
Abstract
It has been proposed that prostaglandin E2 (PGE2) is released from astrocytic endfeet to dilate parenchymal arterioles through activation of prostanoid (EP4) receptors during neurovascular coupling. However, the direct effects of PGE2 on isolated parenchymal arterioles have not been tested. Here, we examined the effects of PGE2 on the diameter of isolated pressurized parenchymal arterioles from rat and mouse brain. Contrary to the prevailing assumption, we found that PGE2 (0.1, 1, and 5 μmol/L) constricted rather than dilated parenchymal arterioles. Vasoconstriction to PGE2 was prevented by inhibitors of EP1 receptors. These results strongly argue against a direct role of PGE2 on arterioles during neurovascular coupling.
INTRODUCTION
Local blood flow is regulated by neuronal activity so as to match changes in the metabolic demand of brain tissue with the supply of oxygen and glucose, a process that is critical to maintain cerebral homeostasis and to prevent the development of ischemic conditions. This linkage between neuronal activity and cerebral blood flow, termed as neurovascular coupling or functional hyperemia, 1 is rapid (seconds) and occurs at the level of the cerebral microcirculation within the brain parenchyma. Local increases in cerebral blood flow are mediated by dilation of parenchymal arterioles, 2 which, unlike surface cerebral arteries (pial arteries), are encased by astrocytic processes (‘endfeet’). 1
Recent evidence indicates that information about neuronal activity is communicated to (1) surface arterioles via astrocytic processes comprising the glia limitans and to (2) parenchymal arterioles through elevation of astrocytic Ca2+ and the subsequent engagement of Ca2+-dependent vasodilatory pathways in astrocytic endfeet.1,3 Two potential Ca2+-dependent astrocytic endfoot targets in particular have received considerable research attention: large-conductance, Ca2+-sensitive K+ (BK) channels, which deliver K+ into the restricted perivascular space and dilate vessels by activating strong inwardly rectifying K+ channels,4–6 and phospholipase A2,3,7 which produces arachidonic acid through the hydrolysis of membrane lipids.
Prostaglandin E2 (PGE2), a metabolite of arachidonic acid produced by the action of cyclooxygenase (COX), has been proposed as a major mediator of neurovascular coupling. 8 A role for PGE2 in neurovascular coupling has largely been inferred from studies on the effects of COX inhibitors on neurally evoked vasodilation in brain slices. 8 However, some studies have found no effect of COX inhibition on neurovascular coupling in brain slices, 6 and others have attributed the reduction of functional hyperemia by COX inhibition to effects on COX2 in neurons. 9 Moreover, whether COX is even expressed in astrocytes has been called into question. 10
In theory, PGE2 could act as a vasodilator or constrictor depending on the nature of the member of the G protein-coupled prostanoid (EP) receptor family. The Gs-coupled EP4 receptor signals through the cyclic adenosine monophosphatedependent protein kinase (PKA) pathway, which is associated with dilation. Whereas the EP1 subtype is thought to couple to Gq-type α subunits and mediates an increase in intracellular Ca2+, consistent with a role in constriction. EP3, a subtype with promiscuous G protein-coupling propensity, has also been linked to elevation of intracellular Ca2+ and thus might mediate a contractile response to PGE2.
A critical test for the potential involvement of PGE2 in neurovascular coupling is the demonstration that PGE2 directly dilates isolated parenchymal arterioles. This evidence is currently lacking. In contrast, external K+, also posited as a mediator of neurovascular coupling, has been shown to be a potent, rapid, reversible dilator of parenchymal arterioles.4–6 Here, we tested the effects of PGE2 on the diameter of isolated pressurized parenchymal arterioles from rat and mouse brain. Contrary to expectation, we found that PGE2 constricted rather than dilated parenchymal arterioles.
MATERIALS AND METHODS
Animals
All experimental protocols used in this study were in accord with institutional guidelines approved by the Institutional Animal Care and Use Committee of the University of Vermont. Male C57BL6 mice and male Sprague-Dawley rats were used. Animals (aged 3 to 4 months) were euthanized by intraperitoneal injection of sodium pentobarbital (mouse: 100 mg/kg; rat: 150 mg/kg) followed by rapid decapitation.
