
Abstract
Oligodendrocytes are one of the major cell types in cerebral white matter. Under normal conditions, they form myelin sheaths that encircle axons to support fast nerve conduction. Under conditions of cerebral ischemia, oligodendrocytes tend to die, resulting in white-matter dysfunction. Repair of white matter involves the ability of oligodendrocyte precursors to proliferate and mature. However, replacement of lost oligodendrocytes may not be the only mechanism for white-matter recovery. Emerging data now suggest that coordinated signaling between neural, glial, and vascular cells in the entire neurovascular unit may be required. In this mini-review, we discuss how oligodendrocyte lineage cells participate in signaling and crosstalk with other cell types to underlie function and recovery in various experimental models of subcortical white-matter injury.
Introduction
Advancement in imaging technology allows us to recognize the prevalence of subcortical white-matter pathology-related small vessel ischemia. White-matter hyperintensity on brain magnetic resonance imaging is an important indicator for cerebrovascular dysfunction of subcortical region. These subcortical ischemic vascular diseases (SIVDs) are complex and heterogenous, but two major categories of SIVDs comprise multiple lacunar strokes and Binswanger disease. They cause progressive cognitive impairment, resulting in subcortical vascular dementia. These diseases are accelerated by several risk factors such as age, diabetes, and hypertension. However, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) syndrome, which is also categorized as SIVD, has little relation to standard vascular risk factors. It is a common monogenic inherited form of degenerative small vessel disease with mutations in Notch3 gene. However, regardless of the potentially different etiologies, most SIVDs may generally involve small vessel stenosis/occlusion, hypoxic conditions that trigger inflammation, disruption of neurovascular matrix integrity, perturbations in blood–brain barrier (BBB) homeostasis, and accumulated white-matter pathology associated with loss of myelin and axon injury.1,2
Oligodendrocytes are one of the major glial cells in cerebral white matter. Oligodendrocytes produce a lipid-rich membrane and form myelin sheaths by encircling axonal bundles. A single oligodendrocyte cell can myelinate up to 50 axonal segments. Myelin has a unique composition and richness in lipids, and its low water content allows the electrical insulation of axons by forming myelin sheath. The unique segmental structure of myelin sheaths can achieve saltatory nerve impulse conduction for the fast nerve conduction. 3 White-matter ischemia is generally severe with rapid cell swelling and tissue edema because of the low blood flow and little collateral blood supply. 4 And, oligodendrocytes are vulnerable to excitatory amino acids and oxidative stress. Since a single oligodendrocyte myelinates multiple axons, damage to only one oligodendrocyte could cause severe dysfunction in white matter. As a compensatory response, residual oligodendrocyte precursor cells (OPCs) may proliferate and differentiate into mature oligodendrocytes to remyelinate injured axons during the chronic phase of SIVDs. Notably, these endogenous compensatory responses not only occur in oligodendrocyte lineage cells alone. Instead coordinated signaling between multiple cell types in the white matter may be required for oligodendrocyte repair and remodeling.
The concept of neurovascular unit is now relatively well accepted for dissecting brain physiology and pathophysiology. The ‘neurovascular unit’ provides a conceptual framework that emphasizes cell–cell interactions between neuronal, glial, and vascular elements.
4
–
10
This concept primarily guides research in neuron-related cell–cell interaction mechanisms in gray matter. But cell–cell signaling between non-neuronal cells should also be critical for white-matter function. As shown in the diagram in Figure 1, cells in cerebral white matter are closely related to each other. For example, oligodendrocytes enwrap axons for efficient conduction of electrical impulses, and oligodendrocyte–axon interaction would be one of the most well-documented aspects of neurovascular unit in white matter. In fact, oligodendrocyte-derived trophic factors such as insulin-like growth factor-1 (IGF-1) and glial cell-derived neurotrophic factor increase axonal length and support neuron survival.
11
Oligodendrocyte precursor cells are also an important component in the neurovascular unit in cerebral white matter. They are known to be adjacent to astrocytes or pericytes, but recent reports suggest that OPCs are also closely located to cerebral endothelial cells.12,13 Although OPCs are most active in the developing stage for maturating into oligodendrocytes to form myelin sheath, non-negligible number of OPCs resides in adult white matter. These remaining OPCs in adult white matter may work on oligodendrocyte renewal and myelin maintenance. Furthermore, OPCs in adult brains actively participate in white-matter repair after injury. But again, OPCs ought to receive support from their neighboring cells to play the assigned roles. In this mini-review, we will summarize the current findings as to how intra/inter cell signaling in oligodendrocytes/OPCs functions in the context of the neurovascular unit under normal and SIVD conditions.
