
Abstract
Chronic consumption of high-fat-and-fructose diets (HFFD) is associated with the development of insulin resistance (InsRes) and obesity. Systemic insulin resistance resulting from long-term HFFD feeding has detrimental consequences on cognitive performance, neurogenesis, and long-term potentiation establishment, accompanied by neuronal alterations in the hippocampus. However, diet-induced hippocampal InsRes has not been reported. Therefore, we investigated whether short-term HFFD feeding produced hippocampal insulin signaling alterations associated with neuronal changes in the hippocampus. Rats were fed with a control diet or an HFFD consisting of 10% lard supplemented chow and 20% high-fructose syrup in the drinking water. Our results show that 7 days of HFFD feeding induce obesity and InsRes, associated with the following alterations in the hippocampus: (1) a decreased insulin signaling; (2) a decreased hippocampal weight; (3) a reduction in dendritic arborization in CA1 and microtubule-associated protein 2 (MAP-2) levels; (4) a decreased dendritic spine number in CA1 and synaptophysin content, along with an increase in tau phosphorylation; and finally, (5) an increase in reactive astrocyte associated with microglial changes. To our knowledge, this is the first report addressing hippocampal insulin signaling, as well as morphologic, structural, and functional modifications due to short-term HFFD feeding in the rat.
Keywords
INTRODUCTION
High intake of hypercaloric diets has increased alarmingly in the past years in western societies. The chronic consumption of diets rich in saturated fats and in processed sugars (high-fat-and-fructose diet, HFFD), particularly high fructose, is strongly associated with a variety of related metabolic diseases including obesity, systemic insulin resistance (InsRes), metabolic syndrome, and type-2 diabetes mellitus.1,2 These diseases have reached epidemic proportions and increased comorbid conditions such as elevated blood pressure, cardiovascular disease, and inflammation, which in turn lead to a reduced quality and expectancy of life.3–5 Obesity caused by hypercaloric diets may be associated with a diminished ability of cells to respond to the action of insulin, leading to InsRes and metabolic syndrome, characterized by hyperglycemia, dyslipidemia, and compensatory hyperinsulinemia.1,6
Several epidemiologic studies have found that patients with obesity and InsRes have an increased risk for diverse cognitive impairments, including cognitive decline, mild cognitive impairment, and even Alzheimer's disease (AD).7–10 Studies performed in animal models of diet-induced obesity and InsRes such as long-term HFFD feeding show a deficient execution in memory and learning tasks.11–16 These cognitive alterations were associated with functional and structural changes in the hippocampus, such as reduced number and complexity of dendritic spines in the CA1 subfield, altered establishment of long-term potentiation and long-term depression, and diminished dentate gyrus (DG) neurogenesis.11–13 The latter was correlated with decreased expression of brain derived neurotrophic factor, cyclic AMP-response element-binding protein and the synaptic proteins synapsin, and stargazin. 11
It has been extensively shown that insulin has a pivotal role in hippocampal neuronal function as a metabolic, growth, synaptic, and survival modulator.17–20 Additionally, insulin also has a role in enhancing memory and establishing long-term potentiation through several molecular mechanisms that include neurotransmitter release, expression and insertion of receptors at the postsynaptic membrane, and expression of postsynaptic proteins, mainly via the P***I3-K/Akt signaling pathway.21–23 It has also been proposed that insulin can modify microtubule stabilization through inhibition of glycogen synthase kinase 3β (GSK3β), a kinase that can phosphorylate the microtubule-associated protein (MAP) tau.24,25 Given these crucial roles of insulin in the hippocampus, it could be speculated that systemic insulin alterations resulting from obesity and InsRes could result in hippocampal insulin alterations that underlie cognitive impairment. In this regard, it has been reported that long-term high-fat (not fructose) feeding in mice produced alterations in Akt and GSK3β signaling in the hippocampus. 26 Since virtually all studies on hippocampal dysfunction due to HFFD administration have addressed the impact of long-term feeding, for more than 4 weeks, the present work aimed to investigate early biochemical and structural changes in the hippocampus after short-term consumption of HFFD. We found that obesity and InsRes caused by 7 days of HFFD intake were sufficient to alter insulin signaling in the hippocampus associated with decreased complexity of neurites, reduced synaptic markers, modifications of MAPs, and increased number of reactive astrocytes and microglia. These findings shed light on the early molecular and cellular changes in response to reduced hippocampal insulin sensitivity associated with excessive lipid and fructose intake.
MATERIALS AND METHODS
All chemicals and salts were purchased from J.T. Baker Chemicals (Phillipsburg, NJ, USA), unless otherwise stated.
Animals and Diets
Animals were handled in accordance with local government rules and the Society for Neuroscience Guide for the Care and Use of Laboratory Animals with approval of the Animal Care Committee of the Instituto de Investigaciones Biomédicas, UNAM. Efforts were made to minimize animal suffering and to reduce the number of subjects used. We used 48 male Sprague Dawley rats weighing 250 to 270g (Harlan Laboratories, Facultad de Quimica, UNAM, Mexico). Animals were individually housed under standard conditions (12 hours light/dark cycles, 22°C). Rats were divided into two groups and fed for 7 days as follows: The control (ctrl) group (n = 20) was fed a standard rodent chow (3 kcal/g; Harlan Laboratories, Indianapolis, IN, USA) and bidistilled water (0 kcal/g), and the experimental group (n = 22) was fed a high-fat diet consisting of standard rodent chow supplemented with 10% lard (5.4 kcal/g; Bio-Serv, Frenchtown, NJ, USA), and 20% high-fructose corn syrup in the bidistilled water (0.83 kcal/ml, commercially available corn syrup). All rats had ad libitum access to their diets and drinking water. Food and water intake were recorded. Food was removed the morning of the studies. After a 5-hour fast, rats were anesthetized with sodium pentobarbital (80 mg/kg) and killed. Rats receiving an insulin bolus (n = 8 per group) were anesthetized and given an intraperitoneal injection of insulin (5 U/kg; Sigma, St Louis, MO, USA) and decapitated after 15 minutes.
