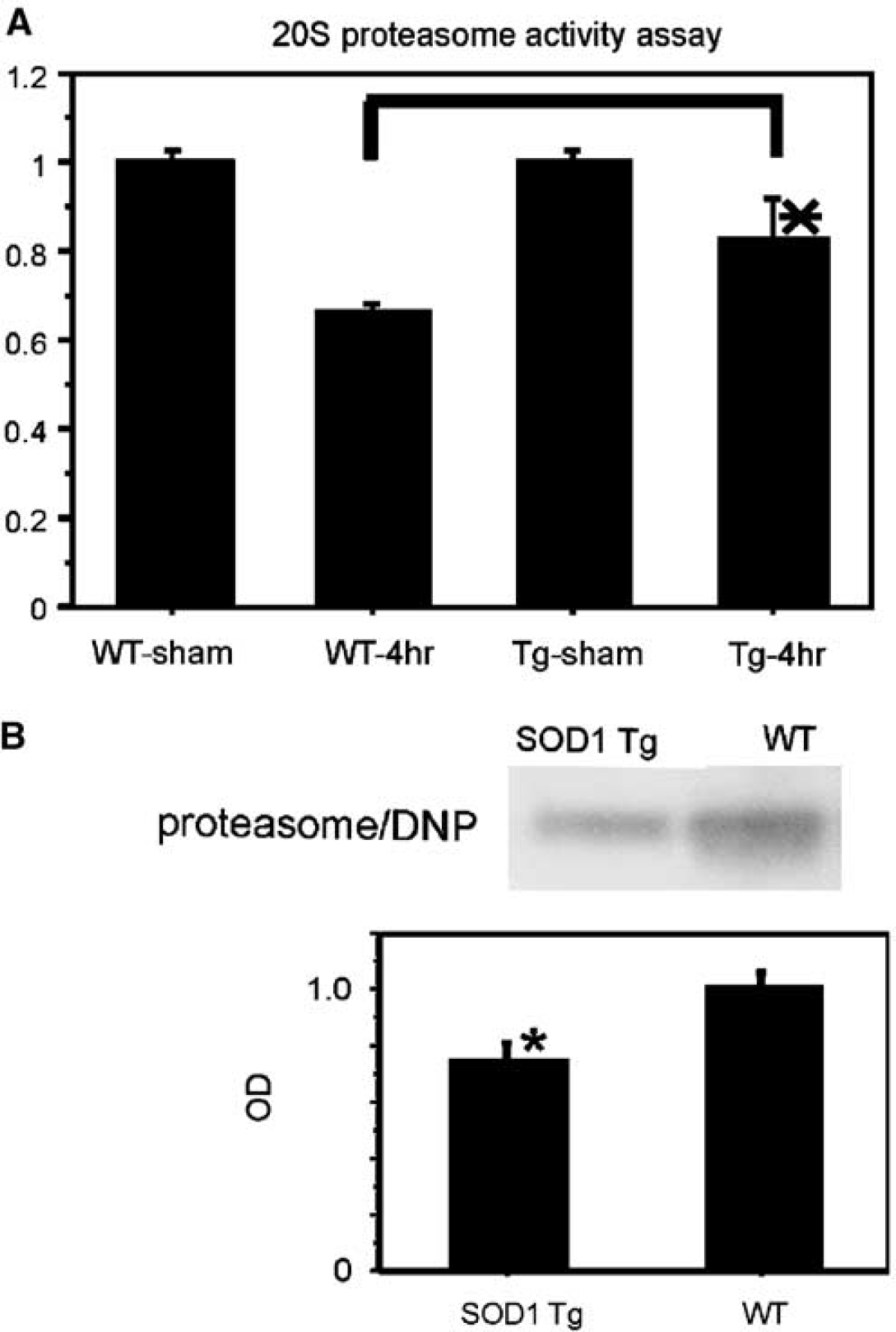
(
Overexpression of SOD1 Promoted the MDM2 Signaling Pathway after Transient Focal Cerebral Ischemia
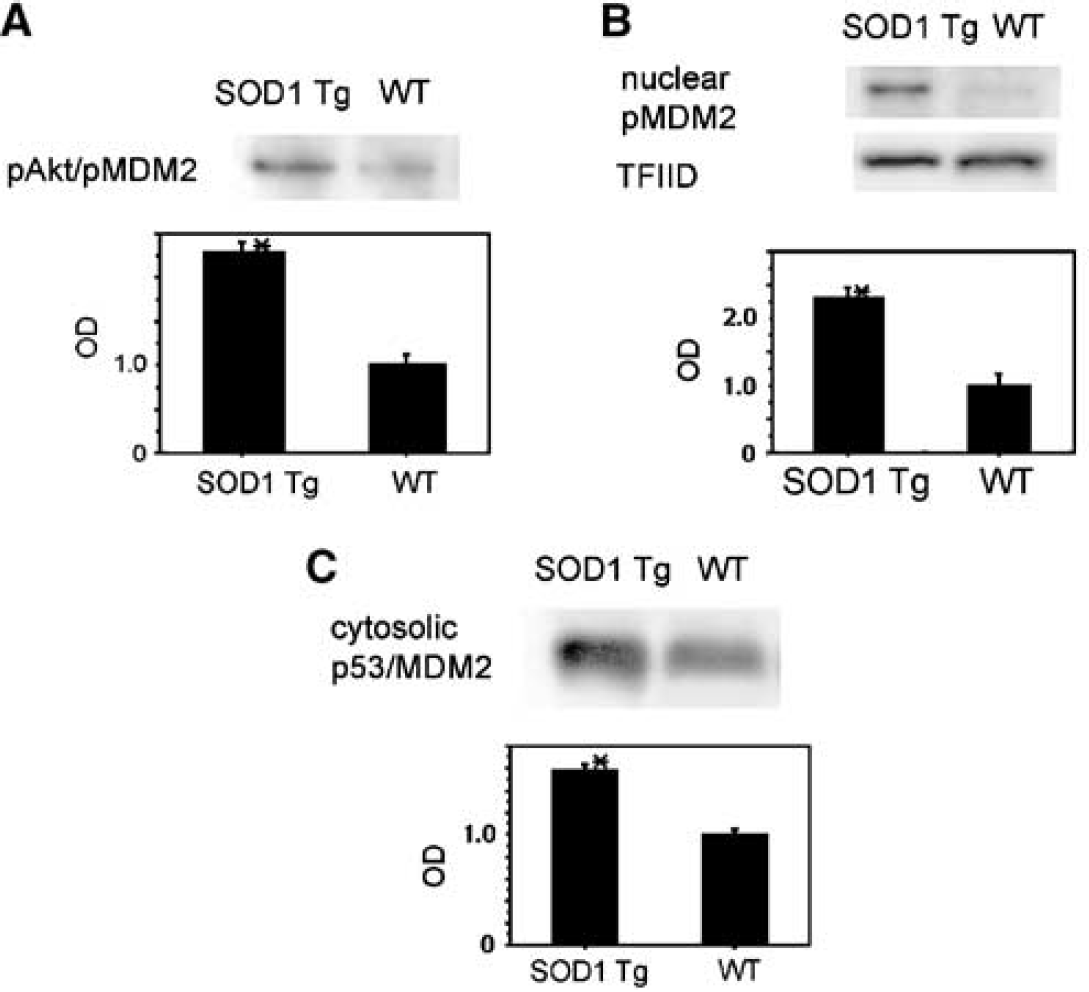
Coimmunoprecipitation showed that pAkt/pMDM2 was promoted in the SOD1 Tg mice compared with the wild-type mice 1 hour after tFCI (n=4; Figure 9A). In our previous study, pAkt expression began to increase 1 hour after tFCI and was promoted in the SOD1 Tg mice compared with the wild-type mice after tFCI (Noshita et al, 2003). Western blot analysis revealed that the nuclear translocated pMDM2 increased in the SOD1 Tg mice compared with the wild-type mice 24 hours after tFCI (n=4; Figure 9B). These results suggest that nuclear translocation of pMDM2 can be detected during the early period of reperfusion injury after tFCI and that PI3-K inhibition and superoxide may prevent it after tFCI. The cytosolic p53/MDM2 complex was promoted in the SOD1 Tg mice compared with the wild-type mice 24 hours after tFCI (n=4; Figure 9C). These results suggest that overexpression of SOD1 promotes nuclear translocation of pMDM2 and binding to pAkt and that it facilitates translocation of the p53/MDM2 complex after tFCI. Superoxide production may prevent p53 degradation that is mediated by inhibition of the MDM2 signaling pathway after tFCI.
