
Abstract
Cerebral small vessel disease (SVD) is among the most frequent causes of both stroke and dementia. There is a growing list of genes known to be implicated in Mendelian forms of SVD. Also, genome-wide association studies have identified common variants at a number of genetic loci that are associated with manifestations of SVD, among them loci for white matter hyperintensities, small vessel stroke, and deep intracerebral hemorrhage. Driven by these discoveries and new animal models substantial progress has been made in elucidating the molecular, cellular, and physiologic mechanisms underlying SVD. A major theme emerging from these studies is the extracellular matrix (ECM). Recent findings include a role of structural constituents of the ECM such as type IV collagens in hereditary and sporadic SVD, the sequestration of proteins with a known role in ECM maintenance into aggregates of NOTCH3, and altered signaling through molecules known to interact with the ECM. Here, we review recent progress in the identification of genes involved in SVD and discuss mechanistic concepts with a particular focus on the ECM.
Introduction
‘Cerebral small vessel disease’ (SVD) refers to a syndrome of clinical, cognitive, neuroimaging, and neuropathologic findings that arise from pathologic processes in cerebral perforating arteries and arterioles, capillaries and venules. Aside from being a major cause of stroke, SVD is now recognized as the leading cause of vascular dementia (VaD). 1 Roughly 80% of 65-year-old individuals and almost all 90-year olds exhibit clinical or radiologic manifestations of SVD earmarking SVD as one of the most common age-related conditions.
Until recently, the molecular, cellular, and pathophysiologic mechanisms underlying SVD have been largely unknown. Mechanistic studies were hampered by a lack of animal models, difficulties in visualizing small blood vessels in vivo, and technological challenges in making brain microvessels accessible to physiologic and biochemical studies. 2 The discovery of genes implicated in hereditary forms of SVD has spurred the development of disease models and of mechanistic studies. Another consequence has been a new etiology-based classification of SVDs with improved options in diagnosing, consulting, and managing patients. 3 In the following, we review current approaches to studying the genetic basis of SVD, highlight recent genetic discoveries, and illustrate how these discoveries have contributed to our understanding of SVD and its major manifestations, stroke and dementia.
Genetic and Epidemiologic Studies on Small Vessel Disease rely on Surrogate Markers
Because cerebral microvessels cannot be directly assessed in living humans, genetic and epidemiologic studies on SVD rely on surrogate markers known to be associated with microvascular disease. 4 Among the most commonly used markers are radiologic white matter hyperintensities (WMH) and lacunar infarcts. They can both be quantified thus offering greater statistical power than binary phenotypes such as stroke or dementia. This increase in power is partially offset by a lack of specificity. Lacunar infarcts may occasionally be caused by other etiologies such as an atheroma, artery-to-artery embolism, or cardiac embolism. 1 Similarly, WMH may be seen with other vascular and nonvascular conditions. The specificity for SVD can be increased by combining multiple radiologic and clinical markers. 5 However, in practice, larger sample sizes are often used to compensate for the limited specificity of individual disease markers. Other markers known to be associated with SVD include deep intracerebral hemorrhage (ICH), cerebral microbleeds (MBs), silent brain infarcts, and microinfarcts. MBs are seen in various forms of SVD and in cerebral amyloid angiopathy. 6 Silent brain infarcts mostly fall in the size range of lacunar infarcts and are thus associated with SVD. However, none of these markers is specific for SVD.
Conventional Risk Factors for Small Vessel Disease have a Genetic Basis
The strongest and most consistent risk factor for SVD aside from age is hypertension. A detailed account of conventional risk factors and their relationships with individual SVD markers is beyond the scope of this review. However, it is important to realize that traits such as blood pressure, diabetes, and smoking each have their own genetic basis. The number of common and rare genetic variants known to be associated with established risk factors for SVD is rapidly growing (see paragraph: Common genetic variants and sporadic small vessel disease).
Small Vessel Disease has a Genetic Basis
The most robust evidence for a genetic basis of SVD comes from magnetic resonance imaging studies on WMH. Estimates of the heritability of radiologic WMH range between 55% and 75%. 7 – 10 Heritability refers to the proportion of variation in phenotype explained by genetic factors. Estimates of the genetic correlation between WMH and blood pressure suggest that the two traits have genetic factors in common. 11 These findings have recently been complemented by genome-wide complex trait analysis. 12 This method estimates the proportion of phenotypic variance explained by multiple genetic variants present on conventional genotyping arrays. Using genome-wide complex trait analysis, the heritability of WMH volume attributable to common genetic variants was estimated to be 21%, with higher estimates for hypertensive (45%) compared with non-hypertensive individuals (13%). 13 A genetic basis of SVD is further suggested by studies on lacunar stroke and deep ICH. The heritability of small vessel-related stroke and deep ICH attributable to common genetic variants is estimated to be 16% and 34%, respectively.14,15 The discrepancy between high heritability estimates for WMH obtained in twin and family history studies and relatively low estimates derived from genome-wide complex trait analysis might indicate that much of the genetic contribution to SVD is because of rare genetic variants. In fact, there is a growing list of Mendelian SVDs, some of which are now recognized as an important cause of juvenile stroke as well as vascular dementia.
Mendelian Forms of Small Vessel Disease
Mendelian forms of small vessel disease and their role in stroke and dementia.