Schematic of the neurovascular unit in white-matter. Neuronal axons, astrocytes, cerebral endothelial cells, pericytes, and oligodendrocytes/oligodendrocyte precursor cells (OPCs) comprise the neurovascular unit in white matter. The concept of neurovascular unit emphasizes the functional aspects of cell–cell signaling, and in fact, the components are closely located and presumably exchange signals to support their function of each other.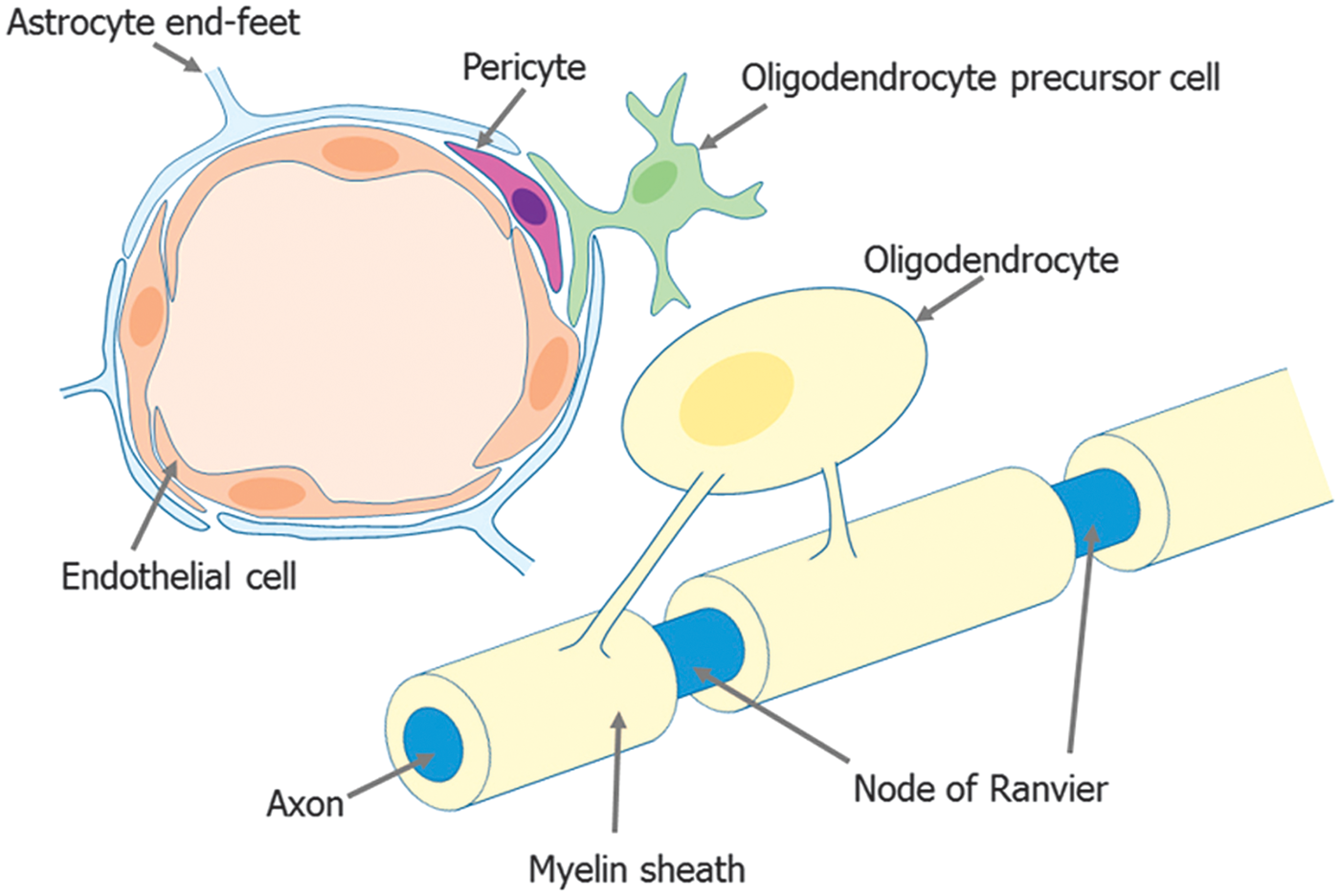
Oligodendrocyte damage and repair in subcortical ischemic vascular disease
Oligodendrocyte lineages cells (oligodendrocytes and OPCs) are vulnerable to ischemic stress. In the exacerbation phase of SIVDs, the structural changes of oligodendrocytes are observed rapidly, and are sometimes accompanied by BBB breakdown. Oligodendrocytes can be easily damaged by oxidative stress, excitatory amino acids, and inflammatory cytokines. In contrast, the number of OPCs would increase in the acute phase, especially in the sublesion area (i.e., ischemic penumbra). This phenomenon is recognized as a compensatory response. In fact, under the chronic phase of white-matter diseases, residual OPCs tend to proliferate, migrate to the legion area, and then mature to compensate the injured axons. Nevertheless, prolonged ischemic conditions generally lead to progressive white-matter damage with loss of myelin and oligodendrocytes. Notably, there