(
Discussion
Ubiquitylated protein aggregation is a characteristic feature of a variety of neurological disorders, including chronic neurodegenerative disorders and an acute pathological state of neuronal cells (Chin et al, 2002; Mengesdorf et al, 2002). Massive accumulation of stress proteins is believed to be one of the key factors in the aggravation of some neurological disorders, and rapid and appropriate degeneration of stress response proteins is required for cell survival (Chin et al, 2002; Yang and Yu, 2003). The ubiquitin–proteasome system plays an important role in controlling various cellular processes such as cell cycle progression, signal transduction, cell transformation and apoptotic cell death (Schwartz et al, 1990; Delic et al, 1993; Grimm et al, 1996; Dimmeler et al, 1999; Orlowski, 1999; Yang and Yu, 2003). In cerebral ischemia in vivo, multiubiquitin chains persistently increased in the area where neurons are destined to die (Ide et al, 1999). In contrast, in ischemic lesions, multiubiquitin chains containing degraded proteins transiently increased in the ischemic-tolerant regions where neuronal cells survived after ischemic insult (Ide et al, 1999). Accumulation of degraded proteins may affect the destination of neuronal cells under a stressful condition such as cerebral ischemia. In hippocampal CA1 lesions after transient forebrain ischemia, this accumulation of multiubiquitin transiently increased concomitantly with the transient inhibition of proteasome activity (Ide et al, 1999; Asai et al, 2002). A proteasome functions in an ATP-dependent manner, and protease activity is reduced under ATP-depleting conditions in vitro and in cerebral ischemia in vivo (Tanahashi et al, 2000; Asai et al, 2002). The 26S proteasome is cleaved after ischemic insult and its reassembly is thought to be impaired after ischemia (Tanahashi et al, 1998; Asai et al, 2002). Cerebral ischemia- and reperfusion-induced proteasomal dysfunction has been suggested to be involved in the accumulation of multiubiquitin chains. The increase in inappropriate multiubiquitin proteins may aggravate apoptotic neuronal cell death after cerebral ischemia (Ide et al, 1999; Asai et al, 2002).
