
Abstract
Lipocalin-2 (LCN2) is a siderophore-binding protein involved in cellular iron transport and neuroinflammation. Both iron and inflammation are involved in brain injury after intracerebral hemorrhage (ICH) and this study examined the role of LCN2 in such injury. Male adult C57BL/6 wild-type (WT) or LCN2-deficient (LCN2-/-) mice had an intracerebral injection of autologous blood or FeCl2. Control animals had a sham operation or saline injection. T2-weighted magnetic resonance imaging and behavioral tests were performed at days 1, 3, 7, 14, and 28 after injection. In WT mice, brain LCN2 levels were increased in the ipsilateral basal ganglia after ICH or iron injection. Lipocalin-2-positive cells were astrocytes, microglia, neurons, and endothelial cells. Intracerebral hemorrhage resulted in a significant increase in ferritin expression in the ipsilateral basal ganglia. Compared with WT mice, ICH caused less ferritin upregulation, microglia activation, brain swelling, brain atrophy, and neurologic deficits in LCN2-/- mice (
INTRODUCTION
Iron has a major role in brain damage after intracerebral hemorrhage (ICH).1,2 Brain non-heme iron increases after ICH in rats and brain iron overload causes brain edema in the acute phase and brain atrophy later after ICH.2,3 An iron chelator, deferoxamine, reduces ICH-induced brain edema, neuronal death, brain atrophy, and neurologic deficits in rats and pigs.2–4 Clinical data also suggest a role of iron in ICH-induced brain injury. For example, clot lysis is associated with perihematomal edema development. 5 Recent studies showed that high levels of serum ferritin, an iron storage protein, are independently associated with poor outcome and severe brain edema in ICH patients.6,7
Lipocalin-2 (LCN2) is an acute phase protein that is upregulated in inflammation, infection, and a variety of injuries. 8 It binds siderophores, which are secreted by microorganisms to scavenge iron. 9 However, there is beginning to be evidence that LCN2 is involved in iron homeostasis. There is evidence that it can be involved in cellular uptake or clearance of iron depending on iron status. 10 Another report has suggested that LCN2 could be a mediator of an alternative, transferrin-independent pathway for cellular iron delivery. 11 In rats, LCN2 is upregulated after ICH and it may potentially have a role in handling iron that is released from the hematoma during clot resolution. 12 However, whether such a role is beneficial or detrimental is uncertain.
As an acute phase protein, LCN2 is also involved in inflammatory responses. Enhanced LCN2 expression was observed in choroid plexus and microvascular endothelial cells in brain parenchyma after peripheral lipopolysaccharide administration. 13 In cerebral ischemia, LCN2 contributed to neuronal cell death by promoting neurotoxic glial activation, neuroinflammation, and blood–brain barrier (BBB) disruption. 14 Lack of LCN2 also reduced inflammation resulting in less secondary damage and better locomotor recovery after spinal cord contusion injury. 15 Inflammation has an important role in ICH-induced brain injury. 16
This study investigates the effect LCN2 deficiency on the expression of ferritin (an iron storage protein), microglia activation, neuronal death, and neurologic deficits in a mouse model of ICH. In addition, the role of LCN2 in iron-induced brain injury was also examined.
MATERIALS AND METHODS
Animal Preparation and Intracerebral Injection
All animal procedures were approved by the University Committee on Use and Care of Animals, University of Michigan. The University of Michigan has an Animal Welfare Assurance on file with the Office for Protection from Research Risks and is fully accredited by the American Association for the Accreditation of Laboratory Animal Care. The studies follow the Guide for The Care and Use of Laboratory Animals (National Research Council) and comply with the ARRIVE guidelines for reporting
Intracerebral injections were performed as previously described. 17 Briefly, mice were anesthetized with ketamine (90 mg/kg, intraperitoneally, Abbott Laboratories, Chicago, IL, USA) and xylazine (5 mg/kg intraperitoneally, Lloyd Laboratories, Shenandoah, IA, USA). Body temperature was maintained at 37.5°C by a feedback-controlled heating pad. The mice were positioned in a stereotaxic frame (Model 500, Kopf Instruments, Tujunga, CA, USA) and a cranial burr hole (1 mm) was drilled near the right coronal suture 2.5 mm lateral to the midline. A 26-gauge needle was inserted stereotaxically into the right basal ganglia (coordinates: 0.2 mm anterior, 3.5 mm ventral, and 2.5 mm lateral to the bregma). Either 30 μL autologous whole blood or 10 μL of FeCl2 (1 mmol/L) was infused at 2 μL/min by a microinfusion pump (Harvard Apparatus Inc, South Natick, MA, USA). Control animals had either a sham operation with a needle insertion or a 10-μL saline injection. After injection, the needle remained in position for 10 minutes to prevent reflux and then it was gently removed. The burr hole was filled with bone wax, and the skin incision was sutured closed.
Experimental Groups
The present study was divided into two parts. In the first part, WT or LCN2-/- mice had ICH (
Magnetic Resonance Imaging and Brain Swelling Measurement
Mice were anesthetized with 2% isoflurane/air mixture throughout MRI examination. Magnetic resonance imaging was performed at days 1, 3, 7, 14, and 28 in a 7.0-T Varian MR scanner (183-mm horizontal bore; Varian, Palo Alto, CA, USA) at the Center for Molecular Imaging of the University of Michigan including a T2 fast spin-echo (repetition time/echo time = 4,000/60 ms) using a field of view of 20 × 20 mm, matrix of 256 × 256 mm, and 25 coronal slices (0.5-mm thick). The images were preserved as 256 × 256 pixel pictures for brain swelling and T2 lesion calculation in NIH image J (Wayne Rasband, National Institutes of Health, USA). The calculation of brain swelling was based on seven every other sections whose center was the anterior commissure layer; the value was ((volume of ipsilateral hemisphere–volume of contralateral hemisphere)/volume of contralateral hemisphere) × 100%.
