
Abstract
Understanding the cellular processes underpinning the changes in binding observed during positron emission tomography neurotransmitter release studies may aid translation of these methodologies to other neurotransmitter systems. We compared the sensitivities of opioid receptor radioligands, carfentanil, and diprenorphine, to amphetamine-induced endogenous opioid peptide (EOP) release and methadone administration in the rat. We also investigated whether agonist-induced internalization was involved in reductions in observed binding using subcellular fractionation and confocal microscopy. After radioligand administration, significant reductions in [11C]carfentanil, but not [3H]diprenorphine, uptake were observed after methadone and amphetamine pretreatment. Subcellular fractionation and in vitro radioligand binding studies showed that amphetamine pretreatment only decreased total [11C]carfentanil binding. In vitro saturation binding studies conducted in buffers representative of the internalization pathway suggested that μ-receptors are significantly less able to bind the radioligands in endosomal compared with extracellular compartments. Finally, a significant increase in μ-receptor-early endosome co-localization in the hypothalamus was observed after amphetamine and methadone treatment using double-labeling confocal microscopy, with no changes in δ- or κ-receptor co-localization. These data indicate carfentanil may be superior to diprenorphine when imaging EOP release in vivo, and that alterations in the ability to bind internalized receptors may be a predictor of ligand sensitivity to endogenous neurotransmitter release.
INTRODUCTION
In vivo competition imaging studies with positron emission tomography (PET) have increased our understanding of numerous central nervous system disorders. The opioid receptor system has been extensively investigated using both selective and non-selective PET radioligands. 1 For example, the μ-opioid receptor has been implicated in a variety of neurological disorders and processes such as pain 2 and substance misuse3,4 using [11C]carfentanil. The μ-, δ-, and κ-opioid receptors have also been associated with both acute 5 and chronic pain,6,7 and seizure activity8,9 using [11C]diprenorphine.
Until recently, the ability to effectively and reproducibly image fluctuations in endogenous neurotransmitters with PET has been mainly limited to dopamine. However, two articles suggest that [11C]carfentanil may also be sensitive to pharmacologically induced endogenous opioid peptide (EOP) release. Mitchell et al 10 showed in humans a significant reduction in [11C]carfentanil binding in the orbitofrontal cortex and nucleus accumbens after consumption of an ethanol-containing drink. 10 Furthermore, Colasanti et al 11 showed a significant reduction of [11C]carfentanil binding in putamen, thalamus, and frontal cortex after acute oral administration of 0.5 mg/kg amphetamine in humans. These data are consistent with preclinical studies showing that single intraperitoneal injection of either ethanol or amphetamine lead to increases in microdialysate β-endorphin concentrations taken from the rat nucleus accumbens12,13 and the central nucleus of the amygdala. 14 However, the findings of Colasanti et al 11 failed to be replicated by another group using intravenous amphetamine administration; 15 therefore, the sensitivity of [11C]carfentanil to EOP release after amphetamine challenge warrants further investigation. In addition to [11C]carfentanil's sensitivity to EOP release, various preclinical16–18 and clinical5,8 paradigms suggest that [11C]diprenorphine may also be susceptible to EOP release; however, the ability of amphetamine to reduce [11C]diprenorphine binding has not yet been tested. Previous studies suggest that because of a proposed high-efficacy and low-occupancy profile of opioid receptor agonists such as methadone, [11C]diprenorphine is not a suitable radioligand for determination of dose–occupancy relationships in vivo.19,20 Moreover, the ability of methadone to reduce [11C]carfentanil binding has also not been assessed.
The occupancy model may not sufficiently describe the changes in binding observed during in vivo competition imaging paradigms; 21 therefore, the cellular processes underpinning these changes warrant further investigation. Alterations in binding observed during pharmacological or physiological studies are manifested as changes in binding potential (BP). As BP is a function of both Bavail and KD, 22 either of these parameters may be driving the ΔBP observed in vivo. Receptor internalization processes have been proposed to contribute to reductions in binding observed during in vivo competition imaging studies with the dopaminergic system 21 by alterations in affinity. 23 The cellular localization of opioid receptors is known to change after exposure to either endogenous or exogenous agonists.24,25 However, how agonist-induced internalization processes effect the binding parameters of [11C]carfentanil and [11C]diprenorphine specifically have not yet been determined. In addition, the cellular composition of the [11C]carfentanil and [11C]diprenorphine binding signal has, to our knowledge, not yet been described.
We have therefore investigated the ability of amphetamine-induced EOP release to reduce [11C]carfentanil and [3H]diprenorphine binding in the rat brain. The ability of methadone to displace [11C]carfentanil binding has also been investigated. To rule out any direct occupancy by amphetamine at [11C]carfentanil-labeled μ- and [3H]diprenorphine-labeled μ-, δ-, and κ-receptor sites, the affinity of amphetamine was also determined using in vitro competition binding. The affinities of a range of EOPs have also been determined using both [11C]carfentanil and [3H]diprenorphine. Furthermore, the ability of both methadone and amphetamine to induce μ-, δ-, and κ-receptor internalization were assessed using double-labeling fluorescence microscopy. The effects of methadone and amphetamine treatment on receptor translocation was also assessed using subcellular fractionation and ex vivo radioligand binding. Finally, the effects of agonist-induced internalization on the μ-, δ-, and κ-receptor affinity and availability was determined for [11C]carfentanil and [3H]diprenorphine using in vitro saturation binding studies conducted in buffer conditions representative of those found throughout the endocytic pathway using the methodology recently published for the serotonin transporter radioligand, [3H]DASB. 26
MATERIALS AND METHODS
An overview of the experimental paradigms, methodologies implemented and the observed outcomes are noted in Table 1. The procedures used were all approved by the Animal Ethical Review Committee of Imperial College London and carried out in compliance with the UK Animals (Scientific Procedures) Act 1986, UK.