In the present study, we have shown for the first time the following: (1) Cytosolic translocation and degradation of p53 were observed after tFCI. (2) Ubiquitylated p53 and multiple ubiquitylated proteins accumulated after tFCI. (3) Proteasome activity was transiently inhibited, and oxidative injury may directly damage the proteasome after tFCI. (4) pMDM2 reacted directly with pAkt and translocated into the nucleus after tFCI. (5) The reaction of pMDM2 with pAkt was involved with PI3-K after tFCI. (6) p53 formed a complex with MDM2 and the complex translocated into the cytoplasm after tFCI. Translocation of the p53/MDM2 complex was involved with PI3-K after tFCI. (7) p53 degradation was facilitated by SOD1 after tFCI. (8) Multiubiquitin accumulation and p53 ubiquitylation were inhibited by SOD1 after tFCI. (9) Inhibition of and oxidative injury to the proteasome were prevented by SOD1 after tFCI. (10) Direct binding of pMDM2 to pAkt, the nuclear translocation of pMDM2, and the cytosolic translocation of the p53/MDM2 complex were promoted by SOD1 after tFCI. Our previous study showed that apoptotic neuronal cell death significantly increased in the cortical penumbra lesion around the ischemic core of the caudate and putamen after tFCI in LY294002-treated mice compared with vehicle-treated mice 24 hours after tFCI. In contrast, apoptotic neuronal cell death was remarkably reduced in the same lesion in the SOD1 Tg mice compared with the wild-type mice 24 hours after tFCI. In our studies, the samples obtained for blotting analyses include this penumbra area. The significant difference in the blot data between these two groups should be based on the mechanistic difference as to how apoptotic neuronal cell death would proceed between these groups. These results suggest that p53 degradation may be regulated by both the MDM2 signaling pathway and the ubiquitin–proteasome system in neuronal cell death after in vivo cerebral ischemia, as has been shown with in vitro studies. Moreover, overexpression of SOD1 may play an important role in regulating these two pathways after tFCI. We hypothesize that superoxide production may prevent p53 degradation via inhibition of these pathways after tFCI (Figure 10).
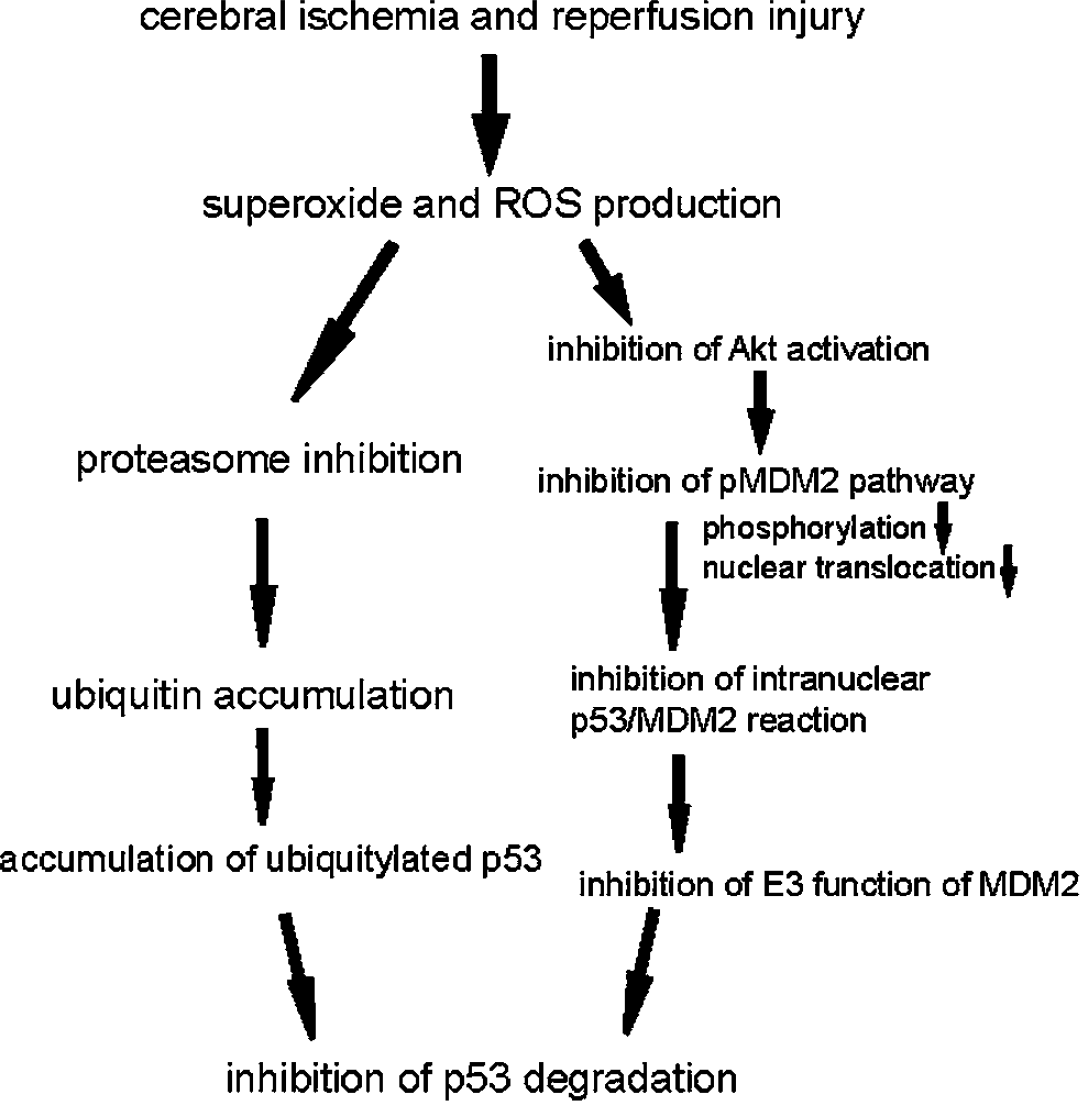
Scheme demonstrating that superoxide production after cerebral ischemia inhibits both the MDM2 signaling pathway and the ubiquitin–proteasome system, which prevents p53 degradation after tFCI.