Serum Glucose, Insulin, and Leptin Determination
Trunk blood was collected from animals after they were killed. Serum was collected by centrifugation and stored at –20 °C. For glucose concentration determinations, a OneTouch Ultra glucose monitor was used (Johnson & Johnson, New Brunswick, NJ, USA). Insulin and leptin concentrations were performed in duplicate by solid-phase 125I radioimmunoassay (Millipore Corporation, Billerica, MA, USA) in serum samples. The sensitivity of the assays was 0.04 ng/mL to insulin and 0.08 ng/mL to leptin. The intraassay and interassay coefficients of variation were 4% and 6%, and 5.1% and 7%, respectively.
Western Blotting
Animals (n = 16 control, n = 16 HFFD) were anesthetized as described and decapitated. We rapidly dissected hippocampus from whole brains that were immediately frozen and stored at –20°C. Then, tissues were homogenized, lysed for 20 minutes and then sonicated in 1-second pulses for 20 times at 4°C in RIPA lysis buffer (50 mmol/L HEPES, pH 7.7; 100 mmol/L sodium chloride; 2 mmol/L PMSF; 1% NP-40; complete inhibitor cocktail from Roche Diagnostics (Indianapolis, IN, USA) and phosphatase inhibitor cocktail from Thermo Scientific, Waltham, MA, USA). Samples were centrifuged at 20,817g for 15 minutes at 4°C and supernatants were collected. Protein concentration was determined using a BCA kit (Pierce, Rockford, IL, USA). Lysates were boiled in Laemmli buffer and fractionated in SDS-PAGE 10% acrylamide gel. Proteins were then transferred onto a nitrocellulose membrane (Amersham, Buckinghamshire, England) under standard conditions. We blocked membranes with 5% nonfat-milk-TBS-T and incubated them with one of the following primary antibodies at 4°C overnight in 3% BSA (bovine serum albumin)-TBS-T: anti-pi R Y1146 (1:1,000; Cell Signaling Technology, Danvers, MA, USA); anti-pi RS-1 Y608 (1:250; BioSource, Carlsbad, CA, USA); anti-pAkt S473 (1:1,000; Cell Signaling Technology); anti-pS6K T389 (1:500; Cell Signaling Technology); anti-pTau S199/S202 (1:2,000; Chemicon, Temecula, CA, USA); anti-SYP (1:1,000; Santa Cruz Biotechnology, Dallas, TX, USA) anti-tubulin (1:30,000; Sigma-Aldrich, St Louis, MO, USA). Next, we incubated membranes with horseradish peroxidase-conjugated secondary antibodies for 2 hours at room temperature in 5% nonfat-milk-TBST-T (anti-mouse IgG or anti-rabbit IgG horseradish peroxidase conjugated; Santa Cruz Biotechnology) and membranes were revealed using a chemoluminiscent substrate ECL (Millipore) on Kodak X-Omat film. Densitometrie analysis of the bands obtained was performed using Photoshop. To normalize the data, we divided the values obtained for each band into their respective tubulin loading control and obtained a ratio. This value was then converted to percent of control for each experiment.
Histology and Immunohistochemistry
Animals (n = 8 control, n = 8 HFFD) were perfused transcardially with phosphate-buffered saline (PBS) 1 × and 4% paraformaldehyde as a fixative (Sigma). Brains were then removed and stored at 4°C in fixative. Afterwards, brains were successively transferred to 20% and 30% sucrose solutions for 24 hours each, and then sectioned coronally throughout the entire hippocampus into 30 μm thick sections using a cryostat at — 20°C. Coronal sections were treated for immunohistochemistry or were stained with cresyl violet or Golgi staining. For immunohistochemistry, free-floating sections were incubated at room temperature for 30 minutes in PBS containing 0.25% Triton X-100 and 0.3% H2O2 and left for 2 hours at 4°C in PBS/5% BSA solution. Slices were then immersed overnight at 4 °C in a PBS/5% BSA solution containing one of the following antibodies: anti-GFAP (1:500; Dako, Glostrup, Denmark), anti-***lbal (1:250; Dako), anti-MAP-2 (1:500; Millipore). After this period, the brain slices were washed three times with PBS, each for 5 minutes, and incubated with the secondary antibody, biotinylated mouse IgG (1:500; Vector, Burlingame, CA, USA) for 2 hours at room temperature. Finally, the slices were processed with the ABC-biotin-avidin-peroxidase kit (Vector) and developed with diamino-benzidine tetrahydrochloride as a substrate for peroxidase reaction. For negative controls, primary antibodies were omitted from the procedure. All images were captured with a digital camera attached to a Zeiss, Axioskop 40 microscope (Zeiss, Oberkochen, Germany).
Microtubule-Associated Protein 2 Densitometry and GFAP Quantification
Four animals per group were used for immunostaining. For MAP-2 densitometry, 3 to 4 100 μm × 100 μm sections were selected from × 20 magnification images of different sections of the stratum pyramidale of CA1 and the granular cell layer of the DG. The mean pixel intensity was automatically calculated using ImageJ (http://imagej.nih.gov/). Glial fibrillary acidic protein (GFAP) immunopositive cells were hand recorded from × 20 magnification images of four different sections of the stratum radiatum of CA1 and the hilus of DG per animal. Cells were counted when complete processes and nuclei were clearly observed, and expressed as the number of cells per mm2. For calculating GFAP area, ImageJ was used. × 40 magnification images were used, and only complete astrocytes were selected. The area of each cell was automatically calculated.