Immunohistochemistry and Brain Atrophy Measurement
Immunohistochemistry and immunofluorescence double staining were performed as described previously. 18 Briefly, mice were euthanized (ketamine 120 mg and xylazine 5 mg/kg intraperitoneally) and subjected to intracardiac perfusion with 4% paraformaldehyde in 0.1 mmol/L phosphate-buffered saline (pH 7.4). Brains were harvested and kept in 4% paraformaldehyde for 24 hours, then immersed in 30% sucrose for 3 to 4 days at 4°C. After embedding in a mixture of 30% sucrose and optimal cutting temperature compound (Sakura Finetek, Inc., Torrance, CA, USA) at a ratio of 1:2, the brain was sectioned on a cryostat into 18 μm slices and then preserved at −80°C.
Hematoxylin and eosin (H&E) staining was used for caudate atrophy measurement at day 28. Three coronal sections from 0.5 mm posterior to the blood injection site were stained and photographed under a microscope (Olympus, BX51, Waltham, MA, USA) with × 1.25 magnification. The bilateral caudate were outlined in NIH image J and caudate size measured as described in the MRI method section. Brain atrophy was calculated as ((contralateral–ipsilateral caudate area)/ contralateral caudate area) × 100%.
Immunohistochemistry was performed as previously described. 19 The primary antibodies were polyclonal goat anti-LCN2 IgG (R&D System, Minneapolis, MN, USA, 1:200 dilution), polyclonal rabbit anti-ferritin IgG (AbD; 1:500 dilution), goat anti-albumin antibody (Bathyl Laboratories, Inc., Montgomery, TX, USA, 1:10,000 dilution) and polyclonal rabbit anti-ionized calcium-binding adaptor molecule 1 (Iba1) IgG (Wako, Richmond, VA, USA, 1:400 dilution). For