Akt, a serine/threonine protein kinase, is a multifunctional regulator of cell survival, cell growth, and glucose metabolism (Datta et al, 1999). Akt contains an amino-terminal pleckstrin homolog domain, which binds phosphorylated lipids at the membrane in response to activation of PI3-K (Franke et al, 1997). pAkt activates many substrates such as Bad, forkhead transcription factor, DAF-16 homologs, the tuberous sclerosis complex-2 tumor suppressor gene (tuberin), and MDM2, via their phosphorylation, and it plays a critical role in the regulation of cell survival after a variety of apoptotic stimuli (Datta et al, 1999; Cahill et al, 2001; Mayo and Donner, 2001). In apoptotic models in vitro, pAkt activated pMDM2, which then promoted the nuclear translocation of MDM2 (Mayo and Donner, 2001; Ogawara et al, 2002). In cerebral ischemia in vivo, pAkt transiently increased during the early period of reperfusion injury after tFCI in ischemic cortical penumbra lesions around the core lesion (Noshita et al, 2001). SOD1 overexpression is involved with the PI3-K pathway and promotes expression of pAkt after tFCI (Noshita et al, 2003). Moreover, overexpression of SOD1 promotes Bad phosphorylation after tFCI (Saito et al, 2003b). In the present study, MDM2 reacted with pAkt and its nuclear translocation was prevented by PI3-K inhibition. These results suggest that the nuclear translocation of MDM2 may be regulated by PI3-K via the reaction with pAkt after cerebral ischemia. Overexpression of SOD1 promoted the reaction of MDM2 with pAkt and the nuclear translocation of MDM2 after tFCI. These results lend support to our previous studies on the Akt cell survival pathways after tFCI, one of which showed that pAkt transiently increased after tFCI in the same mouse ischemia model (Noshita et al, 2001). The time course of the transient increase in pMDM2 was in accord with the increase in pAkt, and pMDM2 expression may be closely associated with pAkt expression after cerebral ischemia. PI3-K inhibition can reduce the expression of pAkt and could affect the interaction of pAkt with MDM2 and its ability to phosphorylate MDM2. However, PI3-K inhibition is also known to aggravate apoptotic neuronal cell death after cerebral ischemia (Noshita et al, 2001). There is a possibility that the decrease in pAkt/pMDM2 via treatment with LY294002 may be a result of the aggravation of neuronal damage by direct PI3-K inhibition or other signaling pathways of the PI3-K signaling pathway. We found that MDM2 formed a complex with p53 in the nucleus, that this complex translocated into the cytoplasm after tFCI, and that superoxide was involved with the interaction of p53/MDM2 after tFCI. In vitro studies, however, showed that MDM2 regulated p53 expression and its function reciprocally in the nucleus, but the precise mechanisms of the interaction of MDM2 with p53 and superoxide remain unclear. These mechanisms are thought to be complicated (Haupt et al, 1997; Oren, 1999; Kawai et al, 2003). Further studies are needed to clarify the role of MDM2 in the p53 pathway in apoptosis in vivo and its relationship to superoxide.