Golgi Staining, Sholl Analysis, and Dendritic Spine Count
Four animals per group were used for Golgi stainings. We used the FD Rapid Golgi Stain kit with the manufacturers' instructions (FD NeuroTechnologies, Columbia, MD, USA). Stained brains were sliced using a Vibratome (VT1000S; Leica, Wetzlar, Germany) at a thickness of 150 μm. For Sholl analysis, traces of CA1 neurons obtained at magnification × 20 were loaded into ImageJ and then the Sholl Analysis plug-in was used for automatic analysis (http://labs.biology.ucsd.edu/ghosh/software/). For dendritic spine count, × 40 magnification images of CA1 neurons were used, and spine was counted in segments of 10 μm of the dendrite manually. Only secondary and tertiary dendrites were used for the analysis, and no more than three 10 μm segments of the same dendrite were counted. Both Sholl Analysis and spine counting were performed in a blind manner.
Statistical Analysis
Results represent the mean ± s.e.m. Comparisons among groups were made using a nonpaired Student's t-test. Correlation of hippocampal weight with caloric intake per rat was assessed by linear regression analysis, and the correlation of insulin and leptin levels was evaluated by the Spearman Rank test (r). A P<0.05 was considered as significant. We used GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA, USA) for graphs and statistical analysis.
RESULTS
Short-Term High-Fat-And-Fructose Diet Feeding Induces Obesity and Systemic Insulin Resistance
Previous studies have shown that rats fed with HFFD for 3 days show hepatic InsRes, while 7 days of HFFD feeding are required for InsRes to be systemic2,27 To assess the metabolic state of the rats used in this study, we examined some nutritional, metabolic, and hormonal parameters. We found that ingestion of HFFD for 7 days produces hyperphagia, obesity, and InsRes (Figure 1). Further, HFFD-fed rats consumed significantly more chow (43.8%, P<0.05) and water (33%, P<0.05) than controls and thus had an increased caloric intake (254%, P<0.001), leading this experimental group to gain more weight (132%, P<0.001) (Figure 1A). We also investigated whether systemic insulin sensitivity was diminished in HFFD-fed animals. Fasting serum glucose levels were evaluated at basal state and after 15 minutes of the administration of an intraperitoneal insulin bolus (5 U/kg) (Figure 1B). While basal glucose levels were not different in both groups, insulin injection significantly reduced blood glucose levels in control rats (24.7%, P<0.05) but not in HFFD rats (9%, n.s.), indicating an impairment in systemic insulin sensitivity in HFFD-fed rats. In agreement with this, we found that serum insulin levels were increased in HFFD rats (55.3%, n.s.). Additionally, we measured leptin levels since it has been reported by some groups that InsRes is accompanied by leptin resistance, and that the ratio of both hormones can be used as a parameter for measuring metabolic alterations in humans and rodents. 28 We found a significant increase in leptin levels in the HFFD rats as compared with controls (94.5%, P<0.001) as well as a positive correlation between the two hormones levels (r = 0.6697, P = 0.0280) (Figure 1C). These data indicate that HFFD-fed rats develop systemic InsRes and elevated levels of leptin, which has also been associated with the development of obesity and InsRes.