may be a significant difference in OPC response to white-matter injury between young and aged brains. In general, younger white matter exhibits more oligodendrogenesis (oligodendrocyte regeneration), and correspondingly, aged brains would show a more severe white-matter damage after injury.14,15 Importantly, these oligodendrocyte/OPC responses under white-matter diseases may be regulated by neighboring cells. Although precise mechanisms in oligodendrocyte function and dysfunction in SIVD conditions are still understudied, several important findings in cell–cell interactions of oligodendrocytes and other cells have been reported.
In vitro model for oligodendrocyte damage.
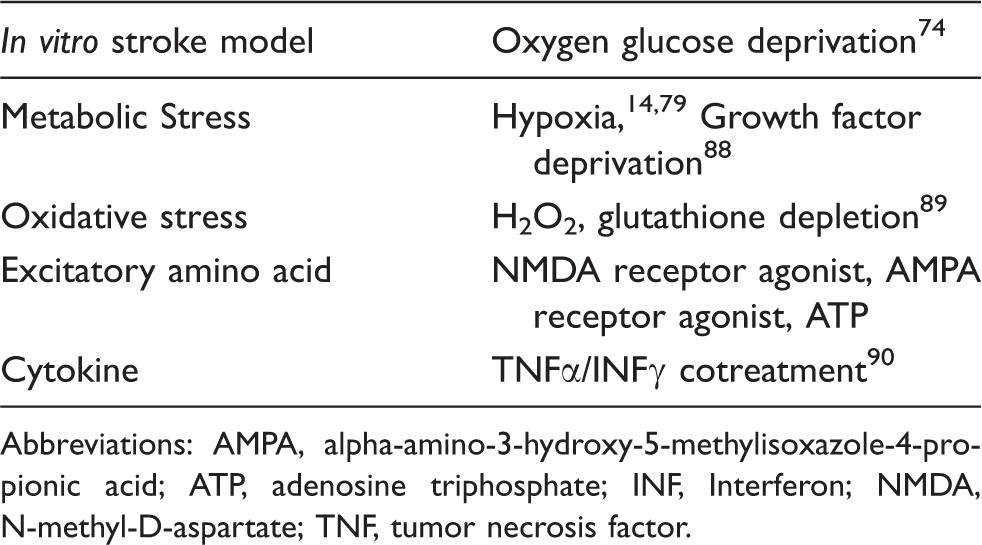
Abbreviations: AMPA, alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; ATP, adenosine triphosphate; INF, Interferon; NMDA, N-methyl-D-aspartate; TNF, tumor necrosis factor.
Intracellular/intercellular signaling pathways that regulate oligodendrocyte function after injury.

Abbreviations: AMPA, alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; bHLH, basic helix-loop-helix; CNTF, ciliary neurotrophic factor; Ezh2, Enhancer of zeste homolog 2; HDAC, histone deacetylase; miRNA, microRNA; NMDA, N-methyl-D-aspartate; OPC, oligodendrocyte precursor cells; PDGF, platelet-derived growth factor; Shh, Sonic Hedgehog.
Roles of astrocyte/endothelium/microglia in OPC maturation in vitro.