immunofluorescence double labeling, the primary antibodies were polyclonal rabbit anti-glial fibrillary acidic protein (GFAP) IgG (Millipore, Billerica, MA, USA, 1:400 dilution), polyclonal rabbit anti-neuronal-specific nuclear protein (NeuN) IgG (Abcam, Cambridge, MA, USA, 1:500 dilution) and polyclonal rabbit anti-CD31 IgG (Abcam, 1:50 dilution), as well as those for LCN2 and Iba1. The secondary antibodies were Alexa Fluro 488-conjugated donkey anti-rabbit mAb (Invitrogen, 1:500 dilution) and Alexa Fluro 594-conjugated donkey anti-goat mAb (Invitrogen, Grand Island, NY, USA, 1:500 dilution). The double labeling was analyzed using a fluorescence microscope (Olympus, BX51).
Western Blotting
Western blot analysis was performed as previously described. 18 Briefly, mice were perfused with 0.1 mmol/L phosphate-buffered saline (pH 7.4) after euthanasia, and the ipsi- and contralateral basal ganglia were sampled. Protein concentration was determined by Bio-Rad protein assay kit (Hercules, CA, USA), and 50 μg protein samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a hybond-C pure nitrocellulose membrane (Amersham, Pittsburgh, PA, USA). Membranes were probed with the following primary antibodies: polyclonal goat anti-LCN2 IgG (R&D System, 1:500 dilution), polyclonal rabbit anti-Iba1 IgG (Wako, 1:1,000 dilution), polyclonal goat anti-ferritin-L-chain IgG (FTL, Abnova, Walnut, CA, USA, 1:2,000 dilution), goat anti-albumin antibody (Bathyl Laboratories, Inc., 1:10,000 dilution), polyclonal rabbit anti ferritin-H-chain IgG (FTH, Cell Signaling, Beverly, MA, USA, 1:2,000 dilution) and polyclonal rabbit anti β-actin IgG (Cell Signaling, 1:5,000 dilution). The