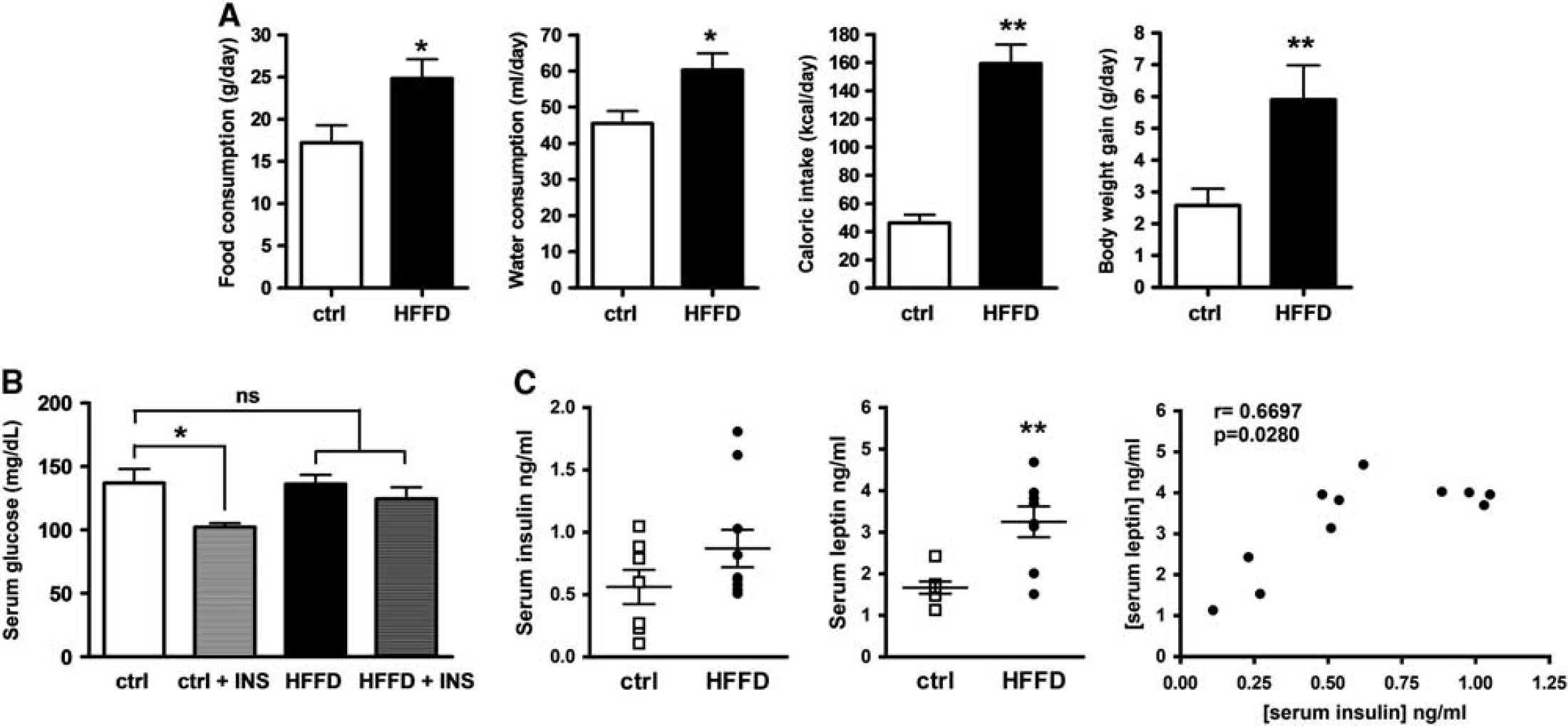
Obesity and insulin resistance parameters. (
High-Fat-And-Fructose Diet Downregulates Insulin Signaling in the Hippocampus
To investigate whether HFFD feeding modulates hippocampal insulin signaling, we examined the phosphorylation state of insulin receptor (IRY1146), insulin receptor substrate-1 (IRS-1Y608), and protein kinase B (AktS473). There was a marked decrease in IRS-1 and Akt phosphorylation in rats fed with HFFD for 7 days compared with control rats (51%, P<0.001 and 25%, P<0.05, respectively) (Figures 2B and 2C), whereas no significant change in IR phosphorylation was found (3%, n.s.) (Figure 2A). These findings collectively indicate that 7 days of HFFD feeding are sufficient to modify hippocampal insulin signaling. Earlier experiments in rats have shown that HFFD feeding activates the mTOR/S6K pathway in muscle, liver, and mediobasal hypothalamus.29,30 Therefore, we analyzed whether HFFD also induces the activation of S6K in the hippocampus. As shown in Figure 2D, we found an enhanced S6K phosphorylation of threonine 389 after 7 days of HFFD feeding (71.9%, P< 0.001).
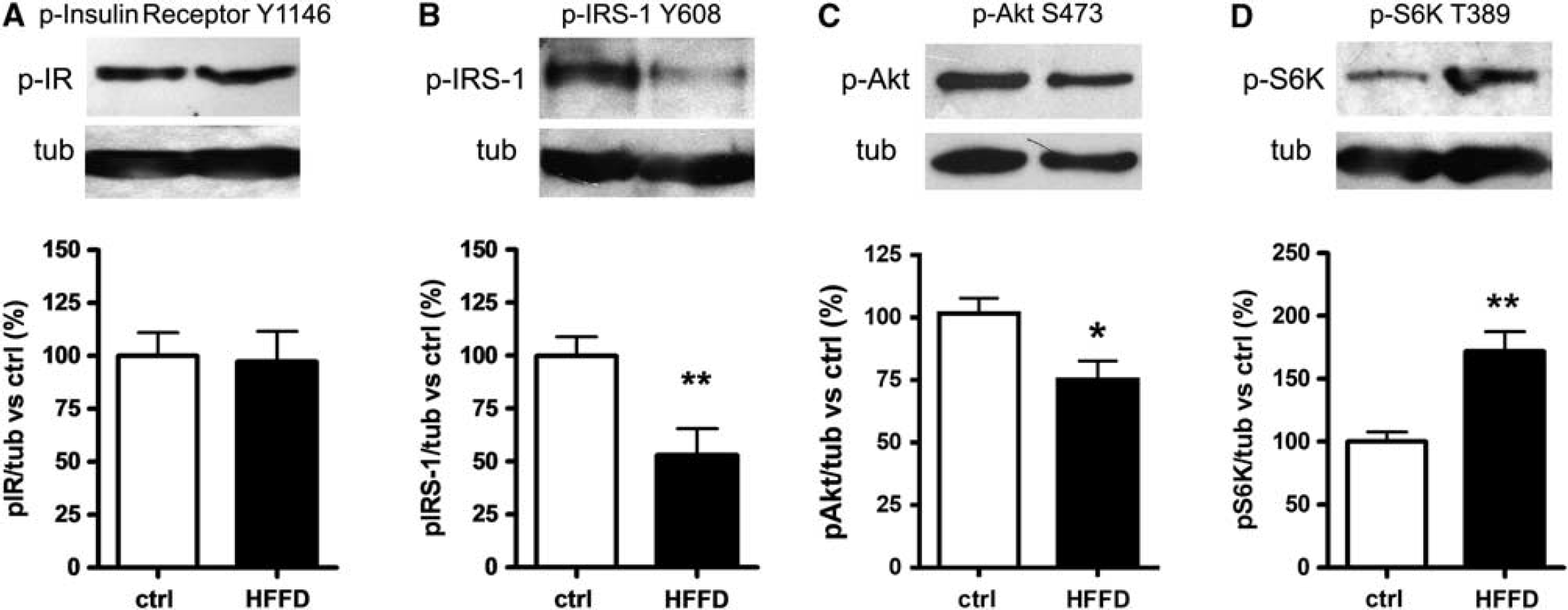
Hippocampal insulin signaling assessment. Western blot of insulin signaling proteins: (
High-Fat-And-Fructose Diet Decreases Hippocampal Weight, Dendritic Branching in CA1 and Induces Cytoskeletal Protein Modifications
To evaluate whether HFFD-mediated blunting of insulin signaling in the hippocampus was accompanied by morphologic alterations, we examined different parameters of hippocampal integrity. As shown in Figure 3A, a significant reduced hippocampal wet weight was observed in the HFFD group (19.5%, P<0.05) as compared with control animals (Figure 3A). This decrease in hippocampal weight positively correlated with the caloric intake of each rat (r 2 0.7622) (Figure 3A), but was not associated with morphologic changes assessed by Nissl staining (Figure 3B). However, a detailed histologic analysis by Golgi impregnation and a Sholl analysis revealed a reduction in the dendritic arbor complexity of the pyramidal CA1 neurons from HFFD-fed animals (Figure 3C). This reduced dendritic branching was observed in both basal and apical dendrites. Furthermore, MAP-2, a protein related with changes in structural stability in dendrites, was significantly decreased in HFFD rats as compared with controls in both CA1 (29.78%, P<0.001) and DG (18.75%, P<0.001) (Figures 3D and 3E). In addition, we found the axonal MAP tau was significantly phosphorylated in a reported GSK3β site, S199/202 (52%, P<0.001), in HFFD-fed rats (Figure 3F).