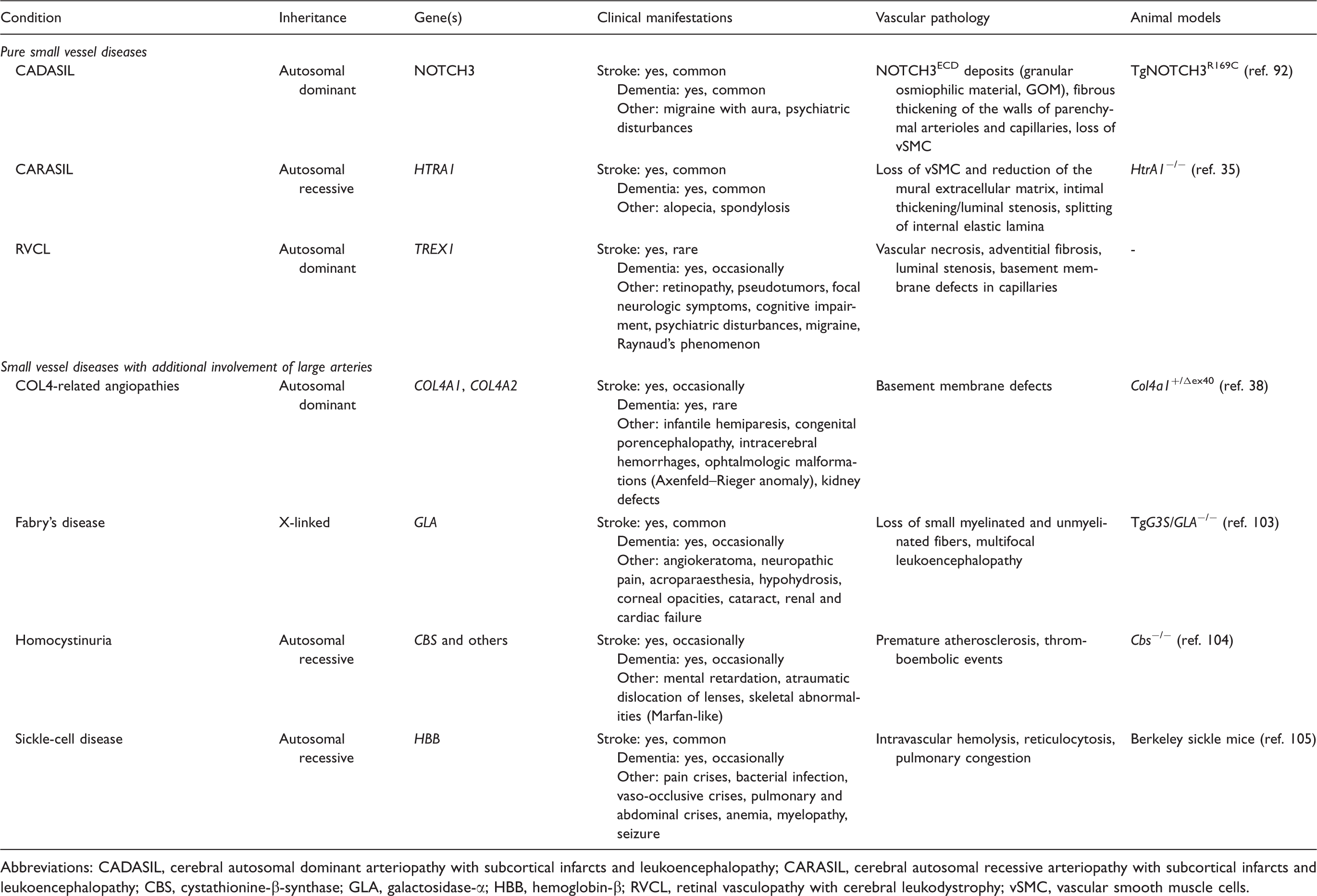
Abbreviations: CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CARASIL, cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy; CBS, cystathionine-β-synthase; GLA, galactosidase-α; HBB, hemoglobin-β; RVCL, retinal vasculopathy with cerebral leukodystrophy; vSMC, vascular smooth muscle cells.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) (OMIM: 125310) represents the most prevalent monogenic SVD with a prevalence of up to five cases per 100,000 individuals.17,18 The condition is caused by mutations in NOTCH3, a transmembrane receptor of the Notch family with a large extracellular domain (ECD) mainly consisting of epidermal growth factor (EGF)-like repeats.19,20 Each of these repeats invariably contains six cysteine residues, which engage in three disulfide bridges required for correct folding of the NOTCH3 protein. 21 More than 230 pathogenic NOTCH3 mutations have been described with the majority representing missense mutations, although deletions, duplications, and splice site mutations have also been reported. 22 Mutations are highly stereotyped in that they predominantly affect cysteine residues leading to an uneven number of this amino acid. The resulting unpaired sulfhydryl group promotes NOTCH3ECD aggregation and accumulation in the extracellular space, a critical event in CADASIL pathogenesis (see section on pathomechanisms).
There have been several reports on cysteine-sparing NOTCH3 mutations in patients with features of CADASIL and there is an ongoing debate on whether these sequence variants are pathogenic. 23 – 25 By applying single-particle analysis to an array of mutated NOTCH3 fragments Wollenweber et al. 26 recently showed that some cysteine-sparing mutants including D80G, R75P, and Δ88-91 self-aggregate in vitro similar to typical CADASIL mutants. It thus seems that in some instances, CADASIL can originate from atypical mutations that do not affect cysteines within NOTCH3ECD.