Overview of experimental approaches used and outcomes obtained
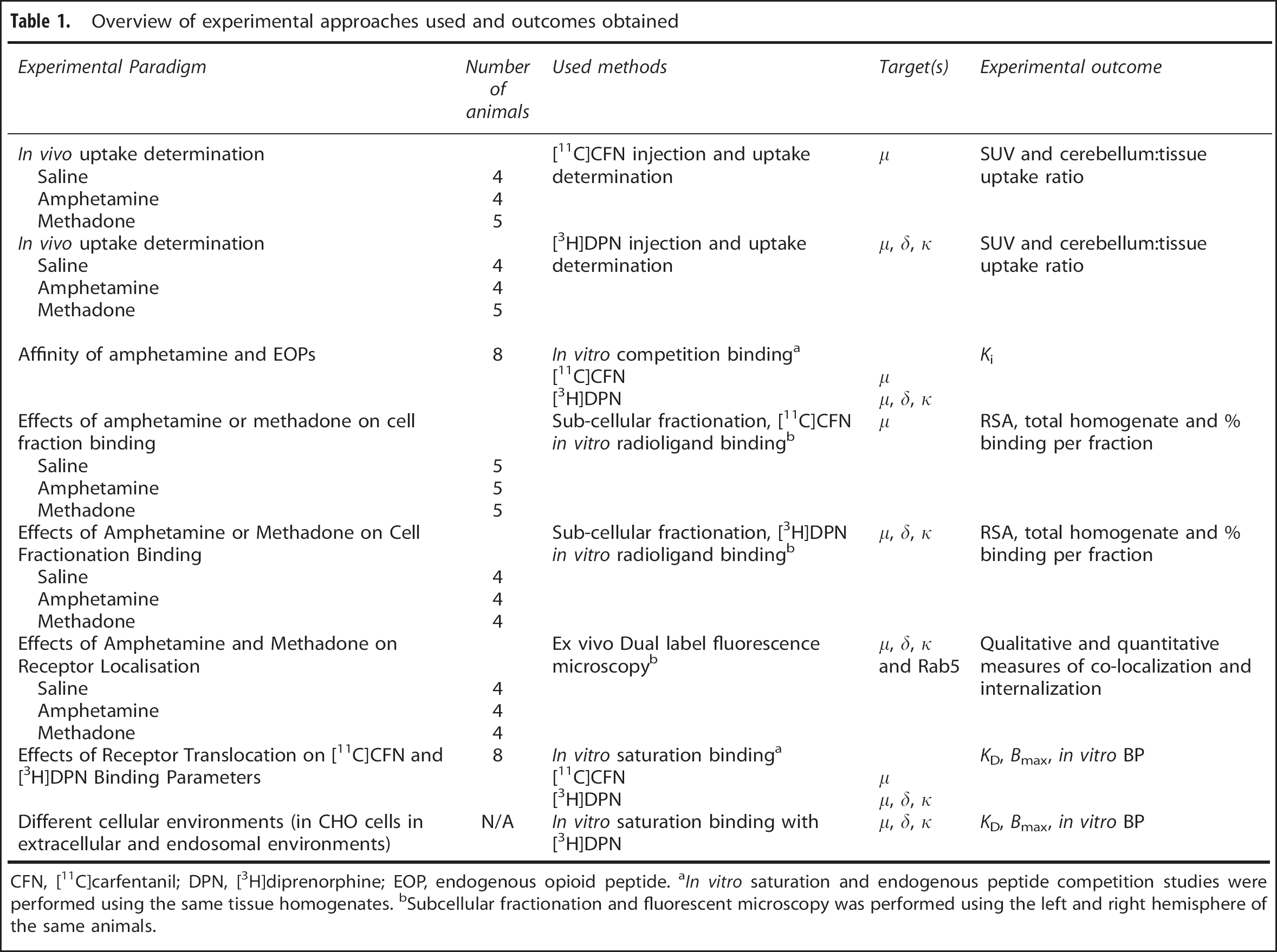
CFN, [11C]carfentanil; DPN, [3H]diprenorphine; EOP, endogenous opioid peptide. aIn vitro saturation and endogenous peptide competition studies were performed using the same tissue homogenates. bSubcellular fractionation and fluorescent microscopy was performed using the left and right hemisphere of the same animals.
[11C]Carfentanil was produced as outlined in Colasanti et al. 11 In total, 21 [11C]carfentanil productions were used (produced at Imanova, Hammersmith Hospital, London), with specific activity and radioactive concentration values ranging from 280 to 411 GBq/μmol and 161–700 MBq/ml, respectively. [3H]Diprenorphine was purchased from Perkin Elmer (Cambridge, UK)—NEN Radiochemicals (Massachusetts, USA) with a specific activity of 50 Ci/mmol and a radioactive concentration of 37 MBq/ml.
In Vivo [11C]Carfentanil and [3H]Diprenorphine Dosing and Uptake Determination
Adult, male Sprague–Dawley rats (~250 g; Charles River) were allowed to acclimatize for a minimum of 48 hours before use for the study with food and water given ad libitum. Subjects were injected with either saline,
Homogenate Preparation for Saturation and Competition Binding
Untreated rat whole brains (minus cerebellum; male; Sprague–Dawley; ~250 g) were weighed and homogenized in 10× weight/volume (w/v) sucrose buffer (0.32 mM sucrose, 5 mM Tris HCl, 1 mM MgCl2 pH 7.4, 4°C) and centrifuged at 32,000 g (20 minutes, 4°C). The supernatant was removed and the pellet washed twice by centrifugation (32,000 g, 20 minutes, 4°C) in Tris buffer (50 mM Tris-Base, 1 mM MgCl2, pH 7.4, 4°C). For all membrane preparations, the final pellets were re-suspended in Tris buffer to ~10 mg/ml.
Competition Radioligand Binding Studies—Affinity of Amphetamine and Endogenous Opioid Peptides
Whole brain (minus cerebellum) P2 membrane homogenates were thawed and diluted to 400 μg membrane in extracellular buffer. Membranes were incubated (37°C) with a single concentration of [11C]carfentanil (30 minutes; 0.3 nM) or [3H]diprenorphine (90 minutes; 0.15 nM) and a concentration range of the following opioid peptides: β-endorphin (10 pM–10 μM), met-, leu-enkephalin, and endomorphin-1/-2 (3 pM–100 μM), Dynorphin-A and B (30 pM–300 μM, [3H]diprenorphine only), and
Subcellular Fractionation Homogenate Preparation
The various fractions generated in these studies and their contents are: (1) P2—plasma membrane and mitochondrial pellet, (2) P3—microsomal pellet and (3) S3—cytosolic fraction. Rat whole brains (minus cerebellum) were taken from animals (male; Sprague–Dawley; ~250 g) previously treated with either saline (n = 9),
The tissues were thawed over ice and suspended in 10× w/v sucrose buffer (0.32 mM sucrose, 5 mM Tris HCl, 1 mM MgCl2 pH 7.4, 4°C), homogenized using a teflon glass homogenizer (20 strokes, on ice) and centrifuged (1,000 g, 10 minutes, 4°C) to generate a crude nuclear fraction. The supernatant was removed and the pellet washed twice by centrifugation (1,000 g, 10 minutes, 4°C) in Tris buffer (50 mM Tris-Base, 1 mM MgCl2, pH 7.4, 4°C). The nuclear pellet was discarded and the supernatant centrifuged to generate P2 (17,000 g, 20 minutes, 4°C). The P2 pellet was further washed twice by centrifugation (17,000 g, 20 minutes, 4°C) in Tris buffer. The supernatant from P2 was centrifuged (100,000 g, 90 minutes, 4°C) to generate P3. The supernatant from P3, S3, was collected as the cytosolic fraction. The P2 fraction was re-suspended in extracellular buffer (50 mM Tris HCl, 140 mM NaCl, 5 mM KCl, 1.5 mM MgCl2, 1.5 mM CaCl2, pH 7.4, 37°C). The P3 fraction, was re-suspended in endosomal buffer (20 mM MES, 10 mM NaCl, 140 mM KCl, 0.5 mM MgCl2, 0.003 mM CaCl2, pH 6.0, 37°C). The concentration of protein in each cell fraction was determined using a Pierce bicinchonic acid colourimetric assay (product number: 23225) and the remainder aliquoted and stored at −80°C for use in subsequent radioligand binding studies.