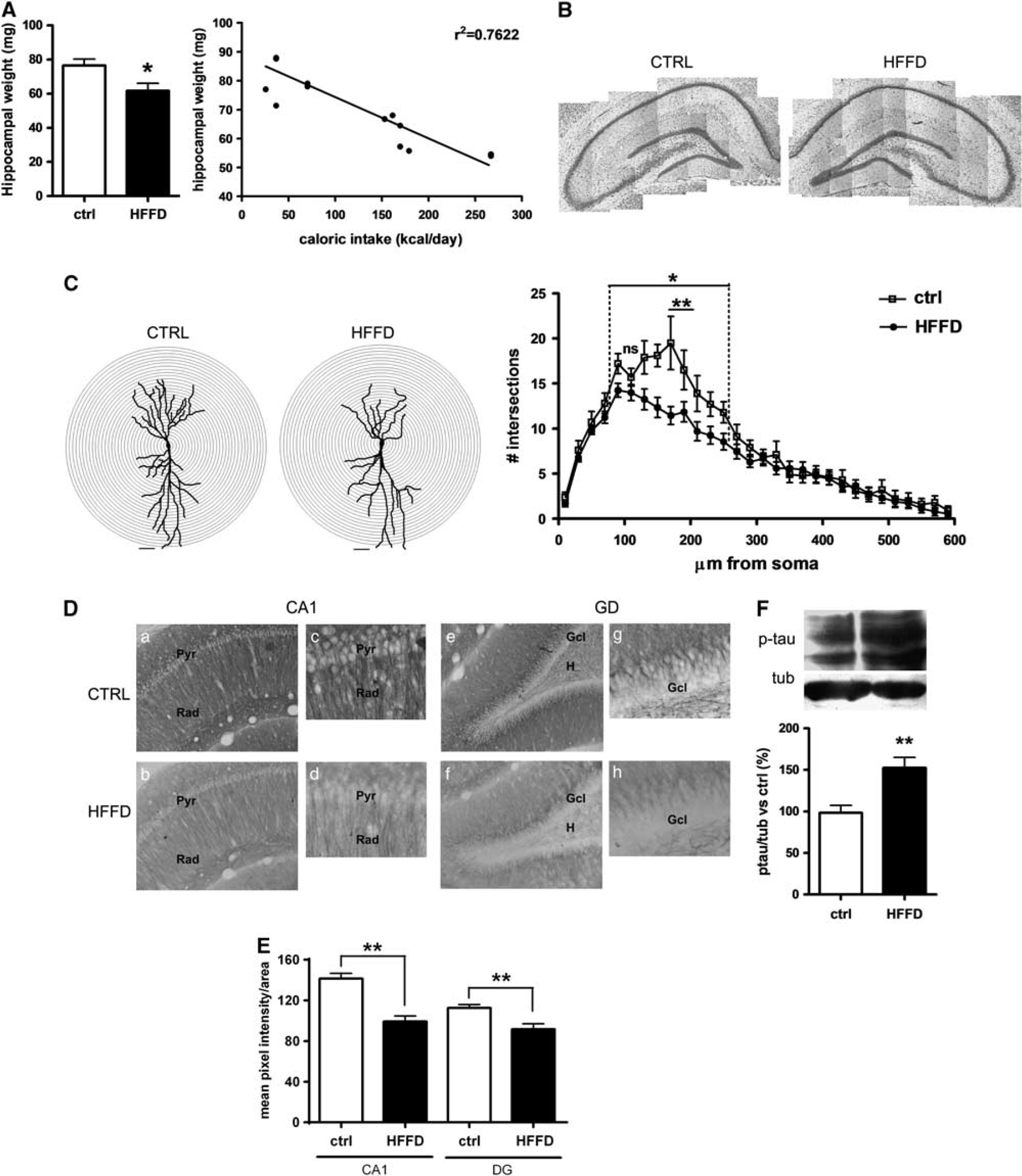
Immunohistochemical and morphologic analysis of the hippocampus. (
High-Fat-And-Fructose Diet Feeding Is Associated with Reduced Synaptic Markers
Since it was found that CA1 neurons of HFFD rats presented a reduction in the dendritic arbor complexity, we wanted to analyze whether this reduction was accompanied by a decrease in presynaptic and postsynaptic elements. To this end, we counted the number of dendritic spines in Golgi-stained CA1 neurons. Quantitative analysis revealed a significant decrease in spines on both apical (81%, P<0.001) and basal (83%, P<0.05) dendrites in the HFFD rats (Figure 4A). Additionally, we found a significant reduction in the presynaptic protein synaptophysin (82%, P<0.05) (Figure 4B).