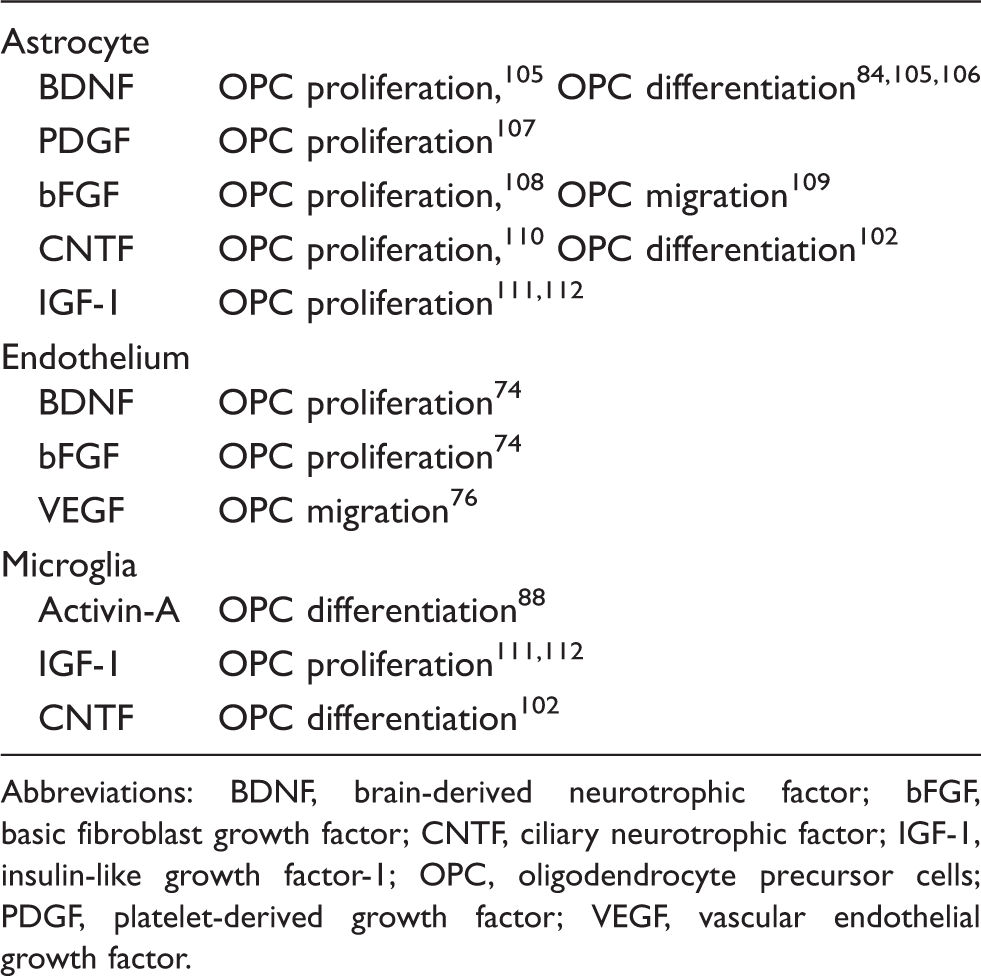
Abbreviations: BDNF, brain-derived neurotrophic factor; bFGF, basic fibroblast growth factor; CNTF, ciliary neurotrophic factor; IGF-1, insulin-like growth factor-1; OPC, oligodendrocyte precursor cells; PDGF, platelet-derived growth factor; VEGF, vascular endothelial growth factor.
Although in vitro models are very helpful and useful to understand the oligodendrocyte pathology, it is also essential to study in vivo models to have a better understanding at defining the roles of cell–cell interaction in white matter. Therefore, in the next section, we will introduce in vivo rodent models of white-matter ischemia and summarize how oligodendrocytes and OPCs behave in these models.