Effects of Amphetamine or Methadone Treatment on Cell Fraction Binding—Fractionation Radioligand Binding Studies
Radioligand binding studies were performed as described above on four independent fractionation procedures from each experimental condition. All fractionation samples from P2, P3, and S3 from saline-, amphetamine-, and methadone-treated animals were diluted to 200 μg/well. Studies were performed using fixed concentrations of [11C]carfentanil (1 nM) and [3H]diprenorphine (5 nM). The specific binding component was determined using an excess of unlabeled naloxone (10 μM). Assays were terminated by filtration and bound radioactivity was determined as described above.
Double Labeling Immunofluorescence—Effects of Amphetamine and Methadone on Opioid Receptor Localization
Rat brain sections (male; Sprague–Dawley; ~250 g) from animals previously treated with either saline,
Saturation Radioligand Binding Studies in Different Cellular Conditions—Effects of Receptor Translocation on [11C]Carfentanil and [3H]Diprenorphine Binding Parameters
Whole brain (minus cerebellum) P2 membrane homogenates were thawed and diluted to 400 μg membrane protein/well in three physiological buffers representative of the extracellular and microsomal conditions (described in Quelch et al 26 ), and incubated (30 minutes for [11C] or 90 minutes for [3H], 37°C) with a range of [11C]carfentanil (0.003–10 nM) and [3H]diprenorphine (0.001–3 nM) concentrations. The specific binding component was determined using an excess of unlabeled naloxone (10 μM). Assays were terminated via filtration through Whatman GF/B filters followed by four 1-ml washes with ice-cold Wash buffer (50 mM Tris HCl, pH 7.4, 4°C). Assays were terminated by filtration and bound radioactivity was determined as described above.
Data Analysis
One- and two-way analysis of variance (ANOVA) with post-hoc Bonferroni's tests were performed using SigmaStat 3.0 (CleCom Birmingham, UK). Student's t-tests and Pearson's correlations were performed using GraphPad Prism 5.0 (La Jolla, CA, USA). Data from statistical analysis were considered statistically significant when P < 0.05. All data are expressed at mean ± s.e.m. For in vivo dosing, investigators were blind to saline, amphetamine, and methadone treatments during analysis of data. For radioligand dosing studies, data were normalized for injected dose, giving standardized uptake values (SUV) by: (Bq/g wet weight tissue) / (injected Bq/g body weight). SUVs were then converted to tissue:cerebellum brain uptake ratios by: (SUV tissue)/(SUV cerebellum). Bmax values in nM (pmol/g tissue) were generated according to: Bmax (fmol/mg protein) × mass tissue (g)/protein content (g/ml), where it is assumed that 1 ml of homogenized wet weight tissue is equivalent to 1 g in weight. In vitro BPs were generated according to: Bmax (nM)/KD (nM). Relative specific activity values were generated according to Laduron: 27 % specific binding per fraction/% protein per fraction. Immunofluorescent cell images were captured using a Leica TCS SP5 confocal microscope (Milton Keynes, UK) and quantification was conducted (mean pixel intensity and co-localization) using the Image J software (NIH) and the JACoP plug-in. 28 Co-localization was determined using Manders M1 overlap coefficient, which is expressed as a ratio ranging from 0 to 1, where 0 is no co-localization and 1 is 100% co-localization. 28
RESULTS
In vivo [11C]Carfentanil and [3H]Diprenorphine Dosing and Uptake Determination
After intravenous administration of saline, the highest uptake of [11C]carfentanil was observed in the hypothalamus (SUV = 1.52 ± 0.67; mean ± s.e.m.), superior colliculi (SUV = 1.40 ± 0.54), and periaqueductal grey (SUV = 1.29 ± 0.31). Lowest uptake was found in the cerebellum (SUV = 0.12 ± 0.01). When comparing tissue:cerebellum uptake ratios (Table 2), there was a significant effect of both treatment (two-way ANOVA: F(2,114) = 11.904, P < 0.001) and brain region (two-way ANOVA: F(12,114) = 5.607, P < 0.001) on [11C]carfentanil binding. A significant reduction in uptake was observed in the hypothalamus, superior colliculi (P < 0.01), and amygdala (P < 0.05) of amphetamine-treated rats compared with saline. [11C]Carfentanil uptake was also reduced in the superior colliculi of methadone-treated animals (P < 0.05) compared with saline. A correlation was observed between the uptake of [11C]carfentanil in brain regions from the saline-treated group and the percentage change after either amphetamine (Figure 1A) or methadone treatment (Figure 1B). Specifically, in regions with the greatest expression of μ-receptors, and therefore [11C]carfentanil tissue:cerebellum uptake ratio after saline treatment, the greatest reductions in uptake were observed after either amphetamine (r 2 = 0.18, P = 0.0011; Figure 1A) or methadone (r 2 = 0.23, P < 0.0001; Figure 1B) treatment.