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) (OMIM: 600142) shares considerable phenotypic overlap with CADASIL including magnetic resonance imaging hyperintensities in periventricular and deep white matter, multiple lacunar infarcts, and vasculopathic alterations of penetrating arteries and arterioles.27,28 Typical features include the recessive mode of inheritance, an early age of onset (20 to 30 years) and the frequent occurrence of spondylosis and alopecia as associated non-neurologic manifestations.29,30 The genetic cause of CARASIL, identified only recently by linkage analysis in five consanguineous families, 31 was mapped to the HTRA1 gene encoding high temperature requirement protein A1, a highly conserved serine protease. 32 Patients carry homozygous missense or nonsense mutations resulting either in dramatically reduced protease activity or in the loss of HtrA1 expression, in agreement with the recessive inheritance mode. Initially thought to be restricted to the Japanese population, in recent years an increasing number of CARASIL cases has been reported from outside Japan indicating a more widespread distribution of this disorder. 33 – 35
The COL4A1/A2-related angiopathies (OMIM: 607595; 614519) represent highly penetrant multi-system disorders with heterogeneous phenotypes. 36 Heterozygous mutations in COL4A1 encoding the α1 chain of type IV procollagen were initially identified as a cause for porencephaly, an infantile neurologic disorder associated with hemorrhages and hemiplegia. 37 Triggered by observations in Col4a1 mutant mice Gould et al.37,38 found that heterozygous COL4A1 mutations in humans are associated with familial SVD and a wide clinical spectrum including lacunar stroke, ICH, WMH, and cerebral microbleeds. Although apparently less frequent, mutations in COL4A2, whose gene product forms heterotrimers with COL4A1 in a 2:1 stoichiometry, likewise cause familial SVD.39,40 The spectrum of COL4A1 and COL4A2 mutations associated with familial SVD is broad (mostly point mutations, but in rare cases also gene duplications) as is the phenotypic spectrum, which also includes intracranial aneurysms, ocular, cardiac, and renal abnormalities. 36 Some patients with COL4A1/A2 mutations and familial SVD display congenital ocular defects characteristic of the Axenfeld–Rieger syndrome. 41 The combination of Axenfeld–Rieger syndrome and SVD has also been described in patients carrying mutations in FOXC1 and PITX2, which encode two physically interacting transcription factors, and common variants at these loci were recently found to be associated with WMH in the general population. 42
Retinal vasculopathy with cerebral leukodystrophy (RVCL) (OMIM: 192315) refers to a group of cerebroretinal syndromes originally described as separate disorders: cerebroretinal vasculopathy (CRV); 43 hereditary endotheliopathy with retinopathy, nephropathy and stroke (HERNS); 44 and hereditary vascular retinopathy (HVR).45,46 They had initially been described as dominantly inherited vasculopathies with similar, but distinct phenotypes including ophthalmologic and neurologic symptoms. Most patients present with retinopathy (mean age of onset ∼45 years), but subsequently develop severe neurologic symptoms including focal neurologic deficits, cognitive impairment, and psychiatric disturbances. 47 CRV, HERNS, and HVR all map to the same genetic locus 3p12.1–p21.3 (ref. 48) and are caused by heterozygous mutations in the DNA exonuclease TREX1. 49 Mutations lead to a frameshift resulting in a protein with truncated or extended C terminus and retained enzymatic activity, but altered subcellular localization.
Common Genetic Variants and Sporadic Small Vessel Disease
Common Variants in NOTCH3 and COL4A1/A2
Genetic association studies in clinic- and population-based samples suggest that common genetic variants in the NOTCH3 and COL4A1/A2 genes also contribute to the more common sporadic forms of SVD. NOTCH3 variants were found to be associated with both the presence and progression of WMH in 4,773 population-based hypertensive subjects from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium. 50 By analyzing more than 1,500 cases and a similar number of controls from the International Stroke Genetics Consortium, Rannikmäe et al. could show that common variants in the COL4A1/A2 gene cluster are associated with deep intracerebral hemorrhage (ICH). 51 The same variants showed suggestive associations with lacunar stroke and WMH volume in symptomatic ischemic stroke cases. Whether common genetic variants at HTRA1, TREX1, and other genetic loci for monogenic SVD are implicated in sporadic forms of SVD remains to be determined.
Genome-Wide Association Studies
Up to now, genome-wide association studies (GWAS) have focused on individual markers of SVD rather than considering multiple markers in aggregate. Hence these studies are discussed separately for each target phenotype (see Figure 1).