Experimental models for subcortical ischemic vascular diseases
Rat Model of Bilateral/Unilateral Common Carotid Artery Occlusion
The two-vessel occlusion by permanent bilateral occlusion of the common carotid arteries has been extensively used as a rat model of white-matter ischemia. 20 This model is characterized by pathologic changes in the corpus callosum, the internal capsule, optic nerve, and optic tract.21,22 The number of oligodendrocytes in white matter decreases after 7 days of ligation, and correspondingly, upregulation of caspase-3 activation in oligodendrocytes is observed. 23 In addition, the animals develop demyelination with axonal damage, 22 which appears similar to that found in human white-matter lesions. The expression level of myelin basic protein, which is a major component of myelins, is also decreased. 24 On the contrary, the number of OPCs is temporarily increased, but at later time point, OPCs seem to be injured as well. 24 Moreover, this model also shows an increase in matrix metalloproteinases (MMPs) and associate with disruption of BBB, 25 which are similar characteristics observed in clinic. Although this model is technically easy to perform and the pathologic changes are well validated, there is at least one potential drawback. Since the visual pathway is injured by carotid artery occlusion, some standard behavioral tests would not be suitable for assessing neurologic function. 21 Unilateral carotid artery occlusion model has also been used for evaluating white-matter ischemic stroke in rats. 26 Similar to the rat bilateral common carotid artery occlusion model, the unilateral model shows myelin/oligodendrocyte loss, immature oligodendrocyte increase, MMPs increase, and BBB damage. But it should be noted that spontaneous hypertensive/stroke prone rats are preferable for the unilateral model because occluding one carotid artery has little impact in other rat strains. Since severe bilateral carotid artery stenosis are rare in the patients, this unilateral carotid artery model in spontaneous hypertensive/stroke prone rats may have a close relation to the cause of white-matter damage in clinic.
Mouse Model of Bilateral Common Carotid Artery Stenosis
The mouse model of bilateral common carotid artery stenosis 27 is now relatively well accepted as a mouse model of white-matter diseases. In this model, micro-coils (0.18 mm diameter coil is generally used) are placed at both common carotid arteries to cause the cerebral hypoperfusion. The animals exhibit similar white-matter pathologies, including demyelination, axonal damage, and oligodendrocyte loss, as observed in clinic. The white-matter lesion is accompanied with BBB breakdown, which is at least partly due to the upregulation of MMP-9. 28 Similar to the rat model of bilateral common carotid artery occlusion, OPC number temporarily increases in acute phase, presumably as a compensatory response. 29 Notably, this OPC response is much lower as compared with older white matter. 14 The visual pathway is almost intact in this model, and therefore, the animals would be usable for behavioral tests. In fact, the 8-arm radial maze, water maze, and Y-maze tests show that the mice with prolonged cerebral hypoperfusion exhibit significant impairments in learning ability. 30 – 32 One potential drawback of this model is the acute decrease in cerebral blood flow by the micro-coil placement, which may not be so consistent with the phenomena in clinic (this point is also applied to the rat model of bilateral common carotid artery occlusion).
Rat/Mouse Model of 2-Vessel Gradual Occlusion Model
This model was recently developed to avoid the acute decrease in the cerebral blood flow observed in the rat model of bilateral common carotid artery occlusion and the mouse model of bilateral common carotid artery stenosis. After the placement of ameroid constrictor device to bilateral common carotid arteries, the device gradually narrows the arteries.33,34 For rats, the inner diameter of ameroid constrictor is 0.5 mm, the outer diameter 3.25 mm, and the length 1.28 mm. Rats with the ameroid constrictor device show selective white-matter pathologies in 4 weeks. The white-matter damage, oligodendrocyte loss, and demyelination observed in this model were milder than ones in the rat model of bilateral common carotid artery occlusion. 33 This model shows recovery of white-matter metabolism in chronic phase than in acute phase (at 28 days). However, oligodendrogenesis and remyelination are still unknown in this model. In addition, ameroid constrictor devices for mice are still under development. Mice with the ameroid constrictor device exhibit multiple cerebral infarctions in both gray and white matter, presumably due to the technical limitation to make appropriate size of ameroid constrictor devices for mice. 34