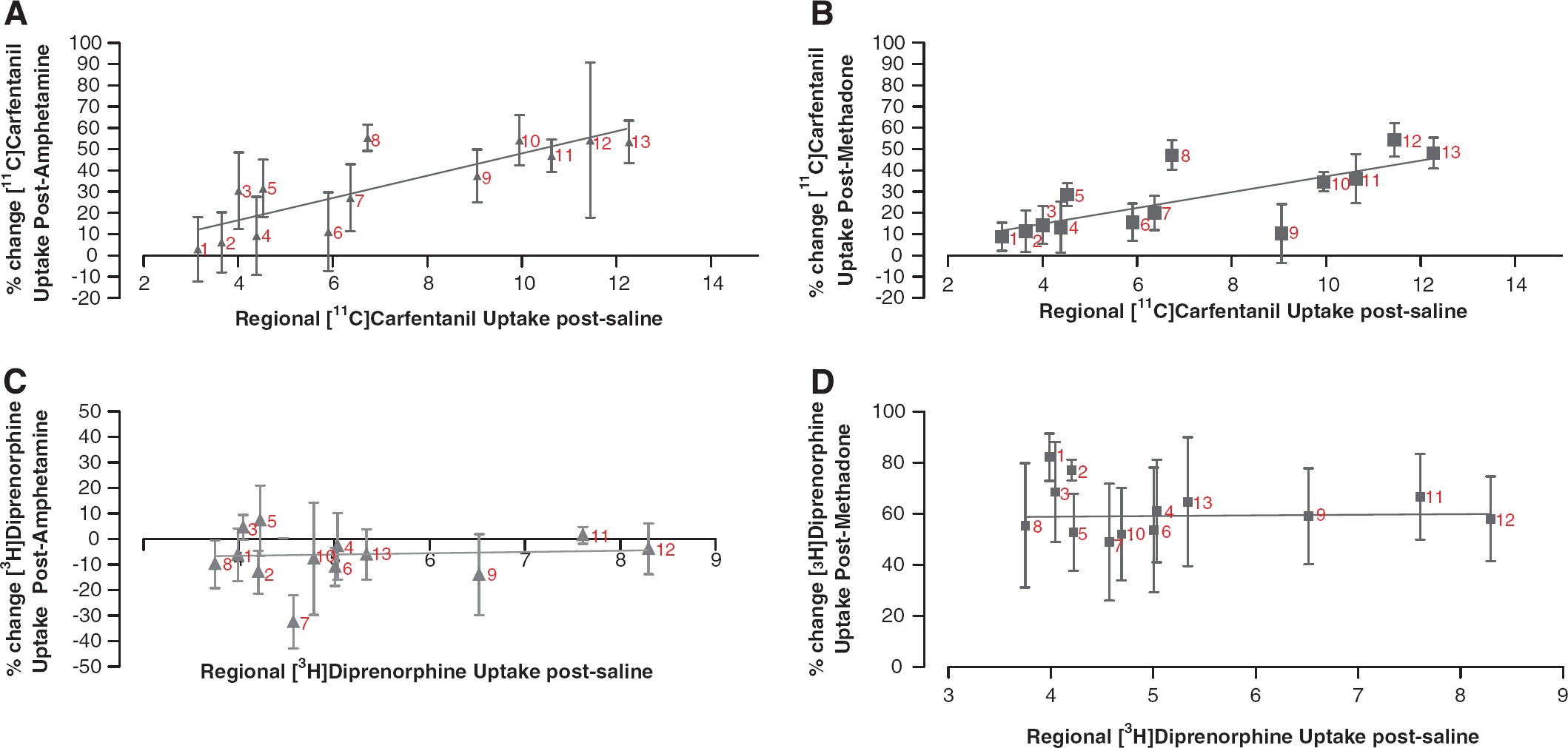
Correlation between regional uptake of [11C]carfentanil or [3H]diprenorphine after either amphetamine or methadone demonstrates reductions in [11C]carfentanil, but not [3H]diprenorphine binding. (
Tissue:cerebellum brain uptake ratios for [11C]carfentanil and [3H]diprenorphine
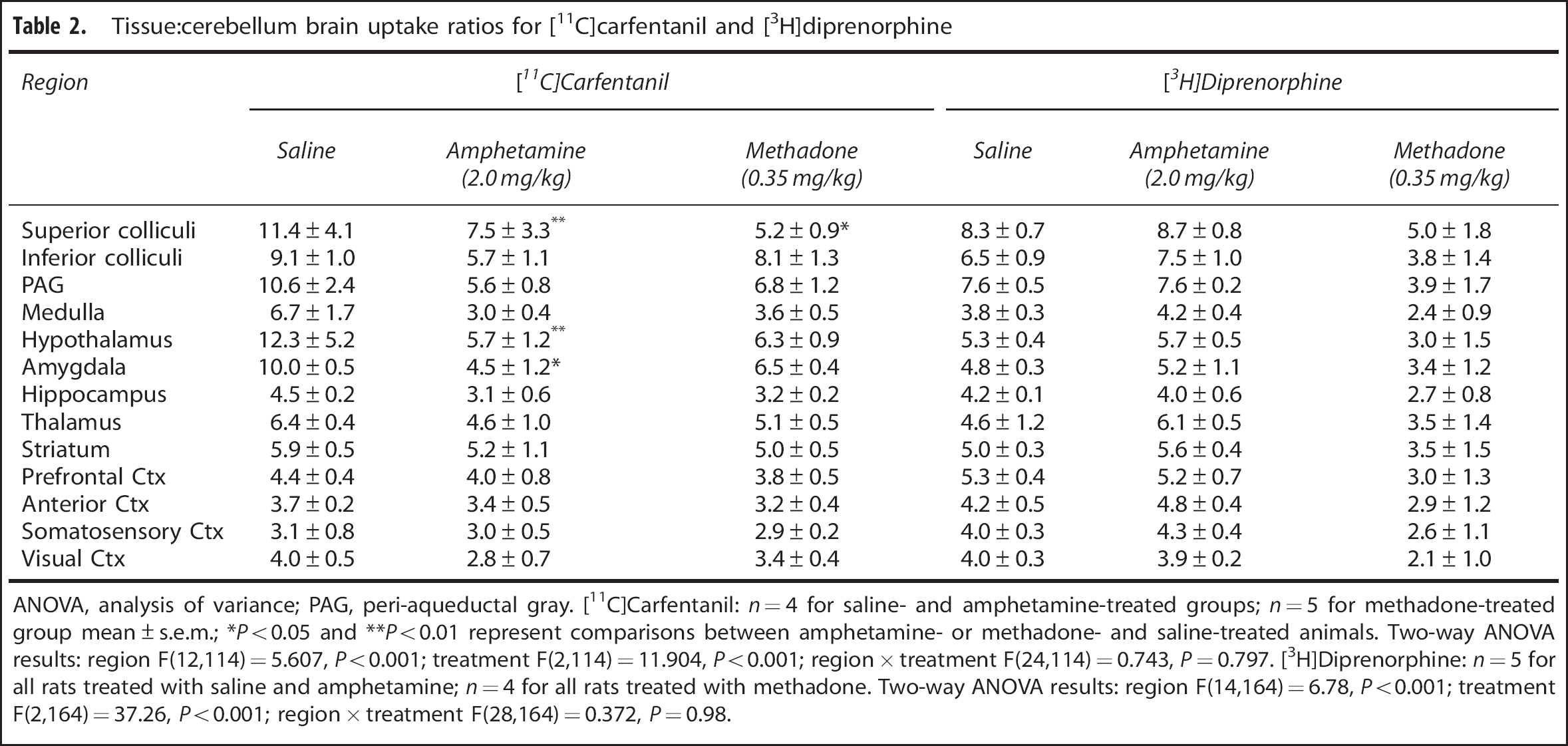
ANOVA, analysis of variance; PAG, peri-aqueductal gray. [11C]Carfentanil: n = 4 for saline- and amphetamine-treated groups; n = 5 for methadone-treated group mean ± s.e.m.; *P < 0.05 and **P < 0.01 represent comparisons between amphetamine- or methadone- and saline-treated animals. Two-way ANOVA results: region F(12,114) = 5.607, P < 0.001; treatment F(2,114) = 11.904, P < 0.001; region × treatment F(24,114) = 0.743, P = 0.797. [3H]Diprenorphine: n = 5 for all rats treated with saline and amphetamine; n = 4 for all rats treated with methadone. Two-way ANOVA results: region F(14,164) = 6.78, P < 0.001; treatment F(2,164) = 37.26, P < 0.001; region × treatment F(28,164) = 0.372, P = 0.98.