Pressurized Parenchymal Arterioles
After euthanasia, the brain was removed and placed into 4°C MOPS-buffered saline. Parenchymal arterioles, arising from the M1 region of the middle cerebral artery and perfusing the neocortex, were dissected. Precapillary arteriolar segments were then cannulated on glass micropipettes with one end occluded in an organ chamber (University of Vermont Instrumentation and Model Facility) and pressurized using an arteriograph system (Living Systems Instrumentation, Inc., Burlington, VT, USA). Parenchymal arterioles were pressurized to 40 mm Hg and superfused (4 mL/min) with prewarmed (35°C to 37°C), gassed (5% CO2, 20% O2, 75% N2) artificial cerebrospinal fluid (aCSF) for at least 1 hour. Only viable parenchymal arterioles, defined as those that developed pressure-induced myogenic tone greater than 20%, were used in subsequent experiments (Supplementary Figure 1A). The average percentage of tone was 36.6 ± 3.4% (
(
Solutions
The composition of MOPS-buffered saline (in mmol/L) was 135 NaCl, 5 KCl, 1 KH2PO4, 1 MgSO4, 2.5 CaCl2, 5 D-glucose, 3 MOPS, 0.02 EDTA, 2 pyruvate, bovine serum albumin (10 mg/mL), pH 7.3 at 4°C. The composition of aCSF (in mmol/L) was 125 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 4 D-glucose, 2 CaCl2; pH was 7.3 when aerated with 5% CO2.
Drugs
Stock solutions of PGE2 (10 mmol/L in dimethyl sulfoxide) were prepared from powder each day of the experiment or stored at −20°C for < 1 month, and were protected from exposure to light in accordance with recommendations of the suppliers. During experiments, PGE2 was superfused immediately after dissolution in aCSF or dilution from dimethyl sulfoxide stock solutions to prevent PGA2 formation from PGE2 dehydration.
Prostaglandin E2 was purchased from different suppliers (Enzo Life Sciences, Farmingdale, NY, USA; Sigma-Aldrich, St Louis, MO, USA; and Tocris Bioscience, Ellisville, MO, USA). No significant differences were observed among the responses to PGE2 from the different vendors in parenchymal arteriolar diameter recording experiments. Prostacyclin (PGI2) was purchased from Enzo Life Sciences. ZM241385 and SC51322 were purchased from Tocris Bioscience. AH6809 and adenosine were purchased from Sigma-Aldrich.
Data Analysis and Statistics