secondary antibodies were goat anti-rabbit IgG and rabbit anti-goat IgG (1:2,000 dilution; Bio-Rad, Hercules, CA, USA). Antigen–antibody complexes were visualized with the ECL chemiluminescence system (Amersham) and exposed to Kodak X-OMAT film. The relative densities of bands were analyzed with NIH Image J.
Behavioral Tests
Forelimb use asymmetry and corner turn tests were used for behavioral evaluation.20–22 For the forelimb use asymmetry, forelimb use during exploratory activity was analyzed in a transparent cylinder (10 cm in diameter and 13 cm in height). The following criteria were used for scoring: independent use of the left or right forelimb for contacting the wall during a full rear to initiate a weight-shifting movement and simultaneous use of both the left and right forelimbs to contact the wall. Behavior score was recorded by determining the number of times the ipsilateral (unimpaired) forelimb (
Statistical Analysis
All the data in this study are presented as mean±s.d. Data were analyzed by Student's
RESULTS
Brain Lipocalin-2 Upregulation after Intracerebral Hemorrhage Intracerebral hemorrhage resulted in LCN2 upregulation in WT mice with many LCN2-positive cells detected in the ipsilateral basal ganglia at 24 hours after ICH. Few LCN2-positive cells were detected in the contralateral basal ganglia of ICH mice or the ipsilateral basal ganglia of sham mice (Figure 1A). Brain LCN2 protein levels in the ipsilateral basal ganglia were significantly higher in ICH mice compared with sham-operated mice at day 1 (LCN2/β-actin ratio: 1.34 ± 0.33 versus 0.85 ± 0.25 in the sham,
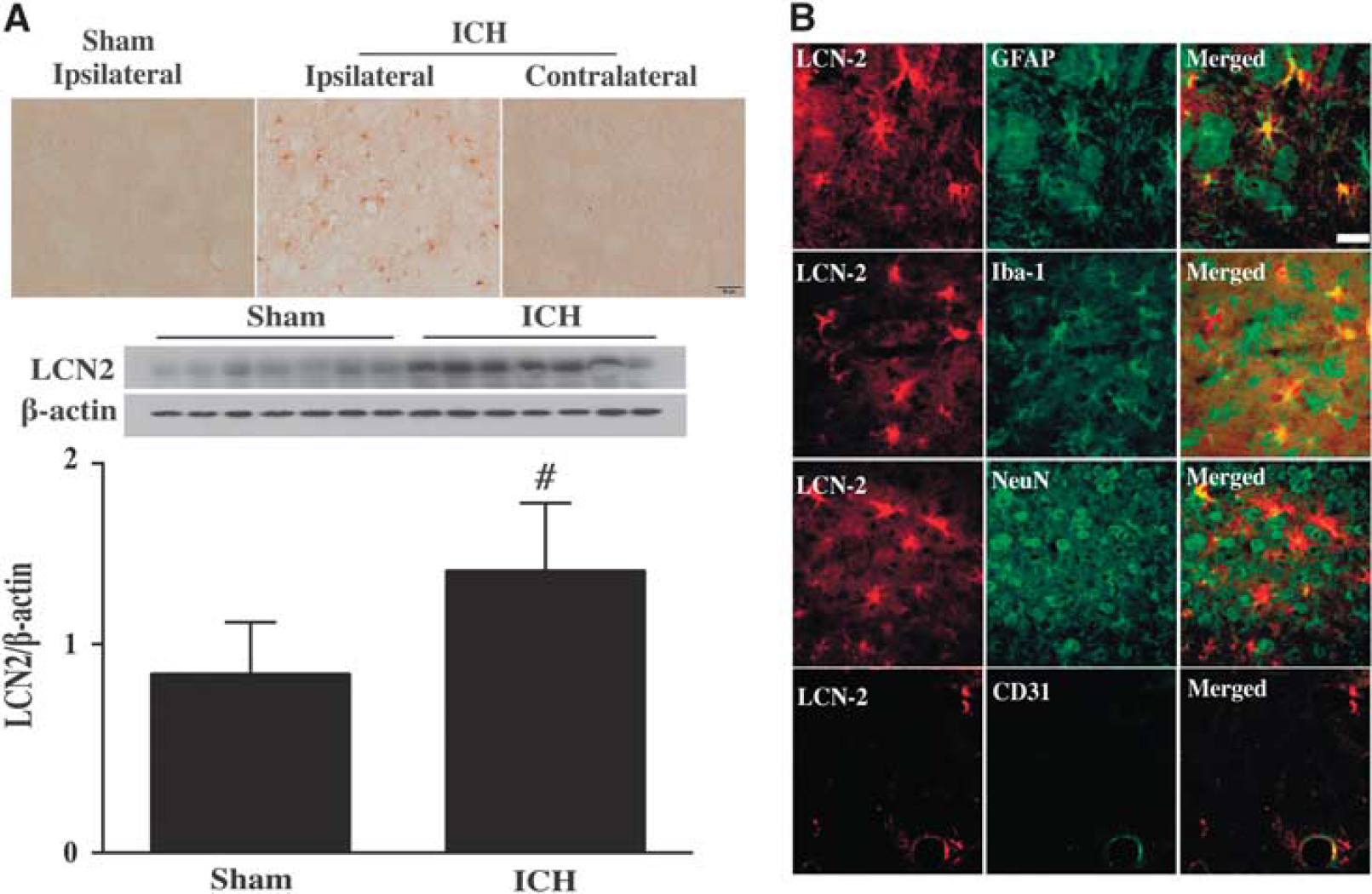
(