Genetic loci implicated in cerebral small vessel disease (SVD). Circus plot of genes and genetic loci associated with SVD. Red symbols mark genes implicated in Mendelian forms of stroke. Green symbols mark genetic loci shown to be associated with one or multiple manifestations of SVD. Loci awaiting confirmation in additional samples are shown in light gray.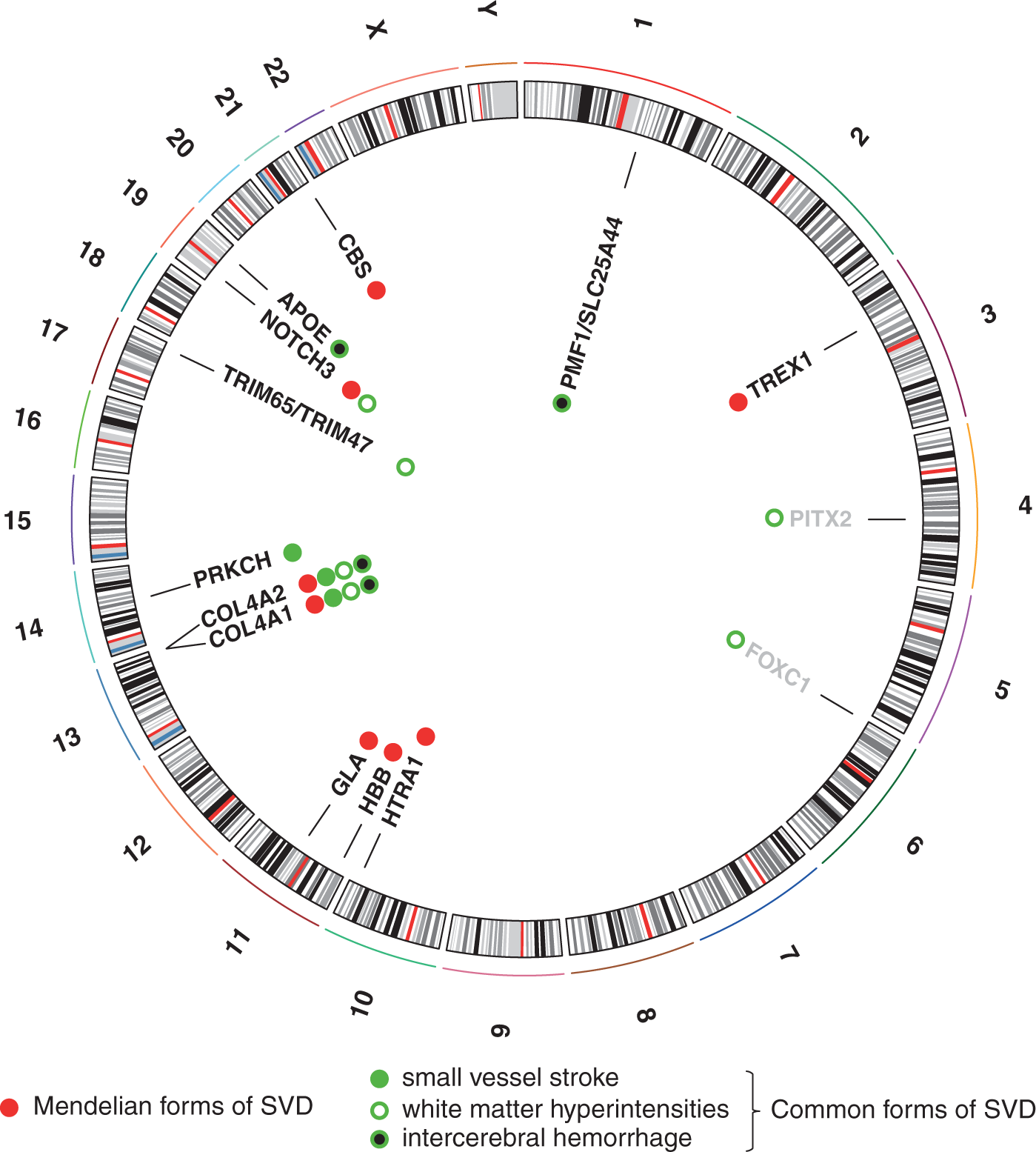
White matter hyperintensities
A study of WMH burden in more than 10,000 stroke-free individuals from the CHARGE consortium revealed a single risk locus on chromosome 17q25. 52 The two lead single-nucleotide polymorphisms (SNPs) belong to a cluster of associated SNPs spanning an ∼100 kb region that contains several genes. Two tripartite motif-containing genes, TRIM65 and TRIM47 are currently considered the strongest candidates for SVD, but other genes in this region (WBP2, MRPL38, FBF1, and ACOX1) must still be seen as potential candidates. Variants at this locus were recently shown to also associate with WMH volume in ischemic stroke patients collected through stroke clinics. Interestingly, in the same sample, no association with small vessel stroke (SVS) was observed indicating that the 17q25 locus acts through mechanisms that are more specific for the pathogenesis of WMH rather than promoting SVD in general. 53 A recent meta-analysis on a much larger set of GWAS data on WMH identified, among known associations, three new loci (2p16, 2p21 and 10q24) that have been implicated in inflammatory and glial proliferative pathways. 54
Small vessel stroke
Although GWAS in ischemic stroke patients have identified several loci for large artery (atherosclerotic) and cardioembolic stroke, the search for risk loci for SVS has been far less successful. 55 This might, in part, relate to insufficient sample size, sample heterogeneity, and the lack of robust standards for classifying patients as SVS. 5 By analyzing 52,608 gene-based tag SNPs from the Japanese Single-Nucleotide Polymorphism (JSNP) database in Japanese patients with cerebral infarction, Kubo et al. 56 identified a non-synonymous SNP in the PRKCH gene encoding protein kinase C-η, that significantly associated with lacunar stroke in two independent samples. Variants at PRKCH were subsequently shown to also associate with subcortical silent brain infarcts 57 and with cerebral hemorrhage in the Chinese population. 58 Protein kinase C-η is expressed in vascular endothelial and other cell types, is known to mediate a variety of signaling pathways, and regulates multiple cellular functions including proliferation, differentiation, and apoptosis. The lead SNP of the initial association is monomorphic in the European population and GWAS in Caucasian samples revealed no signals at PRKCH. Hence, it might be that PRKCH represents a risk locus for SVD-related phenotypes (lacunar stroke and ICH) exclusively in the Asian population. However, this requires confirmation in additional samples.