Changes in arteriolar diameter were calculated as percent change from baseline (change in diameter/initial diameter). Data are expressed as means ± SEMs. Differences between two groups were analyzed using Student's
RESULTS
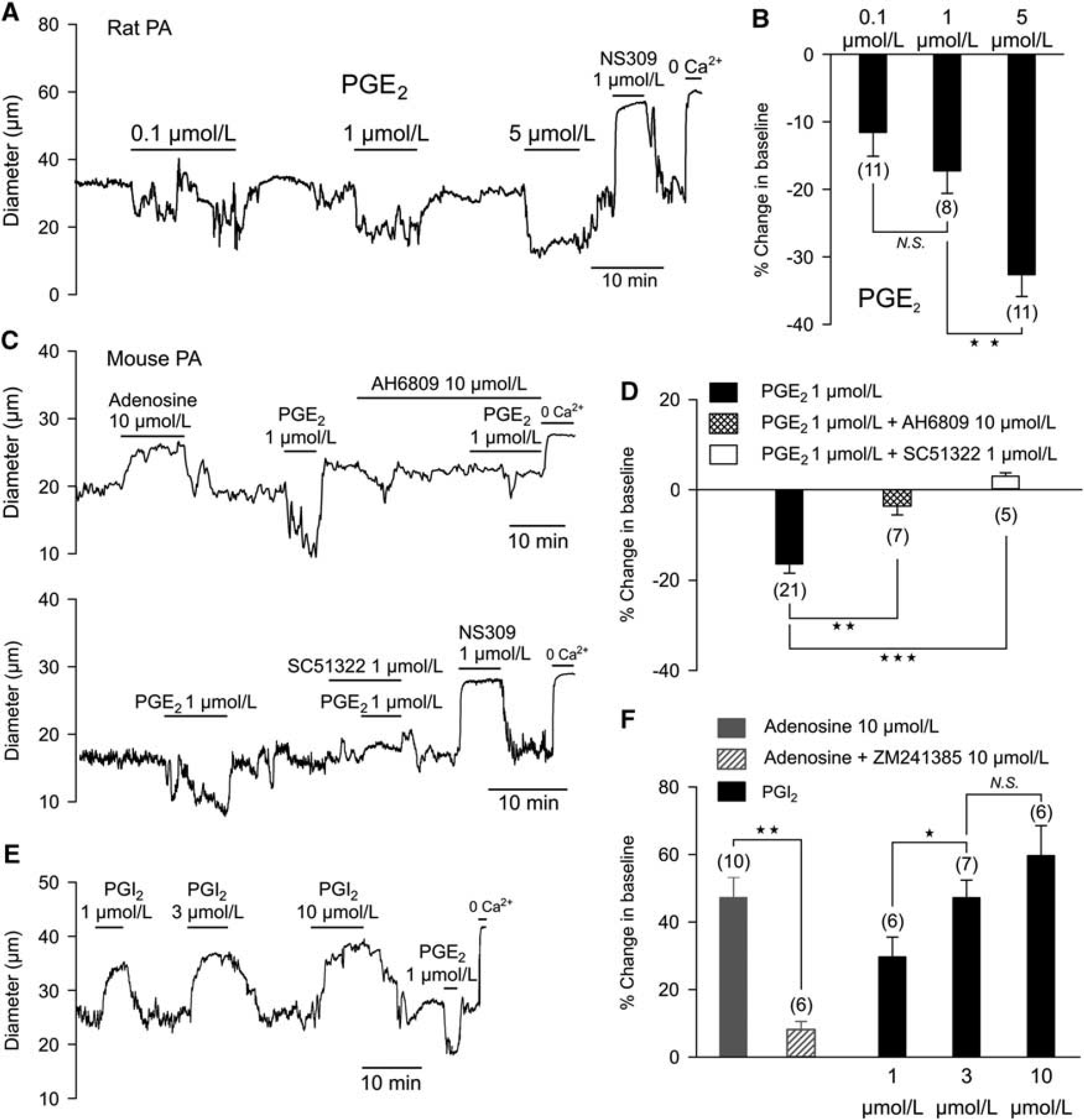
Prostaglandin E2 Constricts Pressurized Parenchymal Arterioles Although PGE2 released from astrocytes has been proposed as a major mediator of neurovascular coupling, whether PGE2 actually dilates parenchymal arterioles has not been experimentally determined. To test this directly, we assessed the effects of different concentrations of PGE2 on the diameter of isolated parenchymal arterioles, preconstricted by elevation of intravascular pressure to a physiologic level (40 mm Hg). Surprisingly, rather than inducing dilation, PGE2 constricted cerebral parenchymal arterioles from rat (Figures 1A and 1B) and mouse (Figures 1C to 1E) in a concentration-dependent manner. Prostaglandin E2 (1 μmol/L) constricted rat and mouse parenchymal arterioles by 18.6 ± 3% (
In mouse, vasoconstriction induced by PGE2 (1 μmol/L) was significantly higher at an intraluminal pressure of 20 mm Hg, than at 80 mm Hg (25.8 ± 2% versus 10.8 ± 1%,
Prostaglandin E2-Induced Constriction of Pressurized Parenchymal Arterioles is Mediated by EP1 Receptors
In studies of newborn and adult pigs, an assessment of PGE2 receptor subtypes along with measurements of the effects of PGE2 on IP3 and cyclic adenosine monophosphate production suggests a predominant expression of EP1 with lesser levels of EP3 in adult brain microvessels. 11 In mouse parenchymal arterioles, we found that vasoconstriction to PGE2 (1 μmol/L) was inhibited by the prostanoid receptor antagonist AH6809 (10 μmol/L), which does not discriminate among EP1, EP2, and EP3 receptors (Figures 1C and 1D). 12 SC51322 (1 μmol/L), a selective antagonist of the EP1 receptor, 12 completely inhibited the constriction to PGE2 (Figures 1C and 1D), indicating that PGE2 acts through EP1 receptors to constrict parenchymal arterioles. This is consistent with the likely coupling of EP1 receptors to G proteins of the Gq class, which typically mediate the contractile response to vasoconstrictor agonists. Prostaglandin E2 did not cause vasodilation in the presence of AH6809 or SC51322.