Intracerebral Hemorrhage-induced Less Brain Ferritin Upregulation in Lipocalin-2 Knockout Mice
Our previous study in rats showed that ferritin, an iron storage protein, is upregulated in brain after ICH.
19
Similarly, in WT mice, numerous ferritin-positive cells were found in the perihematomal area at day 1 after ICH, but not in the contralateral basal ganglia or in the ipsilateral basal ganglia of sham-operated mice (Figure 2A). Western blots showed the protein levels of both FTH and FTL were higher in the ipsilateral basal ganglia versus that in sham mice at day 1 after ICH (
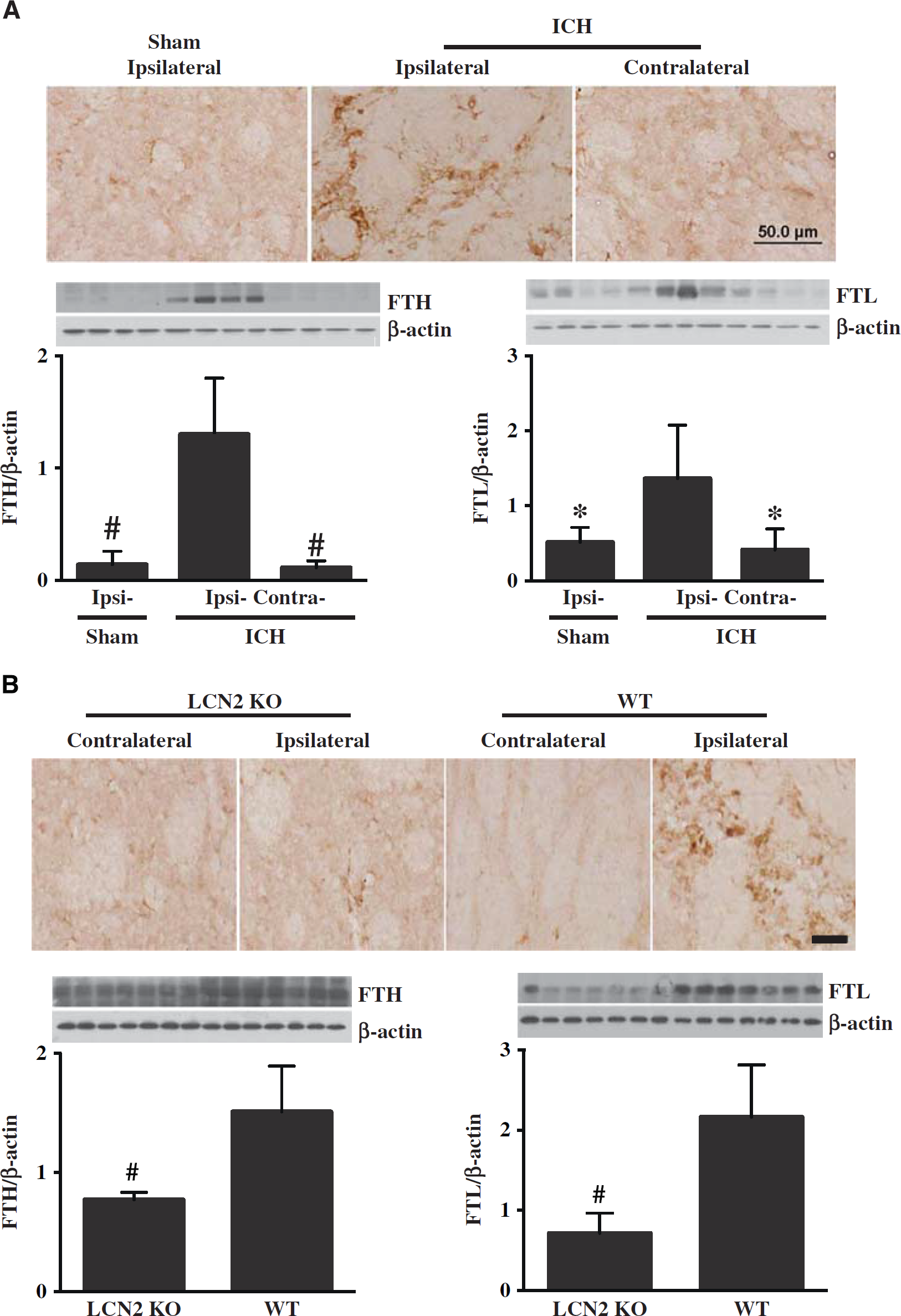
(
After ICH, there were no differences in ferritin immunoreactivity in the contralateral basal ganglia between WT and LCN2-/- mice, but there were far fewer ferritin-positive cells in the ipsilateral basal ganglia of LCN2-/- mice compared with WT mice at day 1 (Figure 2B). Western blots showed both FTH and FTL levels were lower in LCN2-/- mice (FTH/β-actin: 0.77 ± 0.06 versus 1.51 ± 0.38 in WT,
Intracerebral Hemorrhage Caused Less Brain Swelling, Microglia Activation, and Brain Atrophy in Lipocalin-2 Knockout Mice
T2-weighted MRI was used to determine ICH-induced brain swelling in mice. A blinded observer outlined the hemisphere for each animal and measured the volume. Intracerebral hemorrhage caused brain swelling in WT mice at days 1, 3, and 7 compared with sham (e.g., day 3: 8.2 ± 3.8% versus 0.4 ± 0.7%,
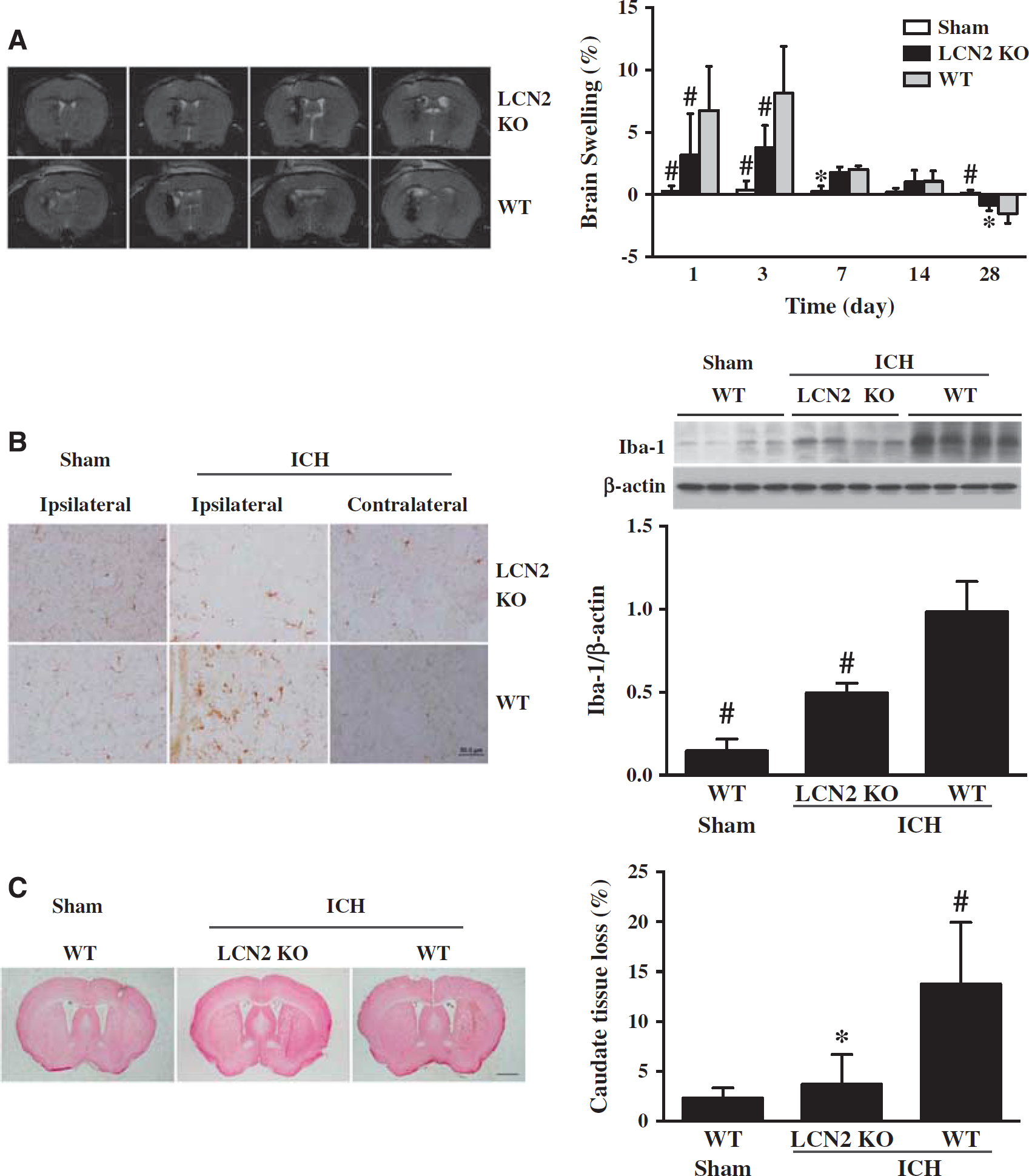
(