Intracerebral hemorrhage
A recent GWAS conducted by the International Stroke Genetics Consortium identified a susceptibility locus on chromosome 1q22, overlapping with the PMF1 and SLC25A44 genes. 59 rs2984613 (minor allele frequency: 32%), the lead SNP at this locus, was associated with a 33% increase in risk of non-lobar ICH. Interestingly, the 1q22 region was also among the top loci in the above-mentioned GWAS on WMH burden from the Cohorts for Heart and Aging Research in Genomic Epidemiology consortium, although it did not quite reach genome-wide significance. 52 PMF1 codes for polyamine-modulated factor 1, which is required for normal chromosome alignment and segregation and kinetochore formation during mitosis; SLC25A44 encodes a mitochondrial carrier protein. This points towards a possible mechanism in energy metabolism, since variation in mitochondrial genes has been implicated in risk of ICH (see below).
Vascular dementia
Small vessel disease accounts for a considerable proportion of VaD cases. There has been a single GWAS on VaD conducted in 5,700 initially dementia-free population-based subjects from the Rotterdam study, 67 of whom developed incident VaD over a mean follow-up of 9.3 ± 3.2 years. 60 A single variant on the X chromosome near the androgen receptor gene reached genome-wide significance with highly variable odds ratios in the discovery and replication cohorts. This locus has so far not been linked to other SVD manifestations and the association with VaD still requires independent replication.
Apolipoprotein E
Genetic variation in the APOE gene encoding apolipoprotein E is the strongest genetic risk factor for both Alzheimer’s disease and cerebral amyloid angiopathy. The ɛ4 allele increases the risk of both conditions. The ɛ2 allele is protective in Alzheimer’s disease but associated with an increased risk of cerebral amyloid angiopathy-related lobar ICH.61,62 The relationship between markers of SVD and APOE genotype was assessed by a meta-analysis of 42 cross-sectional or longitudinal studies. APOE ɛ4 was associated with an increased burden of both WMH and microbleeds, and APOE ɛ2 was associated with an increased WMH burden. In accord with this, a recent GWAS on genetic modifiers in CADASIL found the APOE ɛ2 allele to be an independent risk factor for higher WMH volumes in NOTCH3 mutation carriers (see accompanying manuscript by Gesierich et al. 63 ). This indicates that genetic variation in APOE contributes to manfestations of SVD independent of the presence of amyloid pathology. The biologic mechanisms underlying this relationship are still poorly understood. However, the APOE ɛ2 allele has been associated with vasculopathic changes including vessel dilation, fibrinoid necrosis, microaneurysms, and double barreling. 64
Oxidative Phosphorylation Genes and Small Vessel Disease
Focusing on aggregate measures of genetic variation rather than individual SNPs, Anderson et al. 65 identified several variants within a larger set of oxidative phosphorylation genes, which collectively were associated with increased risk of both lacunar stroke and deep hemispheric ICH. The oxidative phosphorylation genes are encoded by mitochondrial as well as nuclear DNA. Lacunar stroke showed associations with genetic risk scores in oxidative phosphorylation as a whole, complex I, and complex IV, and the latter also associated with deep hemispheric ICH. These findings are complemented by another study that found a genetic score of mitochondrial variants to be associated with WMH volume in patients with ischemic stroke. 66 Collectively, these findings suggest that genetic variation in oxidative phosphorylation influences small vessel pathobiology although the exact mechanisms remain to be determined.
Common Variants Implicated in Risk Factors for Small Vessel Disease
Recent GWAS have identified a large number of genetic loci associated with established risk factors for SVD. There are more than 30 known risk loci for blood pressure-related traits,67,68 more than 100 known loci for type 1 diabetes69,70 or type 2 diabetes, 71 and numbers are steadily growing as sample sizes and genetic data are growing. The impact of these loci on SVD risk has not been systematically explored. However, common variants on chromosome 12q24.12, a known risk locus for hypertension, type 1 diabetes, and hypercholesterolemia have recently been shown to also associate with SVS. 72 Whether the association between common variants at this locus and SVS is mediated by the associated risk factors or independent mechanisms remains to be explored.
Pathomechanisms in Small Vessel Disease
Previous mechanistic studies in humans and experimental models have identified multiple factors and mechanisms that may contribute to vascular and parenchymal injury in SVD.
1
The pathology of inherited SVDs shows considerable overlap with the sporadic disease, although some pathologic features of sporadic SVD such as breakdown of the blood–brain barrier have so far not been demonstrated in the monogenic forms. A novel aspect that has recently emerged mostly from genetics and from mechanistic studies in monogenic SVDs is the extracellular matrix (ECM). The ECM has for long been recognized as a highly dynamic and biologically active compartment that is also implicated in a variety of conditions including diseases of large arteries.
73
However, only recently, the ECM has emerged as a key target relevant to the pathogenesis of SVDs (Figure 2). In fact, a growing body of evidence suggests that the ECM takes center stage in multiple forms of SVD.
