Free accessResearch articleFirst published online 2013-8
Brain Alanine Formation as an Ammonia-Scavenging Pathway during Hyperammonemia: Effects of Glutamine Synthetase Inhibition in Rats and Astrocyte—Neuron Co-Cultures
Hyperammonemia is a major etiological toxic factor in the development of hepatic encephalopathy. Brain ammonia detoxification occurs primarily in astrocytes by glutamine synthetase (GS), and it has been proposed that elevated glutamine levels during hyperammonemia lead to astrocyte swelling and cerebral edema. However, ammonia may also be detoxified by the concerted action of glutamate dehydrogenase (GDH) and alanine aminotransferase (ALAT) leading to trapping of ammonia in alanine, which in vivo likely leaves the brain. Our aim was to investigate whether the GS inhibitor methionine sulfoximine (MSO) enhances incorporation of 15NH4+ in alanine during acute hyperammonemia. We observed a fourfold increased amount of 15NH4 incorporation in brain alanine in rats treated with MSO. Furthermore, co-cultures of neurons and astrocytes exposed to 15NH4Cl in the absence or presence of MSO demonstrated a dose-dependent incorporation of 15NH4 into alanine together with increased 15N incorporation in glutamate. These findings provide evidence that ammonia is detoxified by the concerted action of GDH and ALAT both in vivo and in vitro, a mechanism that is accelerated in the presence of MSO thereby reducing the glutamine level in brain. Thus, GS could be a potential drug target in the treatment of hyperammonemia in patients with hepatic encephalopathy.
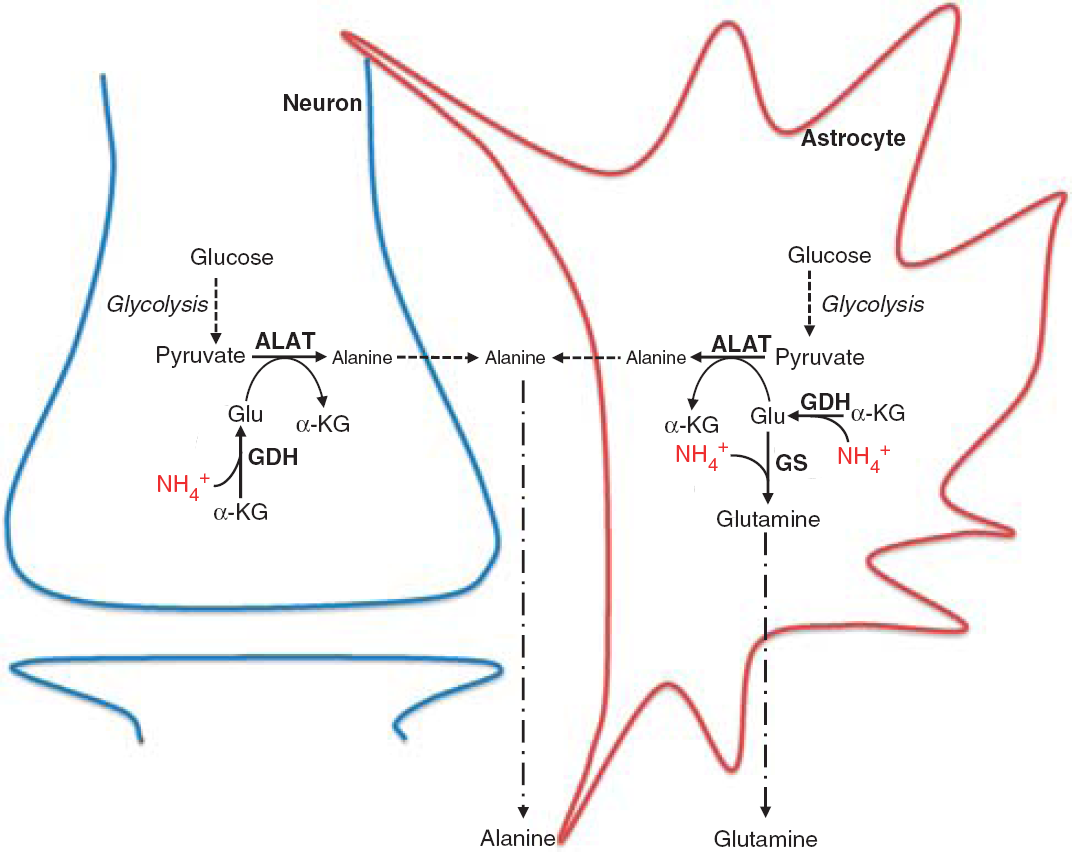
Hepatic encephalopathy (HE) is a neuropsychiatric syndrome associated with liver dysfunction. The cellular and molecular mechanisms for the development of HE remain unclear but the hyperammonemic condition is considered essential for the severity of the disease.1,2 In the brain, the predominant route for ammonia metabolism is by glutamine synthesis via the enzyme glutamine synthetase (GS), which is exclusively located in the astrocytes.3 Consequently, during acute hyperammonemic conditions, brain glutamine levels are increased in HE patients,4–6 which has been suggested as the principal cause leading to astrocyte swelling and cerebral edema.7–9In vivo studies have shown that administration of methionine sulfoximine (MSO), which specifically inhibits GS, ameliorates astrocyte swelling and brain edema observed during acute hyperammonemia.10–13 A decreased ammonia fixation by GS may lead either to a stimulation of an alternative ammonia-scavenging pathway or to a corresponding, increased ammonia concentration. A study using neuronal-astrocytic co-cultures provided evidence for a high incorporation of ammonia nitrogen in both alanine and glutamine,14 suggesting that ammonia might be metabolized not only via GS catalyzed formation of glutamine but also by the concerted action of glutamate dehydrogenase (GDH) and alanine aminotransferase (ALAT) leading to trapping of ammonia in alanine, as illustrated in Figure 1. In addition, de novo synthesis of alanine from glucose was shown to be increased when glutamine synthesis is inhibited by MSO.15 This indicates that a concerted action of