Rat/Mouse Model of Focal Injection of Vasoconstrictor
It is hard to control infarct lesion volume in rodent models of white-matter injury. However, stereotaxic injection of vasoconstrictor agents may manage the infarct lesion to some extent. The direct injection of vasoconstrictor agents into the subcortical white matter has been used to induce focal white-matter stroke both in rats and in mice. 35 – 37 For example, when endothelin-1 is injected into the internal capsule in rats, tissue necrosis and demyelination occur in 14 days, which eventually causes sensorimotor deficits. 35 Also in mice, microinjection of endothelin-1 into the subcortical white matter produces an infarct core as in human subcortical stroke, which accompanies with oligodendrocyte apoptosis, myelin loss, axonal fiber loss, and activation of microglia/ macrophages. 36 After the white-matter stroke, axon initial segment length in peri-infarct cortex shorten and new initial segments from surviving neurons can be observed. 38 This model shows reduction of BrdU-labeled OPCs and oligodendrocytes 1 month after stroke, but underlying mechanisms are still mostly unclear. 36 N5-(1-iminoethyl)-L-ornithine is also used as a vasoconstrictor to induce white-matter ischemia. In mice, N5-(1-iminoethyl)-L-ornithine injection leads to severe oligodendrocyte damage in aged mice, compared with younger animals. 15 A recent study using this model showed that brain-derived neurotrophic factor (BDNF) could influence oligodendrogenesis and remyelination after stroke. 39
Transgenic Mouse Lines for Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy is the most common monogenic inherited form of degenerative small vessel disease, 40 and genetic animal models have been developed to express it. It is linked to mutations in the NOTCH3 gene and the pathologic change of CADASIL is the pathognomonic accumulation of granular osmiophilic material within the tunica media in the proximity to the vascular smooth muscle cell membranes. 41 A recent report shows that not only vascular smooth muscle cells but also pericytes can associate with microvascular change in CADASIL. 42 To understand CADASIL pathologies, Notch3 knockout, knock-in and transgenic mouse models and notch3 mutant zebrafish have been developed.41,43,44 In the zebrafish model, cerebral white-matter changes were microvacuoles in the myelin sheaths associated with focal myelin degradation and occurring in the absence of oligodendrocyte loss and segmental intramyelinic edema was an early changing in CADASIL model mice. 43 Another study showed that Notch3 regulates OPC development and myelin basic protein gene and maintains vascular integrity using zebrafish notch3 mutants. 44 Notch3 may have corresponding roles in OPC development and in encouraging vascular integrity, and also, there is a possibility that notch3 might comprise the communication between OPC and vascular cell. 44
Rat/Mouse Model of Middle Cerebral Artery Occlusion
Strictly speaking, the standard middle cerebral artery occlusion (MCAO) models may not be categorized in animal models for SIVDs. However, the infarctions in the MCAO rats/mice sometimes include subcortical white-matter region, and therefore, white-matter pathophysiology and oligodendrocyte damage/repair mechanisms can be assessed with rat/mouse MCAO models. In MCAO animals, white-matter pathologies appear from the early stage after cerebral ischemia. In general, oligodendrocyte damage, including swelling and vacuolization, occurs after the BBB breakdown, and then, changes in axon/myelin structures are observed due to the fragmentation of myelin. 45 – 47 On the contrary, the number of OPCs increases in the ischemic penumbra after MCAO, which leads to the proliferation of immature (nonmyelinated) oligodendrocytes.46,48,49 The responses of oligodendrocyte lineage cells after ischemia may be affected by comorbidities, such as hypertension or diabetes. In fact, aged stroke-prone spontaneously hypertensive rats exhibit more white-matter damage with less OPC number and larger demyelination. 50 Furthermore, diabetes animals exacerbate the demyelination and delays the remyelination processes. 51
Cell–cell interaction between oligodendrocytes and other cells
As discussed, the concept of neurovascular unit defines the importance for cell–cell trophic coupling in brain. In fact, the dynamic interaction between oligodendrocytes and neighboring cells would support white matter to maintain their highly regulated function. In this section, we introduce how oligodendrocytes communicate with neurons, cerebral endothelial cells, and other kinds of glial cells.