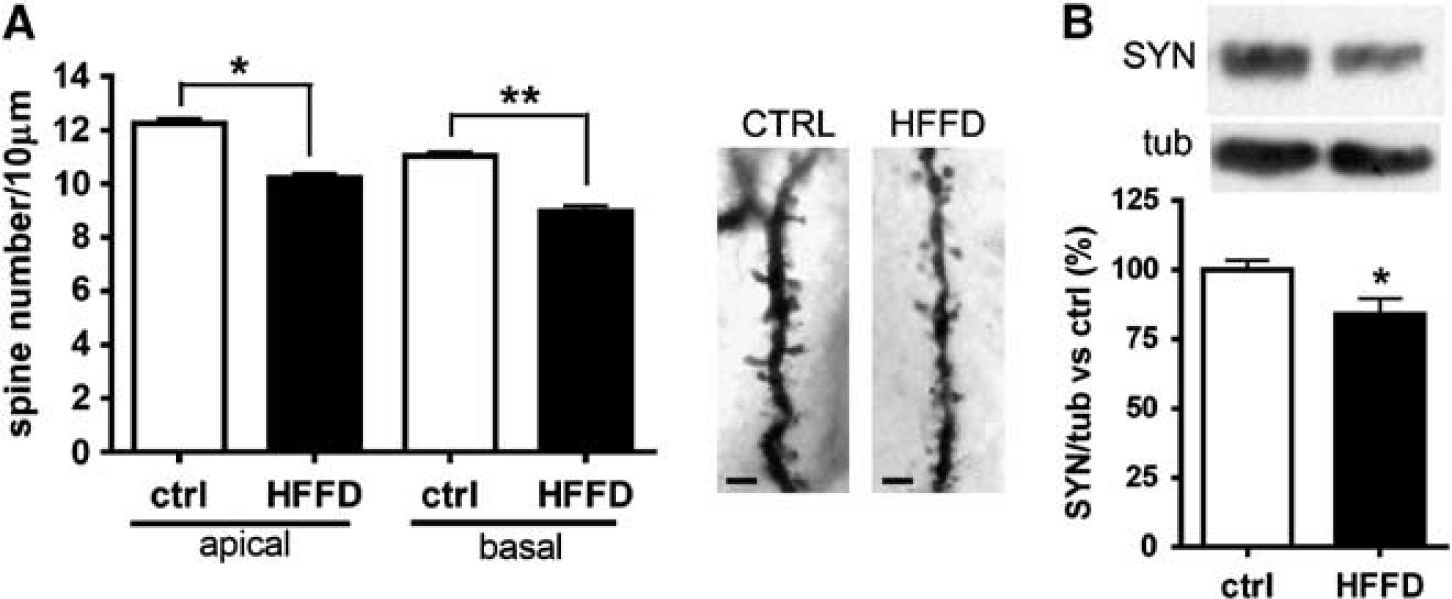
Evaluation of synaptic markers. (
High-Fat-And-Fructose Diet Causes Glial Activation
Given that inflammation and reactive astrogliosis have been found in the hypothalamus in response to obesity, 31 we wanted to evaluate the impact of a short-term exposure to HFFD feeding in astrocytic activation in the hippocampus using GFAP immunohistochemistry to evaluate the number and morphology of astrocytes. Diet-induced obesity was associated with a significant increase in GFAP+ cell number in CA1 (51%, P<0.001) and DG (25.69%, P<0.05) (Figures 5A and B), accompanied by a significant increase in GFAP area in both CA1 (77.7% P<0.001) and DG (62.5%, P<0.05) (Figure 5B). The latter was associated with a change in astrocyte morphology from rounded to stellate shape (Figure 5A). We also found an increase in astrocytic processes extending to the neuronal layers of the DG in the HFFD rats (Figure 5B). Overall, these data suggest a reactive astrocytic response of the hippocampus. Microglial changes were analyzed with Iba 1 staining. The majority of cells showed the characteristic resting microglia morphology consisting in a rod-shaped cell body with many thin processes along all areas of the hippocampus and similarly in both groups of rats. However, it was also observed the presence of few cells that showed the morphology of activated microglia with elongated cell somata and fewer branches mainly located in the CA1 region from HFFD rats (Figure 5C).
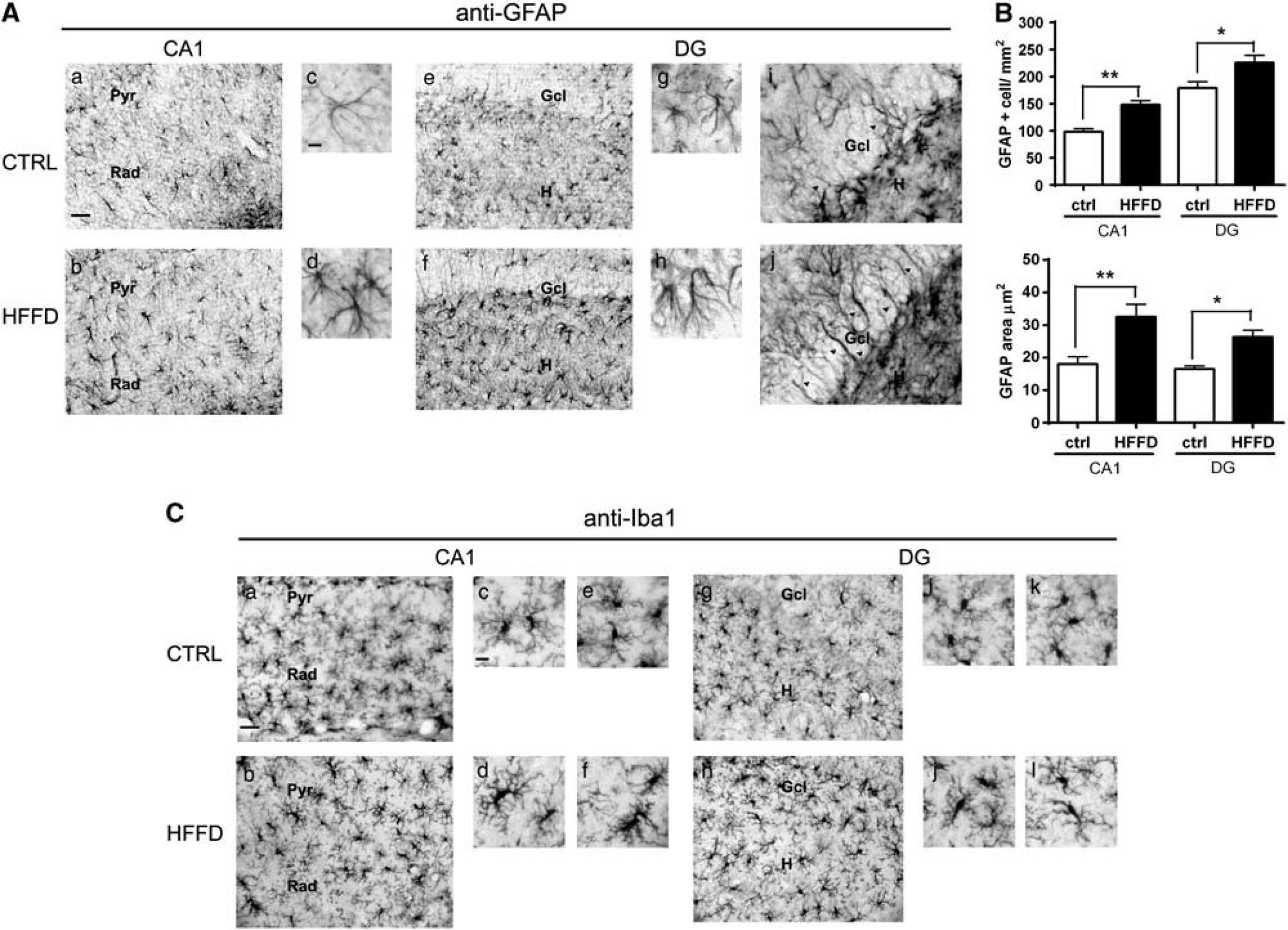
Changes in hippocampal astrocytes and microglia. (
DISCUSSION
The goal of this work was to find biochemical and structural changes in the hippocampus of short-term HFFD-fed rats, since there is no information about the early modifications that occur during acute intake of HFFD in this particular highly plastic region. In this work, we show that HFFD feeding for 7 days reduced hippocampal insulin signaling accompanied by several cytoskeletal modifications, dendritic and synaptic reductions, and astroglial activation.
It is important to mention that the HFFD used in this work substantially reflects hypercaloric western diets consisting of high saturated fats and cholesterol as well as beverages containing high fructose syrup, and therefore is one of the most complete models of metabolic alterations due to hypercaloric diets.
Although it has been thoroughly studied that 7 days of overfeeding rapidly induced hepatic and then systemic InsRes, 2 to our knowledge, this is the first report that shows the impact of an acute HFFD on hippocampal insulin signaling associated with rapid structural changes in this region.