For [3H]diprenorphine, the highest uptake was observed in the superior colliculi (SUV = 130 ± 25.31), periaqueductal grey (SUV = 111.29 ± 20.86), and hypothalamus (SUV = 81.88 ± 13.50) after intravenous administration of saline. The lowest uptake was observed in the cerebellum (SUV = 15.29 ± 2.53). When comparing tissue:cerebellum ratios (Table 2), a significant effect of both brain region (two-way ANOVA: F(14,164) = 6.78, P < 0.001) and treatment (two-way ANOVA: F(2,164) = 37.26, P < 0.01) was observed. No correlation between uptake after saline treatment and reduction in uptake after either amphetamine or methadone treatment was observed (Table 2; Figures 1C and 1D).
Competition Radioligand Binding Studies—Affinity of Amphetamine and Endogenous Opioid Peptides
All peptides displaced [11C]carfentanil binding, with data best fitting to a single-site model of binding (Supplementary Table 1). The following rank order of affinities (Ki) were observed for the endogenous peptides tested at μ-sites: β-endorphin > endomorphin-1 > endomorphin-2 > leu-enkephalin > met-enkephalin.
For [3H]diprenorphine, both Leu- and met-enkephalin, and endomorphin-1 and −2 displayed both a high- and low-affinity binding site, reflecting the non-selective nature of [3H]diprenorphine. The following rank order of affinities were observed for the endogenous peptides tested: dynorphin-B > dynorphin-A > endomorphin-1 > endomorphin-2 > met-enkephalin > leu-enkephalin > β-endorphin (Supplementary Table 1).
No significant reductions in specific binding of either radioligand tested was observed using any of the concentrations of amphetamine investigated; therefore, a Ki of > 100 μM and >300 μM was assumed based on the fit of the data for [11C]carfentanil and [3H]diprenorphine, respectively (Supplementary Table 1).
Effects of Amphetamine or Methadone Treatment on Cell Fraction Binding—Fractionation Radioligand Binding Studies
Total homogenate [11C]carfentanil binding was significantly reduced in amphetamine-treated animals compared with saline-treated animals (P < 0.05; Figure 2A). No difference was observed between treatment groups for [3H]diprenorphine total homogenate binding (Figure 2E). The cellular composition of [11C]carfentanil and [3H]diprenorphine binding have not yet been described. Therefore, relative specific activity values were generated to take into consideration both fraction protein content and percentage of total homogenate [11C]carfentanil or [3H]diprenorphine binding (according to Laduron 27 ). These data show P2 constitutes the largest percentage of total tissue binding with [11C]carfentanil (saline = 93.5 ± 0.5%, amphetamine = 90.1 ± 2.4% and methadone = 91.9 ± 0.5%; Figures 2B, 2C and 2D). Microsomal (P3) and cytosolic (S3) μ-receptor binding also contributed to the total [11C]carfentanil signal in whole brain homogenates, but to a much lesser degree (microsomal: saline = 5.9 ± 0.7%, amphetamine = 7.4 ± 1.4% and methadone = 6.4 ± 1.2%; cytosolic: saline = 1.8 ± 0.5%, amphetamine = 2.5 ± 0.9% and methadone = 2.5 ± 1.5; Figures 2B, 2C, and 2D). A trend for a treatment effect in [11C]carfentanil microsomal (P3) binding was observed (P = 0.08), where both amphetamine (4.1 ± 0.4%) and methadone (4.3 ± 1.1%) values were increased compared with saline (3.4 ± 0.4%; Supplementary Figure 1B).
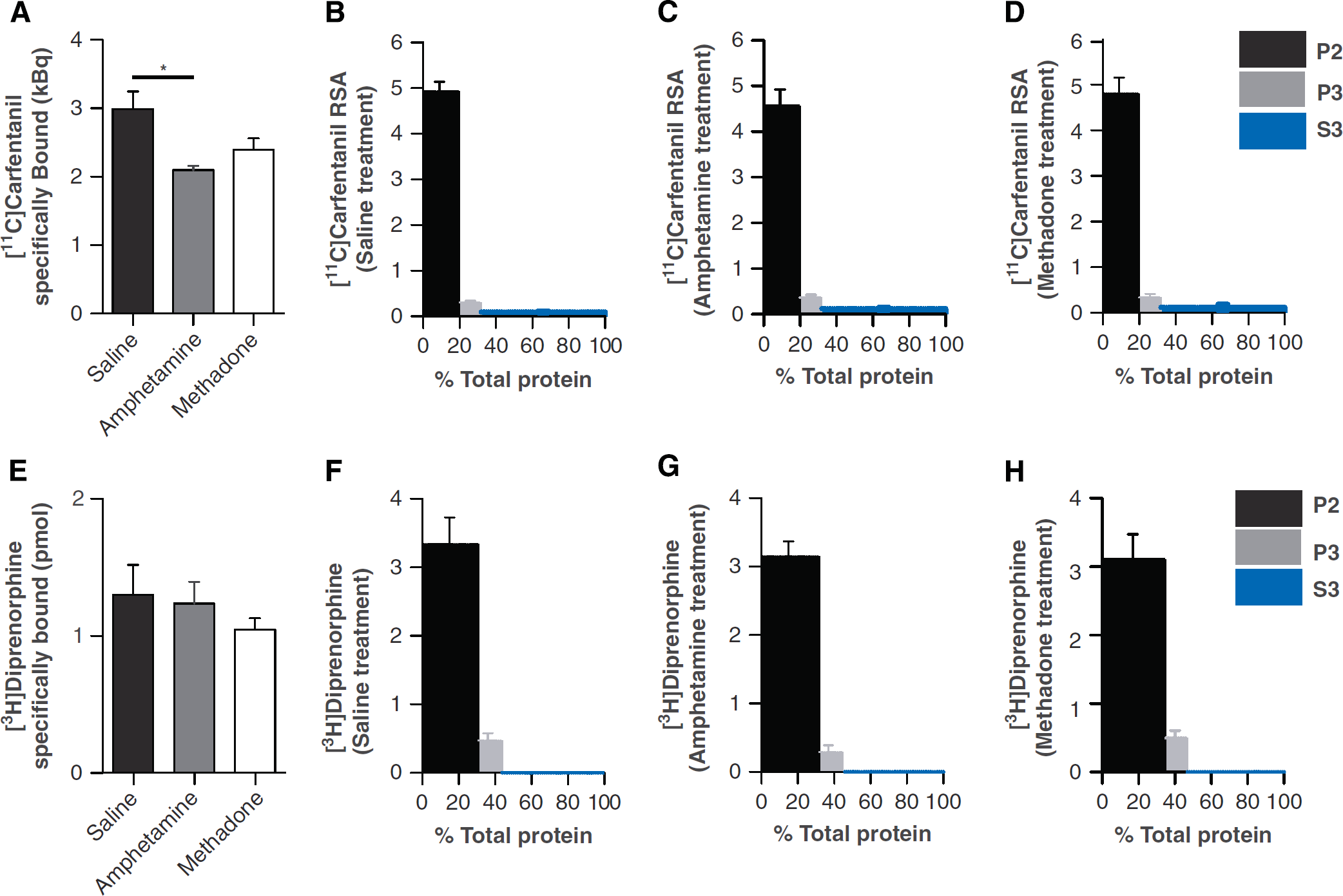
[11C]carfentanil but not [3H]diprenorphine binding is reduced after amphetamine pretreatment in total homogenates. [3H]diprenorphine and [11C]carfentanil relative specific activity (RSA) values per fraction as a function of fraction protein content in saline-, amphetamine-, and methadone-treated rats. RSA values for each fraction and treatment group ([11C]carfentanil
No specific binding was observed in the cytosolic fraction for [3H]diprenorphine (Figure 2F–H). For all treatment groups, P2 constituted the majority of cell binding (saline = 87.01 ± 3.92%, amphetamine = 91.38 ± 3.25%, and methadone = 84.8 ± 4.83%; Figures 2F–H) with the microsomal fraction (P3) also contributing to a much lesser degree to total homogenate binding (saline = 12.99 ± 3.92%, amphetamine = 8.62 ± 3.24%, and methadone = 15.20 ± 4.82%; Figures 2F–H).