Oligodendrocyte–Neuron Interaction
The major role of oligodendrocytes is to myelinate axons to support axonal signal transduction, and oligodendrocyte–neuron interaction is critical for white-matter function. It is well known that oligodendrocytes can signal to neurons via myelin–axon interactions.52,53 In addition, oligodendrocytes may support axonal survival through a myelin-independent mechanism since mouse models of oligodendrocyte injury, such as proteolipid protein (plp1)-null mice 54 and Cnp mutant mice, 55 do not show considerable demyelination but induce significant axon loss. 56 Furthermore, oligodendrocytes also metabolically support neuronal axons. Oligodendrocytes serve as a principal metabolic supplier of lactate, which is integral for axonal energy support, through monocarboxylate transporter 1. 57 In vitro experiments confirm that oligodendrocyte-derived trophic factors, such as IGF-1 and glial cell-derived neurotrophic factor, promote neuron survival and axon outgrowth. 11 In addition to mature oligodendrocytes, OPCs may also support neuronal function. The NG2 ectodomain is shed from OPCs in response to neuronal activity, and then the domain in turn modulates the neuronal network. 58 However, oligodendrocyte lineage cells are not always supportive for neurons, especially after injury. Oligodendrocyte-derived myelin expresses Nogo-A protein, which works as an inhibitory molecule for axonal extension. Under normal conditions, Nogo-A should have an important role in stabilizing axonal status. But under pathologic conditions including white-matter ischemia, the expression level of Nogo-A is increased 59 and the Nogo-A signaling suppresses axonal remodeling/repairing. 60 Oligodendrocyte precursor cells may also inhibit neuronal remodeling under pathologic conditions. The transgenic mice of OPC-specific beta-catenin knockdown exhibit reduced accumulation of activated microglia and reduced astrocyte hypertrophy, which are known to be a major burden for neuronal remodeling/repairing after injury. 61
In turn, neurons/axons may signal to oligodendrocyte lineage cells. For example, laminin is known as pro-myelination signals from neurons to determine proper myelin thickness.52,62 Neuregulin are known as pro-myelination signals from neurons. 52 Neuregulin-1, which is a soluble protein from neuronal cell surface, is also a promyelination signal, but overexpression of neuregulin-1 leads to hypermyelination of axons. 63 Wnt signaling may participate in the neuron-to-OPC signal in myelination, which acts in conjunction with Tcf4 to promote the initial stage of oligodendrocyte differentiation, but prevent subsequent differentiation steps unless downregulated. 52 In addition to extracellular matrix or releasing factors, the electrical activity in neuron/axon would drive myelination. In the developing mouse brain, the majority of divided NG2 cells (also known as polydendrocytes or OPCs) differentiate into oligodendrocytes. But after whisker removal, the density of oligodendrocytes was smaller in the deprived somatosensory cortex (i.e., barrel cortex region). 64 Also, subventricular zone-derived OPCs receive synaptic input in the white matter in a mouse model of remyelination, suggesting that similar to a developmental myelination, remyelination may be partially mediated by neuronal activity. 65 By contrast, some axonal surface ligands suppress oligodendrocyte differentiation/maturation. 52 For example, Jagged in axons inhibits OPC differentiation through the Notch pathway. 66 Other axonal ligands PSA-NCAM 67 or LINGO-168 are also known as inhibitory molecules for myelination. Under normal conditions, these pro- and anti-myelinating signals from neurons are rigorously regulated to maintain proper environments for myelinated axons. But after white-matter ischemia, the balances would be disturbed, resulting in oligodendrocyte loss or myelin damage.
Oligodendrocyte–Endothelium Interaction
One of the well-examined examples of cell–cell interaction in the neurovascular unit is neuron–endothelium trophic coupling in the neurovascular unit. The cell–cell signaling between cerebral endothelial cells and neuronal precursor cells help mediate and sustain pockets of ongoing neurogenesis and angiogenesis in adult brain.69,70 Even under the remodeling phase after brain injury, these close relationships are maintained, and both neurogenesis and angiogenesis occur in the neurovascular niche to promote repairing of the brain.69,71,72 Similarly, in the white matter, the oligovascular niche may mediate the cell–cell interaction between cerebral endothelial cells and oligodendrocyte lineage cells to regulate white-matter homeostasis. In both the developing and adult white matter, some OPCs are placed not far off from cerebral endothelial cells. 12 Therefore, cerebral endothelial cells may take part in the process of oligodendrocyte maturation and remodeling. 73 In fact, during the developmental stage, cerebral endothelial cells support oligodendrocyte generation. 73 In vitro cell culture studies also suggest that endothelial cells show some potentials to enhance OPC proliferation and migration in cell culture studies. 19 For example, cerebral endothelial cells release BDNF and bFGF to promote OPC proliferation. 74 And also, vascular endothelial growth factor-A secreted from cerebral endothelial cells can promote the migration of OPCs without affecting proliferation.75,76 Notably, sublethal oxidative stress reduces the performance of producing growth factors in endothelial cells, and hence, stressed-endothelial cells no longer support OPCs. 74 Therefore, the signaling from cerebral endothelial cells to oligodendrocyte lineage cells may change under pathologic conditions, presumably leading to white-matter dysfunction.