The results of this study show that 7 days of HFFD feeding were effective for inducing systemic InsRes as determined by the presence of moderately higher circulating insulin and leptin levels and a diminished insulin-dependent reduction in blood glucose. In the hippocampus, we did not find changes in the phosphorylation state of the IR, suggesting that they respond to basal insulin levels in a similar way in both HFFD and control rats. In contrast, IRS-1Y608 phosphorylation was importantly diminished in the hippocampus of the HFFD group, although insulin receptor is apparently functional and phosphorylated. Phosphorylation of serine residues of IRS-1 mediated by kinases such as S6K, JNK and PKC, among others, prevents IRS-1 from recognizing the phosphorylated insulin receptor, and therefore gives rise to InsRes in HFFD-fed animals.27,29,30 In line with this, we found an increase in S6K kinase activation, which might in turn phosphorylate IRS-1 in serine residues and therefore blunt insulin signaling in the HFFD-fed rats. mTOR/S6K signaling can be activated by insulin, some growth factors and nutrients. Since insulin signaling is downregulated in our model, we believe that S6K activation is caused by signaling pathways activated by the increase in saturated fatty acids, as has been found in the hypothalamus and the amygdala.29,30 Another interesting finding was a significant decrease in AktSer473 phosphorylation, which is indicative that hippocampal insulin signaling can be compromised at different levels after consumption of an HFFD.
The hippocampus is a highly plastic structure and particularly sensitive to environmental and nutritional stressors. Thus, we were interested in studying neuronal alterations that could be associated with the hippocampal blunted insulin signaling we observed. In line with this, we analyzed several structural changes within the hippocampus. Surprisingly, we found a slight reduction of the hippocampal wet weight in HFFD-fed rats that linearly correlates with caloric intake. It is noteworthy that total brain weight was not different among groups (data not shown). In a previous study, a reduction of hippocampal volume in patients with type-2 diabetes mellitus was reported, while total brain weight remains unaffected, suggesting that the hippocampus presents a particular sensitivity to metabolic diseases. 32 The decrease that we observed in hippocampal weight was not accompanied by evident changes in the area or the general appearance of the hippocampal layers, as showed by the conventional Nissl staining. At this point, it is unclear whether all the hippocampal morphologic changes due to HFFD that we observed are the sole responsible for the reduction of weight in the hippocampus, or other mechanisms such as tissue dehydration, a modification in the composition of the membrane lipids and cholesterol levels, etc. could also be responsible.