Intracerebral hemorrhage induces perihematomal microglial activation. There were more ameboid-shaped Iba-1-positive cells in the ipsilateral basal ganglia of WT mice 24 hours after ICH compared with the contralateral basal ganglia or sham-operated mice (Figure 3B). In contrast to the microglial activation in WT, there were fewer and thinner Iba-1-positive cells in the ipsilateral basal ganglia of LCN2-/- mice after ICH (Figure 3B). These cells morphologically were less dendritic with round shape distinguished from activated microglia. Iba-1 protein levels in ipsilateral basal ganglia after ICH were significantly lower in LCN2-/- mice compared with WT mice (Iba-1/β-actin: 1.12 ± 0.27 versus 0.74 ± 0.18,
Brain atrophy was examined in H&E-stained coronal sections at day 28 after ICH. The ipsilateral caudate tissue loss was calculated as: ((contralateral caudate area–ipsilateral caudate area)/contralateral caudate area) × 100%. There was significantly lower loss in LCN2-/- mice compared with WT mice (3.7 ± 3.0% versus 13.8 ± 6.2%,
Intracerebral Hemorrhage Caused Less Neurologic Deficits in Lipocalin-2 Knockout Mice
Both group showed significant neurologic deficits as assessed by forelimb use asymmetry and corner turn test at day 1 after ICH. However, the LCN2-/- mice had less neurologic deficits for both forelimb use asymmetry score (e.g., day 3: 40 ± 11% versus 60 ± 11%,
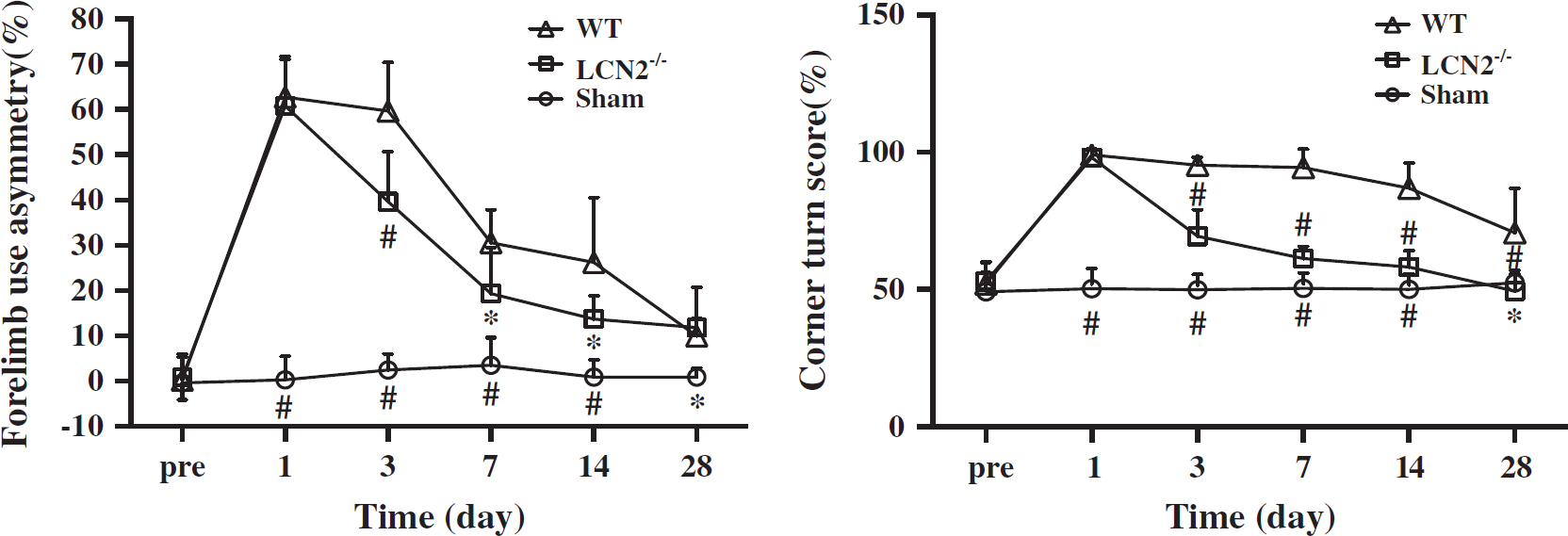
Forelimb use asymmetry and corner turn scores at days 1, 3, 7, 14, and 28 after a needle insertion or 30 μL blood injection into the right basal ganglia of lipocalin-2 knockout (LCN2-/-) and wild-type (WT) mice. Values are mean±s.d.; #
Intracerebral Injection of Iron Caused Lipocalin-2 Upregulation