Double Labeling Immunofluorescence—Effects of Amphetamine and Methadone on Opioid Receptor Localization
Opioid receptor (green) and co-localization (yellow) with Rab5 (red) was investigated in the hypothalamus and striatum after saline, amphetamine or methadone treatment (Figure 3 and Supplementary Figure 2). In methadone- and amphetamine-treated animals, μ-receptor-Rab5 co-localization (yellow) was markedly increased compared with saline in the hypothalamus (Figures 3A, 3B and 3C). Quantitative analysis of μ-receptor-Rab5 co-localization in the hypothalamus (using Manders' M1 overlap coefficients) showed that both methadone (0.65 ± 0.03) and amphetamine (0.64 ± 0.04) pre-treatment significantly increased μ-receptor association with early, recycling endosomes (both P < 0.05) compared with saline-treated animals (0.50 ± 0.03; Figure 3F) in the hypothalamus. Compared with saline-treated animals, amphetamine and methadone treatment did not significantly alter mean pixel intensity associated with μ-receptor immunofluorescence (Figure 3D); however, Rab5 mean pixel intensity was significantly higher in methadone- (26.66 ± 2.26, P < 0.001) and amphetamine- (17.29 ± 1.44, P < 0.05) treated animals compared with saline-treated animals (10.99 ± 1.08; Figure 3E).
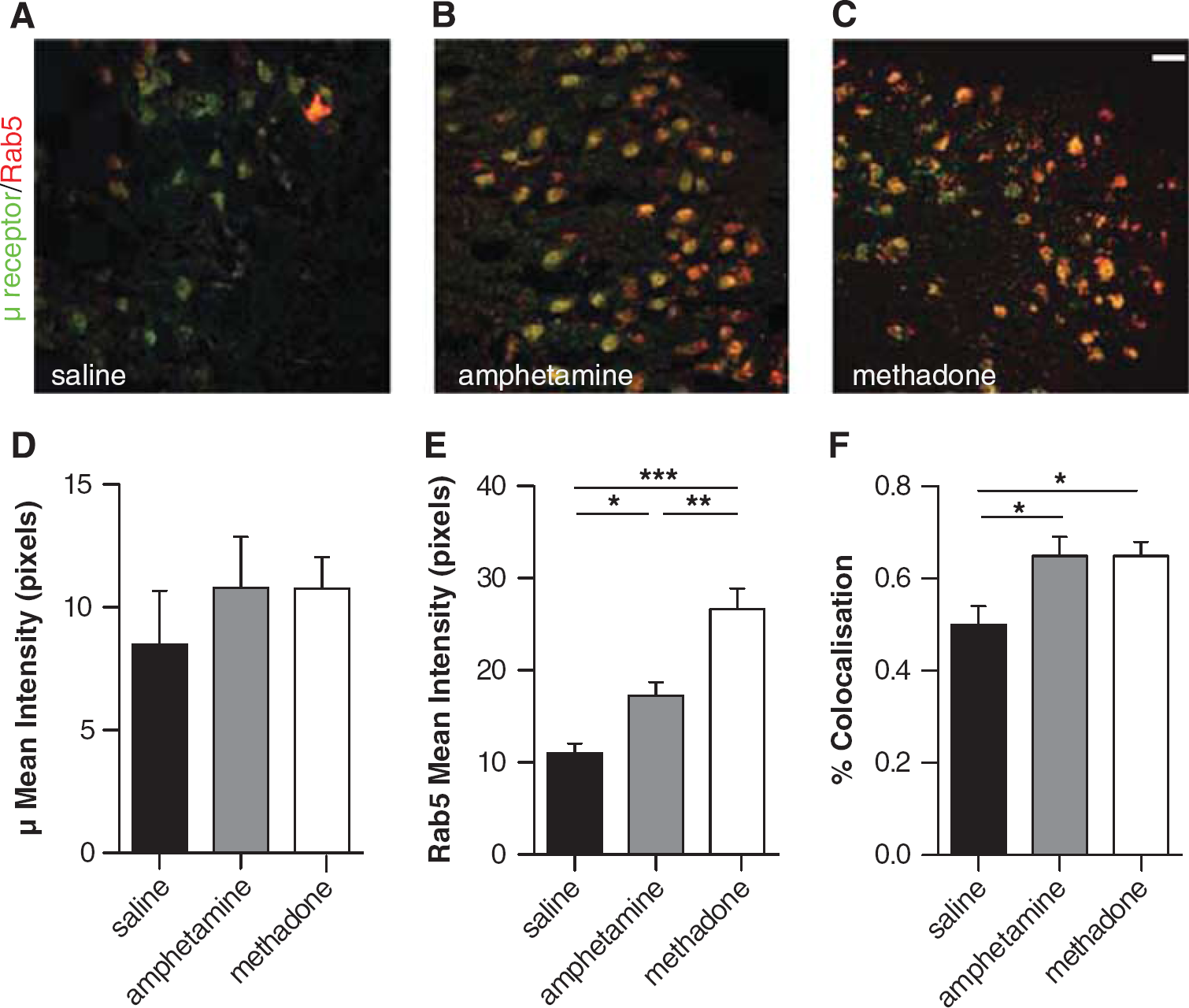
Amphetamine and methadone pretreatment leads to increased μ receptor-Rab5 co-localization in the hypothalamus. μ-Receptor-Rab5 immunofluorescence in the rat hypothalamus after either saline (
No differences in δ- and κ-receptor-Rab5 co-localization were observed after either amphetamine or methadone treatment compared with after saline treatment in the striatum (Supplementary Figure 2), however, similar to that observed in the hypothalamus, a moderate non-significant increase in μ-receptor-Rab5 co-localization was observed (Supplementary Figure 2).