GDH and ALAT constitutes an alternative scavenging pathway when GS is inhibited. Furthermore, increased activity of glycolysis and decreased flux through pyruvate dehydrogenase in brain during acute liver failure16 support the notion of an augmented capacity to synthesize alanine owing to an increased availability of pyruvate, the precursor of alanine. This pathway is likely localized to the astrocytes that possess both high glycolytic and GDH activity.17–19 However, whether this may change during hyperammonemic conditions is at present unknown. The aim of this study was to investigate the metabolic effect of GS inhibition on cerebral ammonia metabolism in vivo and in vitro and to test the hypothesis that such inhibition enhances [15N]ammonia incorporation in alanine during acute hyperammonemia. Rats pretreated with either saline or MSO were administered 15NH4Cl as intravenous infusion and brain tissue contents and incorporation of 15N in the amino acids glutamate, glutamine, aspartate, and alanine were determined. Corresponding in vitro studies were performed in a cell culture model consisting of astrocytes in co-culture with GABAergic neurons to obtain further insight into the cellular localization and biochemical mechanism.
Schematic cartoon depicting the mechanisms involved in incorporation of ammonia in glutamine and alanine. ALAT, alanine aminotransferase; Glu, glutamate; GDH, glutamate dehydrogenase; GS, glutamine synthetase; α-KG, α-ketoglutarate.
MATERIALS AND METHODS
Animals and Chemicals
The protocol involving in vivo studies was approved by the Danish Animal Research Committee. For the in vivo studies, female Wistar rats (body weight 250 to 280 g) were employed. Rats were housed with two animals per cage at 22 ± 2°C and 55 ± 10% relative humidity with free access to water and standard rodent chow. NMRI mice for the in vitro studies were obtained from Taconic M&B (Ry, Denmark). Plastic tissue culture dishes were purchased from NUNC A/S (Roskilde, Denmark), fetal bovine serum from Sigma (St Louis, MO, USA). MSO, culture medium and poly-D-lysine (MW > 300,000) were from Sigma Chemical. Penicillin was from Leo (Ballerup, Denmark). Isotopically labeled ammonia ([15N]ammonium chloride, 98%) was from Cambridge Isotope Laboratories (Andover, MA, USA). All other chemicals used were of the purest grade available from regular commercial sources.
In Vivo Rat Brain Experiments
The rats were treated with intraperitoneal injections of MSO (150 mg/kg) or saline and after 3 hours, 15NH4Cl (2 mmol/kg) was administered as a 15-minute intravenous infusion in a tail vein. This dose of MSO has been shown to decrease GS activity by 64%.10 Immediately after the 15NH4 infusion, the rats were decapitated and a sample of the brain cortex was collected within 40 to 50 seconds and frozen in isopentane containing dry ice. The samples were instantly extracted in 70% v/v ethanol (ice-cold) and the extracts centrifuged (20,000g, 20 minutes). Cell extracts were lyophilized and reconstituted in water for gas chromatography-mass spectrometry (GC-MS) and HPLC analyses. The pellets were dissolved in 1 mol/L KOH and used for determination of protein20 using bovine serum albumin as the standard.