A more detailed histologic examination revealed a significant decrease in the dendritic complexity of CA1 neurons, associated with a significant reduction in MAP-2 immunoreactivity in dendrites on the whole hippocampus of HFFD rats. We also found a reduction in dendritic spine density of neurons in CA1 and a significant decrease in the presynaptic protein synaptophysin. MAP-2 is a microtubule-associated protein that has been shown to be decreased in some models of neurodegeneration and AD.33,34 In particular, it was reported that rats fed with a high-fat diet (soybean oil and cholesterol supplemented, no fructose) for 8 weeks present a diminished staining of MAP-2 in the hippocampus, and that this reduction is correlated with memory errors. 16 Microtubule-associated protein 2 modifies and controls microtubule assembly and spacing within dendrites. Thus, a fine regulation of MAP-2 dynamics can alter hippocampal plasticity. The downregulation of MAP-2 immunoreactivity found in HFFD-fed rats associated with reduced hippocampal insulin signaling may involve an altered protein localization, increased phosphorylation, and/or degradation and diminished gene expression. The last possibilities can be excluded because we did not find changes in total MAP-2 content in the HFFD group (data not shown). It is remarkable that such dramatic changes in neuronal morphology are found with just 7 days of HFFD feeding. Further studies will be required to assess whether these morphologic changes are correlated with synaptic or neuronal dysfunction. Impairment of dendritic spine density and MAP-2 reduction were accompanied by an increased phosphorylation of tau in the S199/202 residue, a reported GSK3β phosphorylation site. It has been reported that long-term HFFD feeding causes GSK3β activation associated with tau phosphorylation at PHF-1 (S396/404) and T231 (ref. 26). Tau phosphorylation sites have been extensively studied in terms of the impairment for microtubule binding and the formation of insoluble neurofibrillary tangles that cause multiple anomalies, from axonal dysfunction to the loss of neuronal integrity and neuronal death.24–26,35
Finally, we examined whether the biochemical, morphologic, and structural changes found in the hippocampus of HFFD rats were accompanied by reactive astrocyte activation. One of the pathologic features of obesity and InsRes is inflammation, and it has been described that central inflammation has an impact on brain function possibly through the import of inflammatory cytokines and immune cells into the central nervous system.31,36–38 Consistent with these reports, we found a significant increase in reactive astrocyte number and size in the hippocampus of HFFD rats, as well as GFAP+ subgranular cells extending vertical processes onto the granular layer of the DG of what we believe, based on morphology and location, is radial glia. In addition, we found slight changes in some microglial cells consistent in increased body size suggesting the onset of a subtle microglial activation. These results are indicative of neuroinflammatory changes within the hippocampus due to acute HFFD feeding.
The structural changes found in the hippocampus may arise from peripheral InsRes as well as alterations in hippocampal insulin signaling. However, an alternative explanation would be that differences in the metabolic balance of carbohydrates, and fats might have a direct effect on hippocampal plasticity and inflammation independently of peripheral metabolic alterations. It is possible that the high fructose intake promotes the increase in circulating triglycerides and accelerates the observed metabolic disturbances in the present model. However, additional studies will be necessary to parse the relative contributions of diet-induced alterations in lipid and glucose metabolism in the hippocampus.
In conclusion, we propose a model in which obesity and InsRes caused by a short-term HFFD feeding produce downregulation of hippocampal insulin signaling (Figure 6). This work provides novel evidence of the early biochemical and structural alterations in functional relevance caused by short-term HFFD feeding in the hippocampus, which ultimately might determine cognitive outcomes and disease susceptibility after longer exposure to a nutritional imbalance.
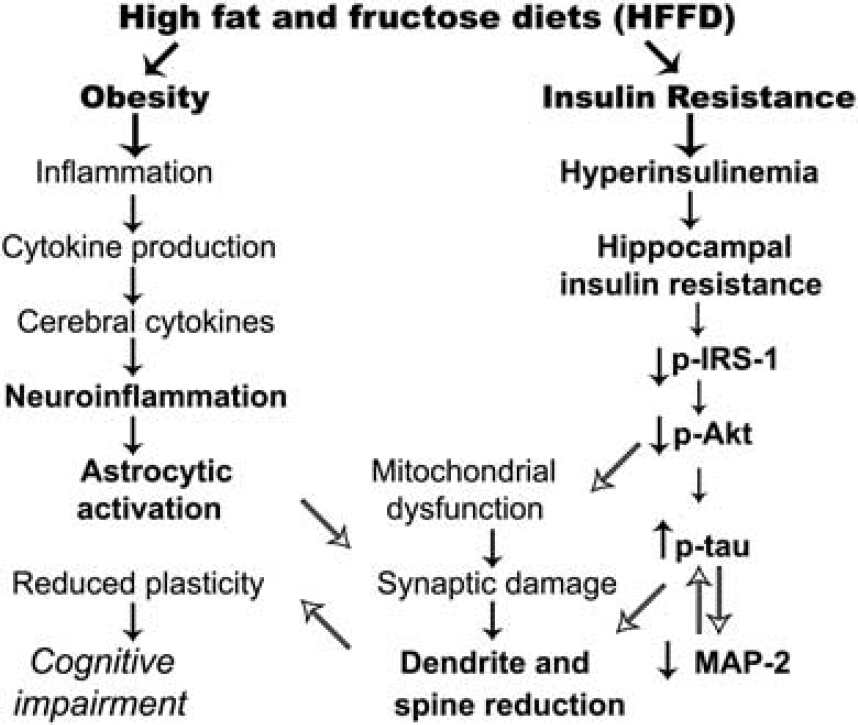
Proposed integrative model of systemic and hippocampal alterations associated with insulin resistance and obesity caused by short-term high-fat and fructose feeding. Parameters/mechanisms assessed in this work are written in bold font, whereas known mechanisms reported in the literature are shown in plain font. MAP-2, microtubule-associated protein 2; IRS-1, insulin receptor substrate-1.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors thank Dr Tzipe Govezensky (UNAM) for helpful suggestions with the statistical analysis, and Dr Roger Gutiérrez-Juarez (Albert Einstein College of Medicine) for reviewing this manuscript.