Given the role of iron in ICH-induced brain injury, the effects of iron on LCN2 were examined. Intracerebral FeCl2 injection caused brain LCN2 upregulation. Lipocalin-2-positive cells were found in the ipsilateral, but not in the contralateral basal ganglia of WT mice after intracaudate injection of iron. Few LCN2-positive cells were found in the saline injection group (Figure 5A). In the ipsilateral basal ganglia, LCN2 protein levels were significantly higher in FeCl2-injected mice than in saline-injected mice as determined by western blot (LCN2/β-actin: 0.94 ± 0.32 versus 0.15 ± 0.05,
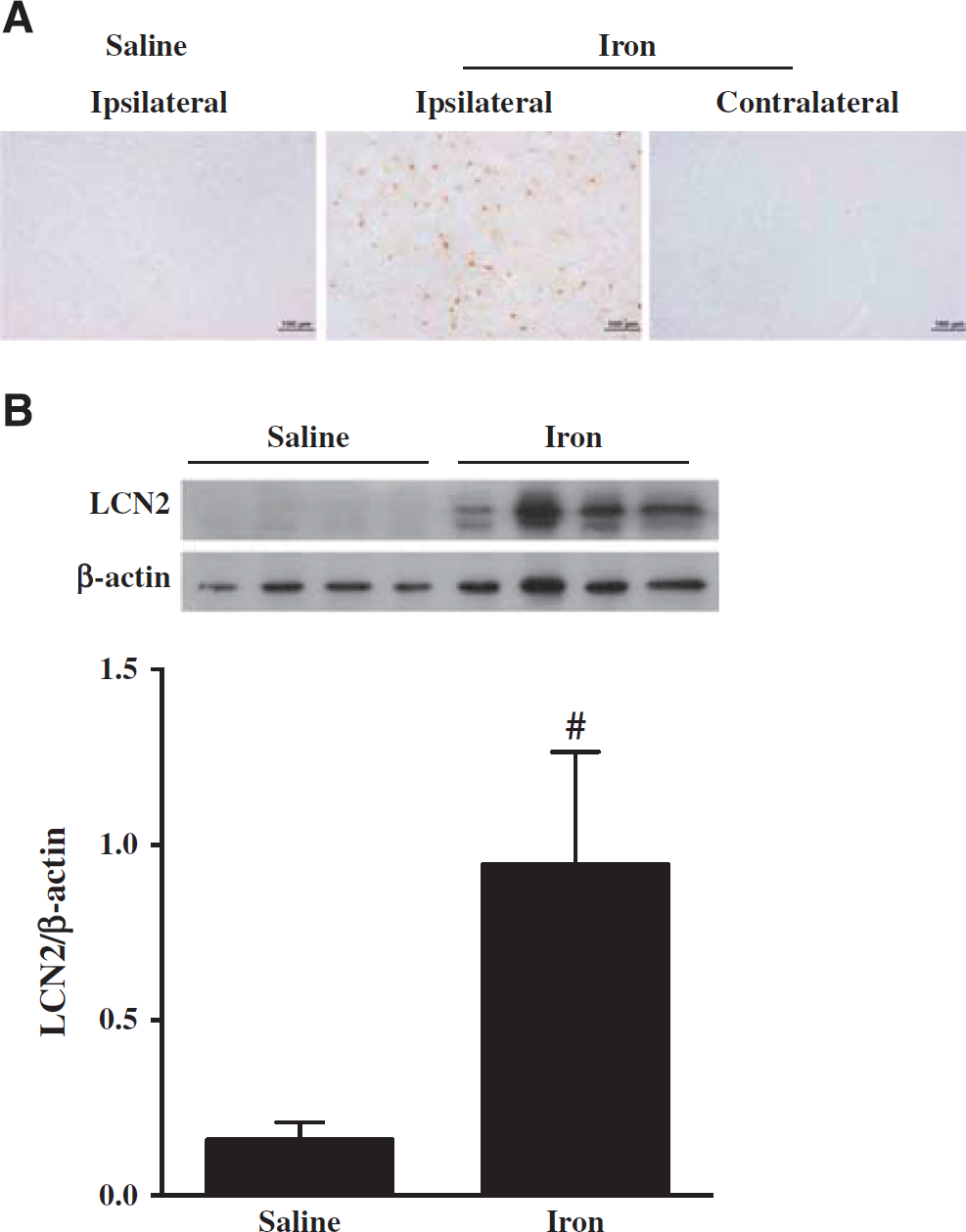
(
Iron-Induced Smaller Magnetic Resonance Imaging T2 Lesions and Less Brain Swelling and Blood–Brain Barrier Disruption in Lipocalin-2 Knockout Mice
Intracerebral injection of FeCl2 caused a MRI T2 lesion, brain swelling, and BBB disruption (Figure 6). Iron caused smaller T2 lesions in LCN2 knockout mice at 24 hours (16.0 ± 4.7 versus 26.6 ± 13.6 mm3 in WT mice,
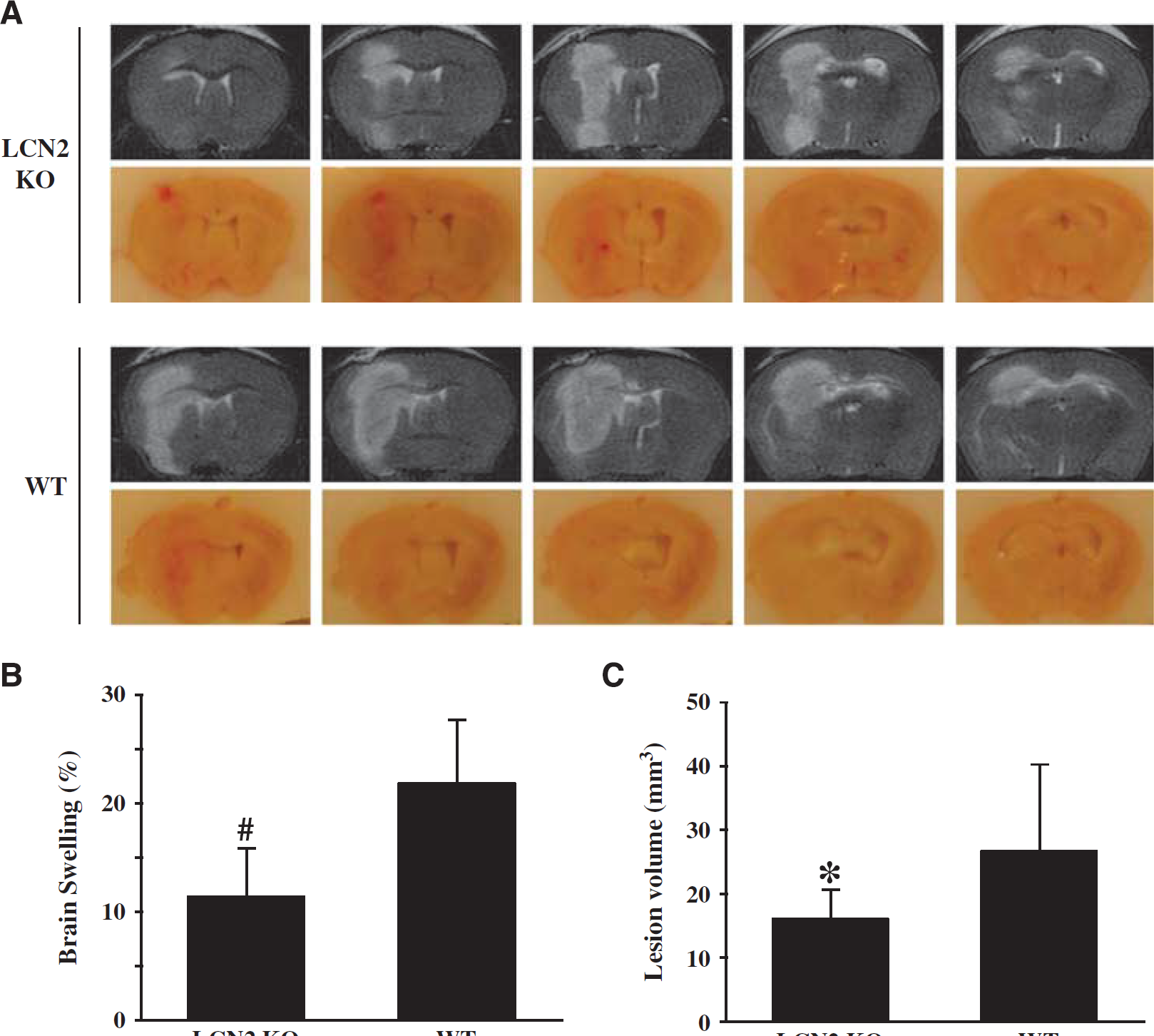
Comparison of brain injury in wild-type (WT) and lipocalin-2 knockout (LCN2-/-) mice at 24 hours after 10 μL FeCl2 (1 mmol/L) was injected into the right basal ganglia. (
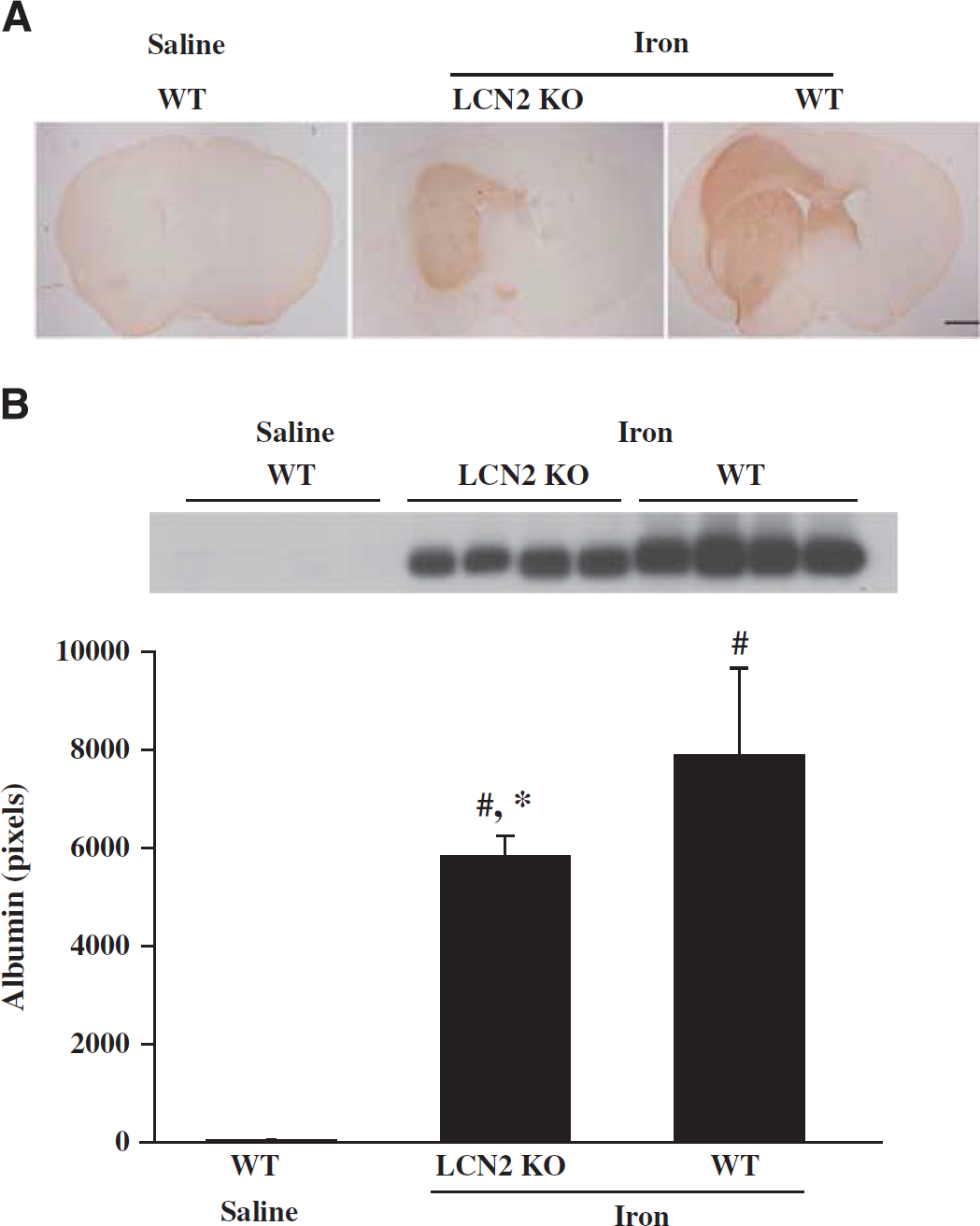
Albumin immunoreactivity (
DISCUSSION
In this study, we evaluated the role of LCN2 in brain injury after ICH in mice. The major findings were (1) Brain LCN2 levels were increased in the caudate after ICH and intracerebral injection of FeCl2; (2) ICH-induced brain ferritin upregulation was less in LCN2 knockout mice; (3) ICH caused less microglial activation, brain swelling, brain atrophy, and neurologic deficits in LCN2 knockout mice; and (4) LCN2 deficiency resulted in smaller iron-induced lesions and less BBB disruption and brain swelling.