The Vasodilator Adenylyl Cyclase-Protein Kinase Pathway is Intact in Parenchymal Arterioles
It has been proposed that PGE2, acting through Gs-coupled receptors (e.g., EP4), stimulates the adenylyl cyclase-PKA pathway to dilate parenchymal arterioles. 8 To confirm that the adenylyl cyclase-PKA pathway is operational in parenchymal arterioles, we tested the effects of the endogenous agents, adenosine and PGI2, which have been shown to relax smooth muscle through this mechanism. Both adenosine and prostacyclin caused rapid and reversible dilation of parenchymal arterioles (Figures 1C, Figures 1E, and 1F). Vasodilation to adenosine was prevented by preincubation with the adenosine A2A receptor antagonist ZM241385 (Figure 1D).
Prostaglandin E2 Constricts Pressurized Mouse Pial Arterioles Previous studies have investigated the effects of PGE2 in isolated cerebral arteries from different species (Supplementary Table 1), the majority of which have shown vasoconstriction. Arteries from guinea pig, dog, pig, and baboon constrict in response to PGE2, whereas feline pial arteries display weak relaxation at lower concentrations and contraction at higher concentrations of PGE2. Studies using rat, rabbit, and postmortem human cerebral arteries have reached very divergent conclusions (Supplementary Table 1). In tests of PGE2 (0.1 and 1 μmol/L) on middle and posterior cerebral arteries from adult mice, we observed only vasoconstriction (Supplementary Figures 1D and E).
DISCUSSION
A number of studies have proposed that astrocyte-derived PGE2 makes a major contribution to functional hyperemia. 8 According to this model, PGE2, generated via a Ca2+-dependent mechanism through the action of astrocytic endfoot COX1 on phospholipase A2-derived arachidonic acid, is released from astrocytes and activates receptors on vascular smooth muscle cells, presumably Gs-coupled EP4 receptors, to promote arteriolar dilation. Much of the evidence for this mechanism comes from inhibitor-based studies using brain slice preparations in which Ca2+ elevation in astrocytes is induced directly through application of a metabotropic glutamate receptor agonist or indirectly through electrical field stimulation of neurons, after which the effects of COX inhibitors on arteriolar dilation are measured. These studies have yielded mixed results.6,7
Several neurovascular coupling agents have been proposed including prostaglandins, epoxyeicosatrienoic acids, adenosine, oxygen, nitric oxide, H+, and K+.1,8 Although mechanisms of functional hyperemia may vary substantially between regions of the brain, one absolute criterion that a putative ‘gliotransmitter’ must satisfy, if it is to couple neuronal activation to vasodilation, is a demonstrated ability to dilate isolated arterioles. Adenosine, prostacyclin, and external H+ and K+4,5,13 are potent vasodilators that induce a rapid dilation when directly applied to isolated arterioles, and therefore fulfill this requirement. Prostaglandin E2 does not. Although the remote possibility that an unknown vasoconstrictor is released from remnant endfoot membranes in response to PGE2 cannot be formally excluded, we consider this highly unlikely given that pial arteries, which lack endfeet also constrict to PGE2 (Supplementary Figures 1D and E) and no dilation was observed in parenchymal arterioles in the presence of an EP1 blocker (Figure 1). Therefore, our results clearly indicate that PGE2 does not dilate cerebral parenchymal arterioles (Figure 1), which are critical to neurovascular coupling, but instead causes vasoconstriction through activation of EP1 receptors.
Interpretation of the effects of exogenous application of PGE2 on arteriolar diameter in brain slices or
In conclusion, our results provide strong evidence that PGE2 is not directly involved in neurovascular coupling, and underscore the need to directly test putative neurovascular coupling transmitters on their target arterioles.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.