Saturation Radioligand Binding Studies in Different Cellular Conditions—Effects of Receptor Translocation on [11C]Carfentanil and [3H]Diprenorphine Binding Parameters
Exposure of whole brain homogenates to acidified, low sodium buffers, representative of the endosomal environment, resulted in a significant reduction in the population of receptors available to bind when compared with exposure of homogenates to buffers representative of cell surface receptor binding conditions (P < 0.05; Table 3). An approximate doubling of affinity was observed for [11C]carfentanil in the endosomal condition; however, this did not reach significance. When affinity and availability values are converted to in vitro BPs (in a manner comparable to derivation of BP in vivo, i.e., Bmax (nM)/KD (nM)), the endosomal environment resulted in an ~48% reduction in the ability to bind compared with the extracellular environment (Table 3). A significant reduction in receptor availability was also observed for [3H]diprenorphine in the endosomal condition compared with the extracellular condition and no significant change in affinity was observed between the conditions tested. When in vitro BP values are generated, a ~50% reduction in [3H]diprenorphine's ability to bind the total available receptor population was observed (Table 3).
Affinity, receptor availability and in vitro BP values for [11C]carfentanil and [3H]diprenorphine binding in tissues after exposure to different cellular environments

ANOVA, analysis of variance. *P < 0.05, **P < 0.01 represent comparison of endosomal to the extracellular (extra) condition. Bmax in nM calculated using Bmax (fmol/mg) × mass tissue (g)/protein content (g/ml). One-way ANOVA results: [3H]diprenorphine KD F(2,9) = 2.01, P = 0.19; Bmax F(2,9) = 13.61, P = 0.002; in vitro BP F(2,9) = 27.43, P < 0.001. [11C]carfentanil KD F(2,9) = 0.80, P < 0.48; Bmax F(2,12) = 4.27, P = 0.04, in vitro BP F(2,12) = 0.47, P = 0.64. aAssumes 1 g of tissue equivalent to 1 ml.
To further investigate the change in affinity observed with [11C]carfentanil in the endosomal condition, saturation studies were conducted in membranes prepared from μ-, δ- or κ-expressing CHO cells using [3H]diprenorphine in both extracellular and endosomal buffer (Supplementary Methods section). The endosomal condition had no significant effect on δ- or κ-receptor affinity for [3H]diprenorphine; however, a significant increase in affinity was observed for μ-receptors (P < 0.05) in the endosomal condition compared with the extracellular (Supplementary Figure 3A). A significant reduction in [3H]diprenorphine Bmax was observed for μ- (P < 0.05), δ- (P < 0.01), and κ- (P < 0.05) receptors in the endosomal condition compared with the extracellular (Supplementary Figure 3B).
DISCUSSION
We have showed that the binding of [11C]carfentanil in the rat brain is sensitive to both amphetamine-induced EOP release and exogenous agonist (methadone) administration. Furthermore, we suggest that [11C]diprenorphine may not be suitable for measuring EOP release using amphetamine as a pharmacological challenge. Both [11C]carfentanil and [11C]diprenorphine would be expected to be less able to bind total Bavail after translocation of their respective opioid receptors to the endosomal compartment. However, reductions in both affinity and Bavail of μ-receptors, once present in the endosomal compartment, may underpin the reductions in binding observed with the μ-selective ligand [11C]carfentanil, after agonist exposure and receptor translocation. Amphetamine pretreatment significantly reduced total homogenate [11C]carfentanil binding; in addition, amphetamine and methadone treatment caused a marked increase in μ-receptor-Rab5 co-localization, but no changes in either δ- or κ-receptor-Rab5 co-localization. Therefore, we suggest that the higher sensitivity of [11C]carfentanil to EOP release and exogenous agonist exposure may be driven by a higher proportion of total receptors bound translocating to the endosomal compartment compared with those bound by [3H]diprenorphine in vivo. Once present in the endosomal compartment, we suggest that μ receptors are less able to bind the respective radioligands by reductions in receptor affinity as well as receptor availability. These alterations in binding parameters are likely to translate to reductions in BP observed in vivo.
The studies presented in this article have been conducted to provide an increased understanding of the behavior of [11C]carfentanil and [11C]diprenorphine in vivo. The use of overexpressing cell lines and transgenic animal strains have been avoided to remove any confounds of poor cell-to-tissue data translation and adaptations to genetic manipulations in vivo. Pretreatment with both the partially selective μ-receptor agonist, methadone and the monoamine-releasing compound, amphetamine, led to a reduction in [11C]carfentanil binding. The magnitude of this reduction in binding post-challenge was correlated with the degree of uptake after saline treatment, i.e., regions with the highest uptake at baseline, and therefore those brain areas with the greatest μ-receptor density were those with the greatest reductions in binding after either amphetamine or methadone treatment. We hypothesize that in addition to direct competition, alterations in receptor binding parameters post internalization are driving the reductions in [11C]carfentanil uptake observed after administration of either amphetamine or methadone.
Direct occupation of either [11C]carfentanil- and [3H]diprenorphine-labeled sites by amphetamine was not expected. However, to eliminate any possible direct effects of amphetamine on [11C]carfentanil or [3H]diprenorphine binding, competition binding studies were conducted. The extracellular amphetamine concentration in the rat caudate putamen after peripheral dosing has previously been determined. 29 After administration (1 mg/kg intraperitoneal; 60–90 minutes), 500 nM of amphetamine was present in micro-dialysate samples, while 300 nM was found to be present in the plasma. Assuming a linear relationship between dose and brain amphetamine concentration, 1 μM amphetamine can be assumed in the brains of the animals used in the studies presented here. Given that the Ki determined is >100 and >300 μM at [11C]carfentanil- and [3H]diprenorphine-labeled sites, respectively, a maximum occupancy of amphetamine of 0.49% and 0.17% can be estimated (if we assume a minimum Ki of 101 μM for [11C]carfentanil and 301 μM for [3H]diprenorphine). As such, it is not expected that any variation in binding after amphetamine pretreatment is because of direct competition of amphetamine at radiolabeled opioid receptor sites.