Role of the extracellular matrix in cerebral SVD. (A) The extracellular matrix (ECM) constitutes an integral part of the vasculature. In large arteries and arterioles, the ECM is a spacious structure in which vascular smooth muscle cells are embedded, whereas in capillaries, it is restricted to the basement membrane (BM), a thin protein matrix separating the endothelium from pericytes, astrocytes, and neurons, which together form the neurovascular unit. (B) The ECM takes center stage in multiple forms of familial SVD. Left panel: COL4A1 and A2 form trimers (α1α1α2). Upon secretion, trimers assemble into a chicken-wire meshwork constituting the major structural component of BMs. Mutations in COL4A1/A2 result in impaired assembly and potentially cytotoxic accumulation of mutant heterotrimers within cells. Alternative mechanisms include the presence of mutant heterotrimers or a deficiency of properly folded heterotrimers in the BM. Both mechanisms could alter interactions with cell surface receptors and signaling molecules such integrins and BMPs; Middle panel: CADASIL-mutated NOTCH3 is efficiently secreted and participates in cell signaling. Instead of undergoing clearance from the extracellular space, the extracellular domain of mutant NOTCH3 multimerizes leading to the formation of large aggregates. Matrix proteins such as tissue inhibitor of metalloproteinases-3 (TIMP3), vitronectin (VTN), and latent TGF-β binding protein-1 (LTBP-1) are recruited into the deposits possibly resulting in an alteration of their physiologic function; Right panel: proposed model by which CARASIL mutations interfere with TGF-β signaling. Upon secretion, latent TGF-β is incorporated into the ECM via interactions of LTBP-1 with matrix proteins such as fibronectin. Mature TGF-β can be liberated from the ECM through cleavage of LTBP-1 by proteases such as HtrA1. CARASIL mutations result in a loss of HtrA1 activity and hence interfere with TGF-β signaling. BMP, bone morphogenic protein; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CARASIL, cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy; SVD, small vessel disease.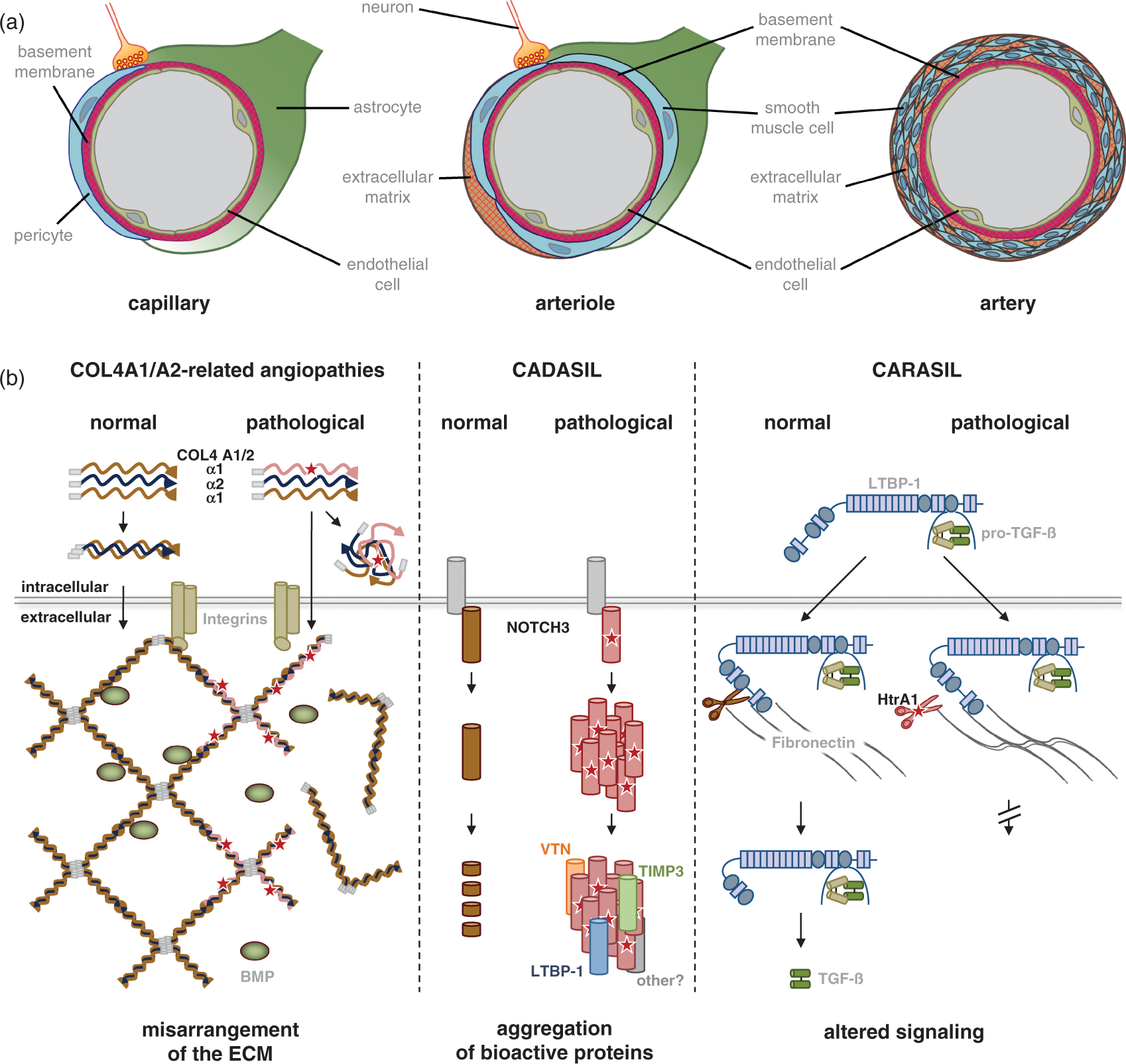
COL4A1/A2-Related Small Vessel Diseases
Collagens type IV α1 (COL4A1) and α2 (COL4A2) represent the most abundant components of basement membranes, which are macromolecular extracellular structures mediating the anchoring of epithelial and endothelial cells to the underlying connective tissue. 36 Apart from providing structural integrity, basement membranes also participate in cell–matrix and cell–cell communication by interacting with integrins and binding growth factors, e.g., of the TGF-β superfamily. 74 The core units of COL4A1/A2 meshworks are heterotrimers (α1α1α2) whose formation is mediated by long stretches of the amino acid triplet Gly-X-Y within the COL4A1 and A2 collagenous domains. Mutations of the glycine residue within this motif are by far the most common cause of COL4A1/A2-related disorders. However, their functional consequences are only partly understood. A simple loss-of-function mechanism is unlikely as mice heterozygous for COL4A1 and A2 null alleles exhibit no overt pathology, 75 whereas strains carrying both COL4A1 and A2 mutations show multiple defects including porencephaly, vascular abnormalities, and intracerebral hemorrhage.37,76,77 Several mutually not exclusive neomorphic pathomechanisms have been proposed, which could explain the remarkable heterogeneity of disease symptoms (Figure 2B). A variety of COL4A1/2 mutations have been shown to result in intracellular accumulation of mutant heterotrimers and in the activation of endoplasmic reticulum stress.39,78,79 Alternatively, intracellular sequestration of heterotrimers may lead to extracellular deficiency of COL4A1/2 and insufficient collagen matrix formation. However, at present, secretion of mutant trimers and their incorporation into the basement membrane network cannot be excluded as an alternative pathomechanism. This could result in the modification of COL4A1/2-dependent interactions with other extracellular components such as bone morphogenic proteins or cell surface molecules. 36 Ultrastructural studies in COL4A1 mutation carriers and COL4A1 mutant mice show profound structural abnormalities with thickening and herniation of the basement membrane.37,80 In accord with this, COL4A1/A2 mutations are assumed to have profound effects on ECM function.
Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy
NOTCH3 belongs to the Notch family of cell signaling receptors and has been shown to be critical for arterial differentiation and maturation of vascular smooth muscle cells in mice. 81 However, several studies failed to demonstrate a loss-of-function mechanism of mutant NOTCH3.82,83,84 Instead, studies performed in mice showed that the arterial defect in NOTCH3 knockout mice can be rescued by CADASIL-mutant NOTCH3. 85 Moreover, aspects of human CADASIL pathology are recapitulated in mouse strains carrying a CADASIL-mutant NOTCH3 allele, but are not seen in heterozygous or homozygous null strains.81,85 These findings have directed mechanistic studies to a neomorphic effect (Figure 2B). A toxic gain-of-function mechanism is supported by the stereotyped nature of CADASIL mutations (see above) and by data showing that CADASIL-mutant NOTCH3ECD aggregates accumulate in the extracellular space of small arteries, arterioles, and capillaries: First, ultrastructural examination of microvessels from CADASIL patients reveals pathognomonic granular osmiophilic material that locates to the vicinity of vascular smooth muscle cells and is NOTCH3-positive on immunogold labeling; 86 Second, NOTCH3 immunostaining of CADASIL microvessels reveals massive immunoreactivity that is specific for this condition; 87 Third, immunoblotting of brain homogenates as well as homogenates from isolated arteries demonstrates accumulation and aggregation of NOTCH3ECD;86,88 And fourth, aggregation and accumulation of mutant NOTCH3 can be recapitulated in vitro using scanning for intensely fluorescent targets, a confocal method allowing the detection of single-protein particles in solution. When the multimerization behavior of NOTCH3-EGF 1 – 5 a fragment encompassing the mutational hot spot for disease-associated mutations, was analyzed, mutant NOTCH3 showed a much stronger tendency to self-aggregate than the wild-type protein. 89 Moreover, mutant NOTCH3 was shown to form mixed multimers with wild-type NOTCH3 and Thrombospondin-2, a known interactor of NOTCH3, providing direct evidence for a pathologic co-aggregation mechanism. 90 These findings were recently extended by the demonstration of a co-aggregation between mutant NOTCH3 and latent TGF-β-binding protein (LTBP-1), a key regulator of the TGF-β pathway (see below). Hence, NOTCH3ECD accumulation and deposition, an early process in CADASIL patients 91 as well as in mouse models, 92 is now considered a critical step in the pathogenesis of CADASIL (Figure 2B). 93
The pathologic processes triggered by NOTCH3ECD aggregation are poorly understood. Recent evidence suggests a recruitment of additional proteins into NOTCH3 deposits as evidenced by the co-aggregation of mutant NOTCH3 and other proteins.90,94 Arboleda-Velasquez et al. 95 used laser capture microdissection of brain vascular cells and subsequent mass spectrometry analysis to detect a number of proteins differentially expressed in normal and CADASIL patient vessels, among them the ECM proteins collagen 18 α1/endostatin and clusterin. By combining biochemical fractionation of brain and artery samples with proteomics and immunohistochemical methods, Monet-Lepretre et al. 88 identified a variety of proteins enriched together with NOTCH3ECD. The most prominent enrichment was found for ECM proteins suggesting a key role of the ECM in CADASIL. From the list of differentially expressed proteins, tissue inhibitor of matrix proteinases 3 and vitronectin were examined in more detail and shown to colocalize with NOTCH3ECD deposits and to interact with NOTCH3 in vitro. Importantly, tissue inhibitor of matrix proteinases 3, a major regulator of metalloproteases in the brain, remains biologically active, which might, in part, explain the fibrotic changes within CADASIL arteries. Using similar approaches and autopsy material from patients representing a wide spectrum of NOTCH3 mutations, Kast et al. 94 recently demonstrated a massive build-up of major ECM proteins in CADASIL arteries, among them fibronectin, fibrillin-1, and LTBP-1, a key regulator of TGF-β bioavailability. LTBP-1 was found to both accumulate and colocalize with NOTCH3 deposits. Moreover, elevated levels were also detected for LAP (latency-associated peptide), the TGF-β pro domain. TGF-β is a strong activator of fibrotic processes and LTBP-1-LAP complexes serve as a reservoir of inactive TGF-β in the ECM from which the mature ligand can be released by a variety of processes. 74 These data not only support the critical role of ECM proteins in mediating NOTCH3ECD toxicity, they further indicate a role of deregulated TGF-β signaling in CADASIL patients, which could contribute to vessel degeneration.
Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy
A key role of the TGF-β signaling pathway in SVD is further suggested by observations in CARASIL patients as well as cellular and animal models of this condition. A link with TGF-β signaling was already suspected when HTRA1, which is expressed in multiple TGF-β-relevant tissues including the vasculature, skin, and bone, was identified as the affected gene. 31 TGF-β is also abundantly expressed and has multiple biologic functions including a regulatory role in vascular development. 73 Consistent with their role in vascular biology, members of the TGF-β signaling pathway are implicated in Marfan syndrome and related vascular conditions. 96 HtrA1 has been suggested to inhibit TGF-β in various experimental systems. 97 – 100 In accord with this, Hara et al. 31 found signs of increased TGF-β signaling in CARASIL-affected vessels as well as patient skin fibroblasts. This led the authors to propose the lack of TGF-β processing as a critical disease mechanism. 101 Contrasting these findings, a more recent study in HTRA1-deficient mice and CARASIL patient cells revealed a facilitating role of HtrA1 on the TGF-β pathway. 35 The same study showed that LTBP-1 is a substrate for HtrA1. These findings suggest that HtrA1-mediated proteolysis of LTBP-1 promotes release of TGF-β from the ECM (Figure 2B). Loss of HtrA1 function either by gene ablation in mice or by CARASIL mutations in humans likely attenuates LTBP-1 processing and TGF-β signaling. Hence, a lack of LTBP-1 cleavage and subsequent reduction in TGF-β release might represent the critical step in CARASIL pathogenesis. Opposing roles of TGF-β have been previously reported in other vascular conditions such as Marfan syndrome and related conditions. 96 This might relate to differential effects of TGF-β in different cell types including endothelial vs. vascular smooth muscle cells, but could also be a consequence of genetic modifiers of the TGF-β pathway. 102
In summary, genetics holds great promise in providing mechanistic insights into SVD. Current efforts such as the imputation of GWAS data to 1000 Genomes data together with whole-exome sequencing and eventually massive parallel sequencing of the entire genome will likely identify multiple additional variants, both common and rare, in yet unknown genes for SVD. However, functional follow-up of such variants in cellular and animal models remains the key for a better mechanistic understanding of SVD and for the development of novel therapeutic approaches to stroke and dementia.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the German Research Foundation (DFG, OP 212/1-1; SFB 1123 B3), the German Federal Ministry of Education and Research (BMBF), the Vascular Dementia Research Foundation, the Fondation Leducq (Transatlantic Network of Excellence on the Pathogenesis of Small Vessel Disease of the Brain), the Corona Foundation, the Dr Werner Jackstädt-Stiftung (S 134–10.065) and a European Union FP7 ERA-NET NEURON grant (01 EW1207).
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: M Dichgans is a consultant of Bayer Vital GmbH; Boehringer Ingelheim Pharma GmbH & Co. KG, Biologische Heilmittel Heel GmbH, Bristol-Myers Squibb GmbH & Co. KGaA, Ever Neuro Pharma GmbH, Daiichi Sankyo Europe GmbH and has received honoraria from the German Center for Neurodegenerative Diseases (DZNE), Georg Thieme Verlag KG, UpToDate, W. Kohlhammer GmbH, Lundbeck GmbH, Sanofi-Aventis Deutschland GmbH and Shire Deutschland GmbH. M Dichgans received research funding from Bayer Vital GmbH, Eisai Medical Research Inc., Eisai Ltd., Essex Pharma GmbH, Ferrer International and ICON Clinical Research GmbH.
Authors’ contributions
CH, RM, and MD wrote the paper.