Similar to axon–oligodendrocyte interaction, endothelium–oligodendrocyte would be a two-way interaction. Oligodendrocyte lineage cells are known to release several trophic factors, which may regulate vascular system in white matter. 73 For example, OPC-derived TGF-beta strengthens the BBB tightness during development. Also in the adult white matter, oligodendrocytes may contribute to vascular remodeling via secreting MMP-9 during the chronic phase after white-matter damage. 77 However, under the cerebral hypoperfusion stress conditions, OPC-derived MMP-9 may initiate the deleterious cascade in white matter to cause BBB damage, myelin degradation, and behavioral deficits. 13 These bidirectional and biphasic trophic coupling between cerebral endothelial cells and oligodendrocyte lineage cells may have a critical role in white-matter function, and therefore, deeper understanding of the mechanisms would lead us to novel therapeutic targets for SIVDs.
Oligodendrocyte–Astrocyte/Microglia Interaction
Oligodendrocyte lineage cells are categorized in glial cells in central nervous system, and the interaction with other glial cells, such as astrocytes and microglia, would also be very important for white-matter physiology and pathophysiology. Particularly, astrocytes secrete many kinds of soluble factors to regulate the surrounding microenvironments. In fact, in vitro cell culture experiments show that astrocyte-derived soluble factors protect OPCs against ischemic stress. 78 In addition, erythropoietin from astrocytes protects OPCs against hypoxic and reoxygenation injury in vitro. 79 Besides through soluble factors, direct cell–cell signaling via gap junctions may also be relevant 80 because astrocytes are found in close apposition to oligodendrocytes in vivo. 81 In fact, astrocytes can attenuate oligodendrocyte death through an alpha6 integrin–laminin-dependent mechanism. 82 Moreover, OPCs seeded on astrocytes exhibit higher motility than OPCs on laminin-coated plates after IL-1alpha/bFGF treatment. 83 Also in vivo, astrocyte-derived BDNF may support white-matter myelin sheaths in the cuprizone-induced demyelination model. 84 Although mechanisms in which oligodendrocyte lineage cells affect astrocyte function remain unclear, a recent report showed that OPCs inhibit astrocyte activation after brain injury. 61 Therefore, astrocyte and oligodendrocyte/OPC would help each other to regulate white-matter function.
Microglia (and macrophage) may also have essential roles in regulating function of oligodendrocyte lineage cells. For instance, microglia can organize the phagocytosis of myelin debris and apoptotic cells during demyelination to support remyelination. 85 As for microglial action on OPC regulation, microglia may exhibit opposite roles, depending on the context. Microglia is accumulated in lesion area, and those microglia may ingest OPCs. 86 However, in the experimental allergic encephalomyelitis model, activated microglia could increase oligodendrogenesis. 87 The phenotypic heterogeneities in microglia may be related to the complex microglial roles on OPCs after brain injury. Pro-inflammatory (so-called M1-type) microglia dominantly exist in the early stage after demyelination and switch to anti-inflammatory or immunoregulatory (so-called M2-type) at the initiation of remyelination. In fact, a recent study reported that M2-microglia produced activin-A to drive oligodendrocyte differentiation. 88 Notably, aging can affect the switch from M1 to M2 phenotype in microglia, and this delay in the M1-to-M2 transition may correlate to the disturbance of OPC differentiation. 88
Conclusion
Subcortical ischemic vascular diseasess are clinically important because cerebral white-matter integrity is closely correlated with the cognitive function. Oligodendrocytes comprise the major cell type in white matter, and function of oligodendrocytes (and their precursor cells) has critical roles in facilitating the repair of damaged white matter. But as the concept of neurovascular unit suggests, oligodendrocytes may not stand alone; rather they actively interact with neighboring cells to allow the integrated control of white-matter function. In this mini-review, we have tried to summarize these mechanisms of cell–cell interaction in physiologic and pathophysiologic conditions of white matter. Cell–cell trophic coupling between oligodendrocytes and OPCs with other neural, glial, and vascular elements present in white matter should be essential for homeostasis and remodeling after injury. Further elucidating these underlying mechanisms may help identify novel therapeutic approaches to protect white-matter function in SIVD patients.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Acknowledgement
This study was supported by National Institutes of Health.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
All authors contributed to (1) conception and design for this review manuscript, (2) drafting and revising the article, and (3) final approval of the version to be published.