In this study, we first examined 20S proteasome activity after tFCI. We selected the 20S proteasome subunit, which exists in the core of the 26S proteasome, because it functions as a central active site (DeMartino and Slaughter, 1999). The carbonyl residues oxidized by oxidative stress were directly observed by the DNP reaction after tFCI. We previously showed that in situ mitochondrial superoxide production remarkably increased 1 and 2 hours after tFCI in the MCA territory and that SOD1 overexpression dramatically reduced this production (Saito et al, 2003b). We speculate that superoxide may spread diffusely into the cytosolic space after reperfusion after tFCI. These superoxide radicals can diffusely form the highly reactive hydroxyl radicals via the Fenton reaction or form peroxynitrite with nitric oxide. These ROS can directly and nonselectively damage a variety of proteins during the early period of reperfusion injury. Detection of the DNP-binding site is an appropriate method for examination of oxidative damage to an individual protein (Levine et al, 1994). The peak time point of oxidative injury to the proteasome was in accord with the peak time point of the inhibition of proteasome activity, and its damage was reduced by SOD1 overexpression. Moreover, accumulation of multiubiquitin increased according to proteasome inhibition. Overexpression of SOD1 attenuated this accumulation after tFCI. We suggest that superoxide production and its ROS products may be directly involved in proteasome inhibition after tFCI and accumulation of multiubiquitin, which was promoted by proteasome inhibition after cerebral ischemia.
p53 ubiquitylation is controlled by MDM2, a specific E3 ubiquitin ligase for p53, and by proteasome activity. As shown in the scheme in Figure 10, superoxide production and ROS production during reperfusion after stroke may damage the mechanisms of both the MDM2 signaling pathway and the ubiquitin–proteasome system, and, as a result, may prevent p53 degradation via ubiquitylation. Accumulation of multiubiquitin proteins was observed even in whole cytosolic proteins. SOD1 overexpression inhibited them as well as p53 ubiquitylation. These results suggest that superoxide and ROS may inhibit the degradation of regulatory proteins after oxidative injury to both the E3 ubiquitin ligase and the proteasome, and that this accumulation may aggravate neuronal cell death after reperfusion injury after cerebral ischemia. Moreover, there is a possibility that the effects of superoxide and ROS might be extensively involved in the mechanisms of other diseases, such as neurodegenerative diseases, which are caused by the aggregation of ubiquitylated proteins. However, little is known about the precise mechanisms of the relationship between E3 ubiquitin ligase and the proteasome or the relationship between superoxide and protein degradation systems, and our technical procedure for the evaluation of the p53/ubiquitylation complex did not show direct evidence of p53 ubiquitylation. Further studies are needed to detail the mechanisms of the degradation and ubiquitylation of p53.
Conclusion
In summary, our results imply that p53 degradation is controlled by the MDM2-mediated ubiquitylation pathway and the ubiquitin–proteasome system after tFCI, and that superoxide and ROS may promote apoptotic progression by preventing p53 degradation via inhibition of both these mechanisms after tFCI.
Footnotes
Acknowledgements
We are grateful to Dr. Charles J Epstein, Department of Pediatrics, University of California, San Francisco, for the breeding pairs of SOD1 transgenic mice. We thank Liza Reola, Bernard Calagui and Maricela González for technical assistance, Cheryl Christensen for editorial assistance and Elizabeth Hoyte for figure preparation.