Co-cultures of Neurons and Astrocytes
Co-cultures of cortical neurons and astrocytes were prepared from cerebral cortices of mouse fetuses, 15-day-gestation, as previously described by Leke et al.21 Cells were seeded in poly-D-lysine coated 6 well plates (2 mL/well) at a density of 2.75 × 106 cells/mL in a slightly modified22 Dulbecco's minimum essential medium containing 10% (v/v) fetal calf serum. The cells were cultured for 7 to 8 days, a period during which they exhibited an abundant proliferation of astrocytes as well as neuronal migration as observed by light microscopy.
Incubation Experiments in Cell Co-Cultures
Cultures were preincubated in the presence or absence (controls) of 10 mM MSO for 1 hour at 37°C followed by incubation with NH4Cl in the concentrations of either 1 mM, 2.5 mM or 5 mM for 1 hour at 37°C. Subsequently, the culture medium was aspirated and the cultures were rinsed twice in phosphate-buffered saline (PBS; 137mM NaCl, 2.7mM KCl, 7.3 mM Na2HPO4, 0.9 mM CaCl2, 0.5 mM MgCl2, pH 7.4, 37°C). Cultures were incubated for 2.5 hours with [15N]ammonia (1 mM; 2.5 mM; 5 mM) alone or together with 10 mM MSO. The incubation period was terminated by removing the medium from the cells, which were then rinsed twice with ice-cold PBS. Subsequently, the cells were extracted in 70% v/v ethanol and centrifuged (20,000g, 20 minutes, 4°C) to separate the soluble extract from the insoluble components. The pellets were employed for protein determination as described above. Cell extracts were lyophilized and reconstituted in water for GC—MS and HPLC analyses. The incubation media were also lyophilized and reconstituted in water for determination of relevant amino acids.
GC-MS and HPLC Analyses
The percentage distribution of 15N-labeling relative to the total amount of metabolite was determined using GC–MS. Amino acids were extracted into an organic phase of ethanol and benzene, dried under nitrogen, and reconstituted in N,N-dimethylformamide before they were derivatized with N-methyl-N-(tert-butyldimethylsilyl)trifluoroacetamide in the presence of 1% tertbutyldimethylchlorosilane modified after the method of Mawhinney et al.23 Mass spectrometric analysis was performed on a GC-MS system consisting of a Shimadzu GC–MS QP2010Plus mass spectrometer coupled to a Shimadzu GC2010 gas chromatograph (Shimadzu Corp., Japan). All labeling data were corrected for natural abundance of 15N by subtracting the mass distribution of a standard containing the relevant metabolites. Isotopic enrichment was calculated according to Biemann.24
Amino acids were separated and quantified by reversed-phase HPLC on an Agilent Eclipse AAA column (4.6 × 150 mm, particle size 5 μm; Agilent Technologies, Santa Clara, CA, USA) employing precolumn, online o-phthaldialdehyde derivatization and fluorescence detection (excitation 350 nm; detection 450 nm), mobile phase A (citrate (0.04 mol/L)-phosphate (0.2 mol/L) buffer containing 4.8% acetonitrile (VWR/Prolabo, Radnor, PA, USA), pH 5.9), mobile phase B: 90% acetonitrile. The gradient of mobile phase B increased from 0% to 50% in 35 minutes, with a total run time of 43 minutes.
Data Analysis
Data are presented as mean ± s.e.m. and differences between groups were analyzed statistically using either one-way ANOVA followed by Turkey post hoc test or unpaired two-tailed Student's t-test. A P-value < 0.05 was considered to indicate statistical significance.