Expression of LCN2 was found in astrocytes, microglia, neurons, and endothelial cells after ICH in mice. Although LCN2 is considered as being mainly expressed in astrocytes,12,14,23,24 where its secretion may be neurotoxic, 24 other cell types may express LCN2. Thus, inflammatory cells, such as microglia and neutrophils15,25,26 can also express LCN2. The effects of LCN2 on microglia are complex. For example, LCN2 can increase the M1-related gene expression in cultured mouse microglia after 8 to 24 hours. Lipocalin-2 can also be secreted from M1-polarized microglia. Mice lacking LCN2 have a markedly reduced M1-related gene expression in microglia after lipopolysaccharide injection. 26 Expression of LCN2 was sensitive to cytotoxic agents, and inflammatory activation of microglia can lead to LCN2 upregulation. Meanwhile, LCN2 expression in BV-2 microglia induces cell morphologic changes. It induces a round shape with a loss of processes (the ameboid form) indicating activation. 25 In our present study, microglia activation after ICH was lower in LCN2 knockout than in WT mice. This difference in microglia activation may contribute to the difference in brain injury between WT and LCN2 knockout mice after ICH. Endothelial LCN2 may be involved in angiogenesis. Recent study showed LCN2 increased tube formation and cell migration in rat brain endothelial cells via iron and ROS-dependent mechanisms. 27
The expression of ferritin after ICH is time dependent. In our previous paper in a rat model of ICH, FTL protein levels at day 1 were very low. 19 However, we still found iron- and ferritin-positive cells in the perihematomal zone as early as the first day. The protein level of FTH could also be detected at 24 hours. This indicates that cellular iron accumulation might start very early in the perihematomal zone after ICH. The source of iron could come from the serum and red blood cell lysis after ICH. In our mouse ICH model, we found that the FTL levels significantly increased at 24 hours. This more pronounced early upregulation compared with rat might be species related.
As well as inflammation, iron has a key role in ICH-induced brain injury. The current study showed that intracerebral injection of ferrous iron caused less brain injury, brain swelling, and BBB leakage in LCN2 knockout mice. Our previous findings showed that LCN2 was upregulated in a rat ICH model and that treatment with a ferric iron chelator, deferoxamine, attenuated ICH-induced LCN2 upregulation and brain injury.12,28 Together, these results indicate that LCN2 has a role in iron-mediated brain injury after ICH. Until now, the detailed mechanism of iron delivery through LCN2 has not been fully elucidated. In the previous literatures, LCN2 was considered to be a mediator of an alternative, transferrin-independent pathway for cellular iron delivery. 11 Iron is suggested to bind an LCN2-associated small molecular weight siderophore, be transferred into cells through 24p3R, a LCN2 cell-surface receptor, and then be released resulting in an increased intracellular iron concentration.10,29 The LCN2 deficiency can block the pathway of LCN2-reliant intracellular iron transportation, as suggested by the reduced iron-induced ferritin synthesis, and alleviate the brain injury. However, studies also showed that LCN2 can regulate the intracellular iron concentration and LCN2 deficiency can increase the cellular iron levels in sepsis. 30 Thus, the role of LCN2 in iron transport still need further study.
Advances have been made recently in identifying mammalian siderophores(s). In particular, recent evidence indicates that 2,5-DHBA (2,5-dihydroxy benzoic acid) is an important mammalian siderophore.31,32 The role of 2,5-DHBA and the enzyme, 3-hydroxybutyrate dehydrogenase-2, involved in 2,5-DHBA biosynthesis needs to be evaluated in ICH.
The BBB disruption can contribute to brain edema after iron injection or ICH. The current study found less BBB leakage and brain swelling in LCN2 knockout mice compared with WT controls. Matrix metalloproteinase-9 (MMP-9) may contribute to the effect of LCN2 on BBB disruption. It is well known that MMP-9 can lead to BBB disruption33,34 and LCN2 reduces MMP-9 degradation by forming an MMP-9/LCN2 complex.35,36 Increasing the stability of MMP-9 may enhance the effect on BBB disruption. In addition, LCN2 may contribute to BBB disruption by enhancing microglia activation. Inflammation is an important regulator of BBB disruption in brain injury.
The current study shows that ICH-induced brain tissue loss is less in LCN2 knockout mice. The difference between LCN2 and WT mice reaction to ICH might relate to role of LCN2 in iron handling. Lipocalin-2 is considered to regulate iron uptake by binding to siderophores, with the resultant complex undergoing endocytosis after binding to cell-surface receptors, such as 24p3R.10,37,38 Neurons, as well as astrocytes and endothelial cells, express the LCN2 receptor, 24p3R. 14 Based on this evidence, LCN2 knockout may decrease cellular iron uptake, alleviating iron-induced cell structure disruption and cell death. Indeed, we found ICH-induced upregulation of ferritin is less in LCN2 knockout mice, suggesting reduced cell iron concentrations.
In contrast to the effects of LCN2 deficiency on ferritin expression after ICH, we found no effect of the KO on ferritin levels in the contralateral hemisphere reflecting the very low levels of LCN2 in the normal brain. Targeting LCN2 after ICH may, therefore, have limited effects on normal brain tissue.
In conclusion, both ICH and intracerebral injection of iron caused upregulation of LCN2 in mouse brain and ICH/iron-induced brain injury was less in LCN2 knockout mice. The results suggest a role of LCN2 in brain injury after ICH.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.