Regions where the greatest uptake of [11C]carfentanil and [3H]diprenorphine were observed are comparable with the distribution of opioid receptors previously described, i.e., high in the amygdala and hypothalamus, low in the cerebellum.30,31 Importantly, regions with the largest reductions in binding post amphetamine treatment (hypothalamus and amygdala, for example) are those associated with high levels of pro-opiomelanocortin expression32,33 and therefore potential β-endorphin release. The ability of amphetamine to increase central enkephalin and β-endorphin levels has previously been characterized.13,34 Furthermore, other compounds with affinity at monoamine transporters, such as cocaine, have been shown to increase β-endorphin concentrations in plasma of drug-naive participants. However, the effect of monoamine manipulation on other μ-receptor opioid peptides such as endomorphin-1 and −2 has not been determined.
Human and animal studies19,20 suggest that [11C]diprenorphine may not be suitable for determining opioid agonist occupancy profiles in vivo. The data presented here are in agreement with this, as we found no significant reduction in [3H]diprenorphine binding after methadone treatment. Methadone has previously been shown to reduce [18F]cyclofoxy binding. 35 In addition, buprenorphine has also been shown to reduce [11C]carfentanil in a dose-dependent manner. 36 However, the ability of methadone to reduce [11C]carfentanil has not, to our knowledge, been examined. We show that 0.35 mg/kg methadone leads to a significant reduction in [11C]carfentanil binding in various highly expressing μ-receptor regions at a dose tolerable in conscious animals and with no observable alterations in respiration rates. 37 Therefore, we propose that [11C]carfentanil may be superior for determining subtle changes in μ-receptor occupancy compared with [11C]diprenorphine.
To further characterize the cellular basis of the changes in binding observed with methadone and amphetamine, subcellular fractionation and ex vivo radioligand binding was performed. Animal numbers were reduced by using whole brain (minus cerebellum) homogenates. This technique allows for (1) the cellular composition of the [11C]carfentanil and [3H]diprenorphine binding signal to be determined, i.e., what proportion of total region binding is emanating from membrane bound, microsomal and cytosolic opioid receptors at baseline and (2) how pharmacological intervention may alter receptor trafficking and therefore overall radioligand binding in vivo. However, because of the wide expression of opioid receptors throughout various brain structures, whole brain fractionation procedures may reduce the ability of this assay to detect subtle changes in receptor translocation in discrete brain regions. Fractionation binding from all three treatment conditions suggests that the majority (92–94%) of cell [11C]carfentanil binding would be due to μ-receptors located at the plasma membrane. Amphetamine pretreatment significantly reduced total homogenate binding. However, μ-receptor grayscale pixel intensity from confocal microscopy studies was not altered by any of the treatments tested. Together, these data suggest that the reduction in binding is not likely due to increased degradation but rather an alteration in the ability of a fraction of μ-receptors in the total homogenate to bind [11C]carfentanil. A trend for an increase in microsomal binding was also observed after either amphetamine or methadone pretreatment. This may either represent an increase in synthesis of μ-receptors, bound in vesicles ready to be transported to the plasma membrane or, more likely, a translocated pool of receptors from the plasma membrane after agonist exposure. These findings are further supported by ex vivo microscopy studies showing a marked increase in Rab5-μ-receptor co-localization in the hypothalamus. Confocal microscopy image analysis from these studies suggest that the increase in co-localization observed after amphetamine or methadone treatment is driven by an upregulation in the number of Rab5-positive recycling endosomes. No significant differences were observed with either [3H]diprenorphine total homogenate binding or cell fraction binding after amphetamine or methadone treatments compared with saline. In addition, no changes in δ- and κ-receptor cell localization were observed after either treatment. These receptors represent ~60% of total [3H]diprenorphine binding in rat brain. 30 Together, these data may suggest (1) that amphetamine pretreatment does not lead to release of EOPs selective for all three opioid receptors, only those with preferential μ-affinity, and (2) the effect of μ-receptor translocation after either amphetamine or methadone treatment on [3H]diprenorphine binding is masked by a larger population of receptors remaining in their native cellular position.
Both [11C]carfentanil and [3H]diprenorphine exhibited a reduced ability to bind the total receptor population in endosomal cellular conditions compared with the extracellular conditions. In vitro binding studies performed in μ-, δ- or κ-expressing CHO cells indicate that the ability of μ-receptors to bind radioligands under endosomal-like conditions is significantly reduced by alterations in both receptor affinity and availability. Together, these data suggest that a reduced ability to bind total Bavail in subcellular compartments is a characteristic that may be shared by all three opioid receptors. However, the ability of μ to bind its respective radioligands in the endosomal compartment may also be reduced by alterations in receptor affinity; a characteristic which could account for the reductions in BP observed in vivo after pretreatment with challenge agents such as amphetamine and methadone. The effects of changing ionic conditions on opioid receptor binding has not been extensively investigated. However, several groups have reported an increase in antagonist and a decrease in agonist binding after changing sodium chloride and pH levels in assay buffers.38–40 These data suggest that in the buffer system implemented here, a shift from 140 mM in the extracellular buffer to 10 mM in the endosomal buffer sodium chloride would lead to a decrease in receptor availability for both [3H]diprenorphine and [11C]carfentanil. This sodium chloride-mediated decrease in agonist receptor availability is consistent with data presented here with [11C]carfentanil.
Summary
The studies presented in this manuscript using radioligand binding and immunofluorescence show that acute amphetamine exposure leads to release of EOPs selective for μ-receptors. This release results in a reduction in [11C]carfentanil but not [3H]diprenorphine binding, thereby showing that a pharmacological challenge capable of imaging EOP release now warrants further implementation. In addition, we have shown that amphetamine pretreatment reduces total [11C]carfentanil binding, in the absence of significant alterations in μ-receptor immunoreactivity, and causes a significant increase in μ-receptor endosomal localization, where we expect μ-receptors to be significantly less able to bind their respective radioligands. Therefore, we suggest receptor internalization processes contribute significantly to the reductions in [11C]carfentanil binding observed in vivo after amphetamine pretreatment. In addition, we suggest that [11C]carfentanil may be superior to [11C]diprenorphine for detecting small changes in μ-receptor occupancy with opioid agonists, as a significant reduction in [11C]carfentanil binding was observed after methadone exposure and not [3H]diprenorphine. Alterations in μ-receptor cellular localization and the ability of μ to bind [11C]carfentanil after receptor internalization likely contribute to the reductions in binding observed in these studies.
These data illustrate that the internalization hypothesis, originally proposed to contribute to changes in D2/3 receptor radioligand binding, 21 may also be applied to other receptor systems. Identification of alterations in binding ability after translocation to subcellular compartments and a better understanding of target protein trafficking after endogenous agonist release or exogenous ligand dosing may be a crucial requirement when predicting sensitivity to endogenous release of PET radioligands in vivo. Therefore, the methodologies developed in this manuscript warrant further application to other PET radioligand targets to translate in vivo competition imaging paradigms to other neurotransmitter systems.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We acknowledge the Radiochemical Sciences group at Imanova Limited for provision of [11C]carfentanil and use of facilities and Dr Alessandro Colasanti for discussions and comments on the data contained in the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.