RESULTS
In Vivo Effect of MSO on Rat Brain Metabolism of 15NH4+
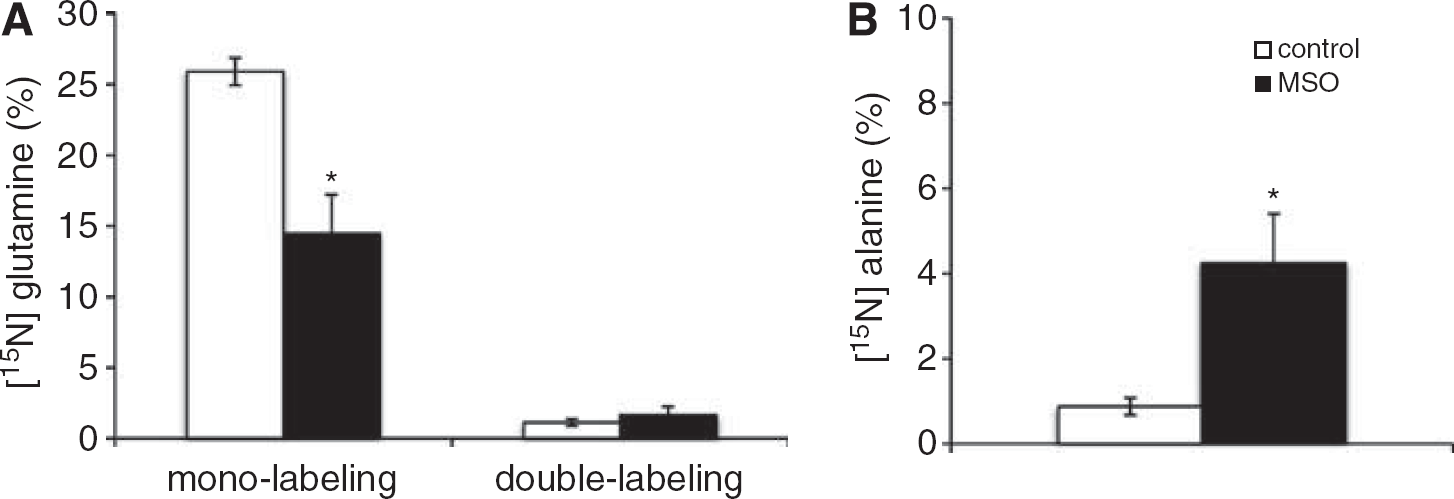
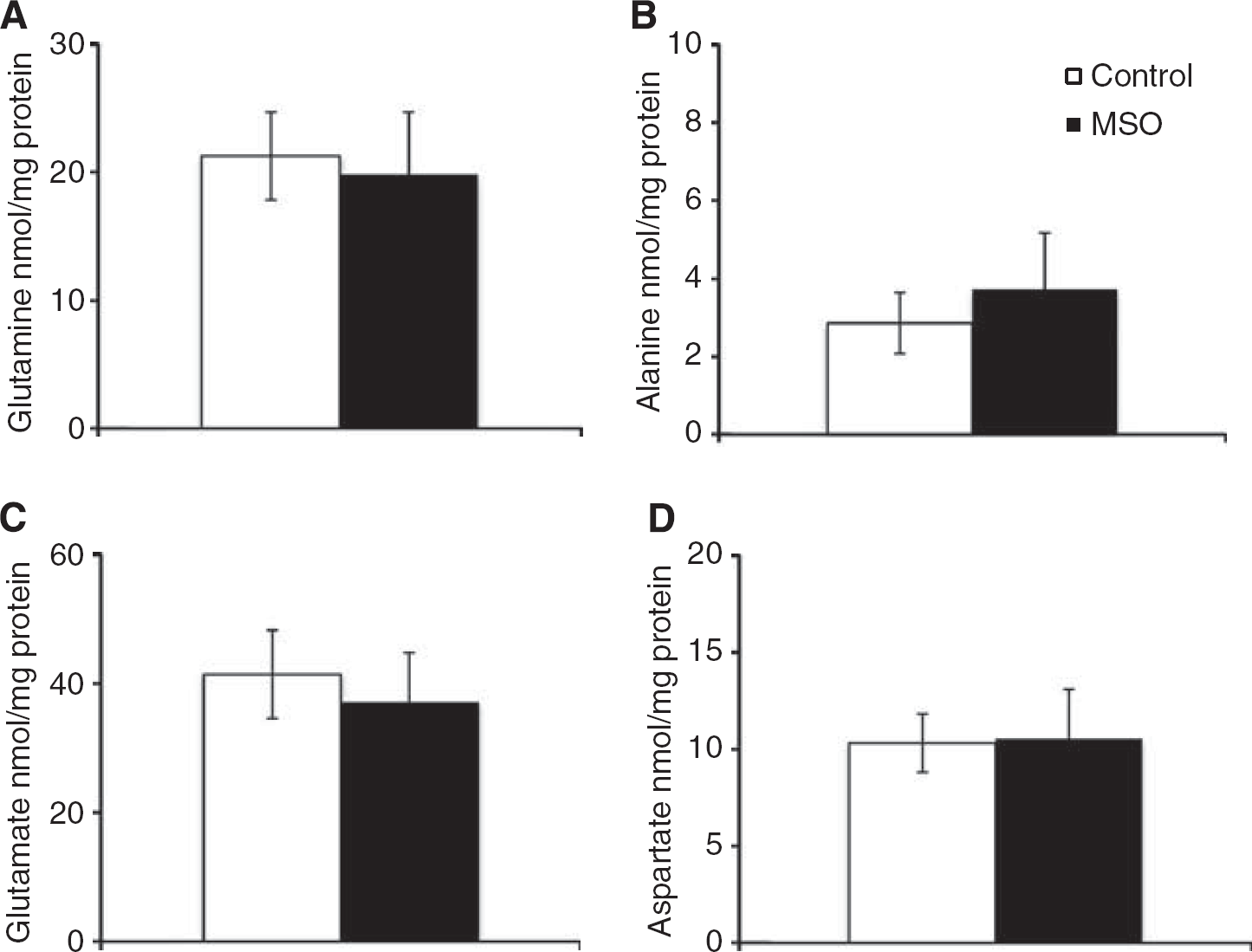
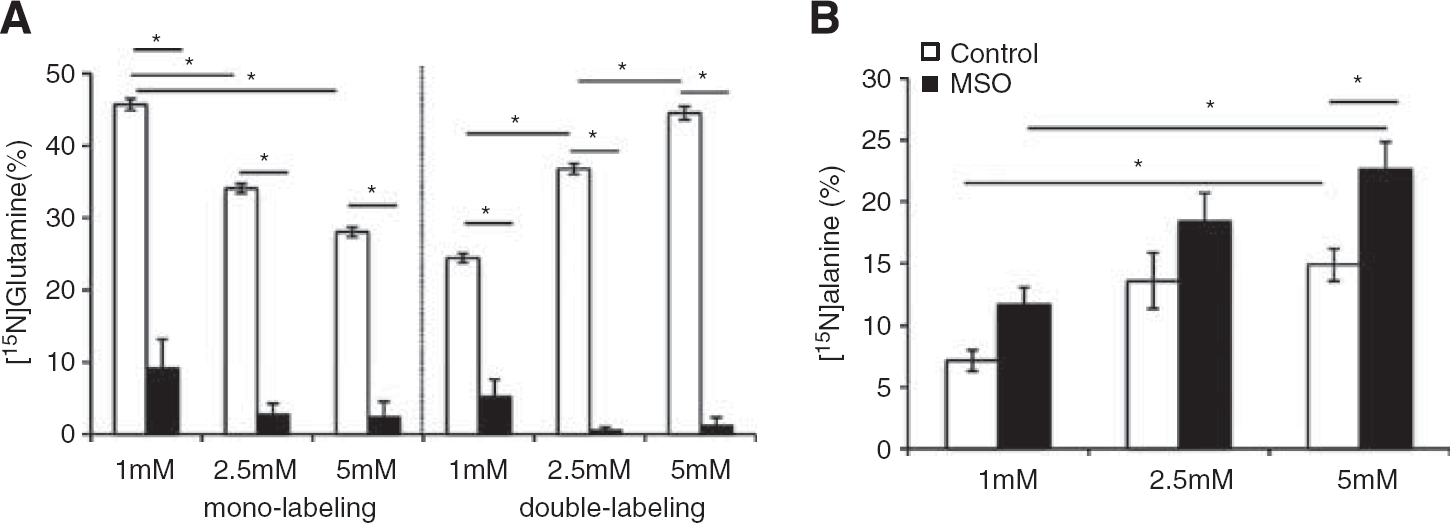
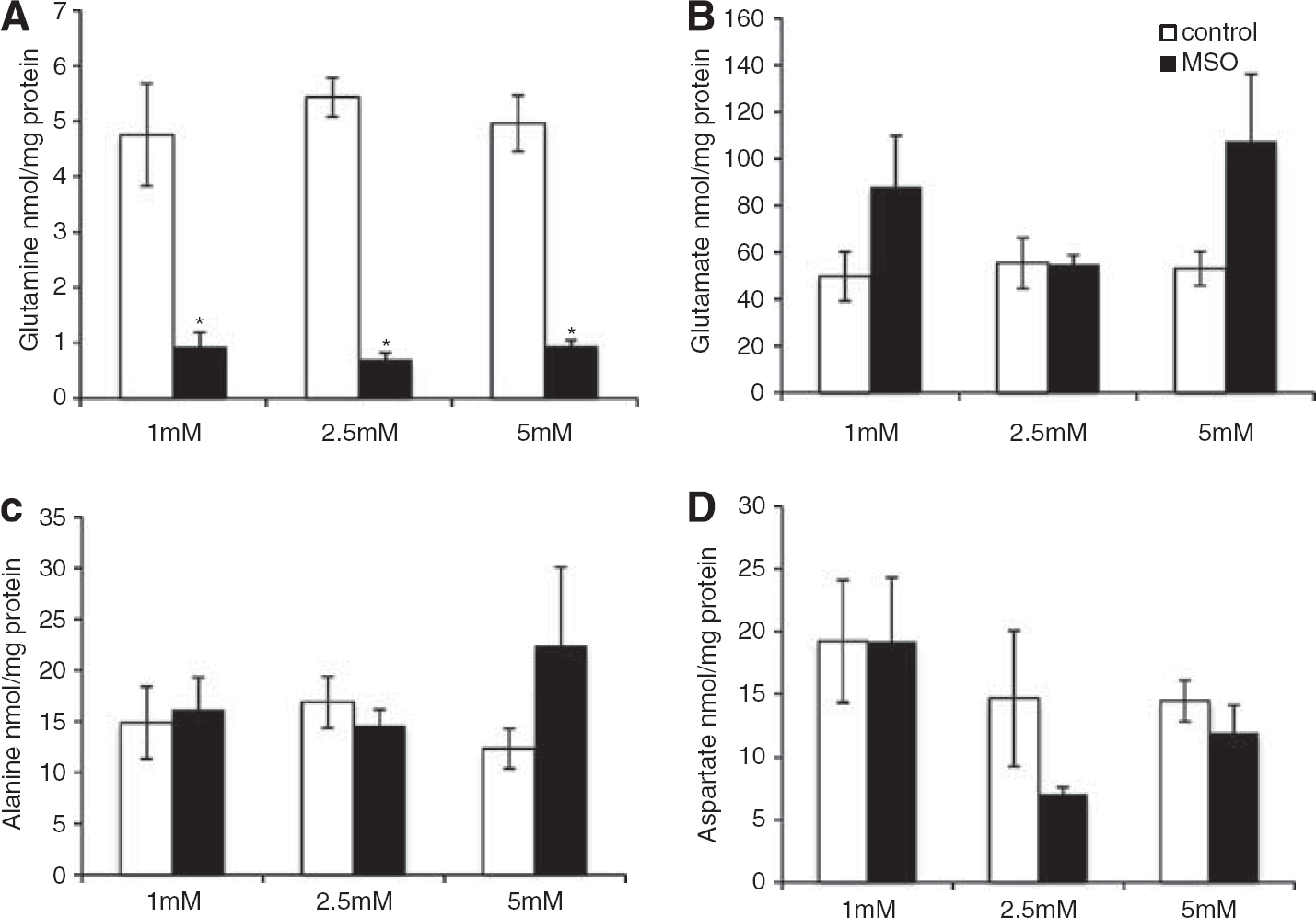
The 15N-labeling of glutamine and alanine from 15NH4+ in brain cortex (Figures 2A and 2B) shows that MSO treatment decreased 15N mono-labeling in glutamine two-fold, while that of alanine was increased four-fold. Glutamine double labeling was not affected by MSO. Glutamate and aspartate did not display any 15N-labeling in MSO-treated rats or in saline-treated rats (data not shown). Double labeling of glutamine is formed only from labeled glutamate, thus, the lack of labeled glutamate confirms the cellular compartmentation of the major glutamate and glutamine pools in brain suggested by Berl et al.25 Treatment with MSO did not change the contents of glutamine, alanine, glutamate and aspartate in brain cortex as shown in Figures 3A—3D. The content of glutamine in the brain is five-fold higher than the alanine content, thus the amount of 15N-label in the alanine pool (nmol labeling/mg protein) relative to the amount of 15N label in the glutamine pool is 5.4% in the rat brain after MSO treatment.
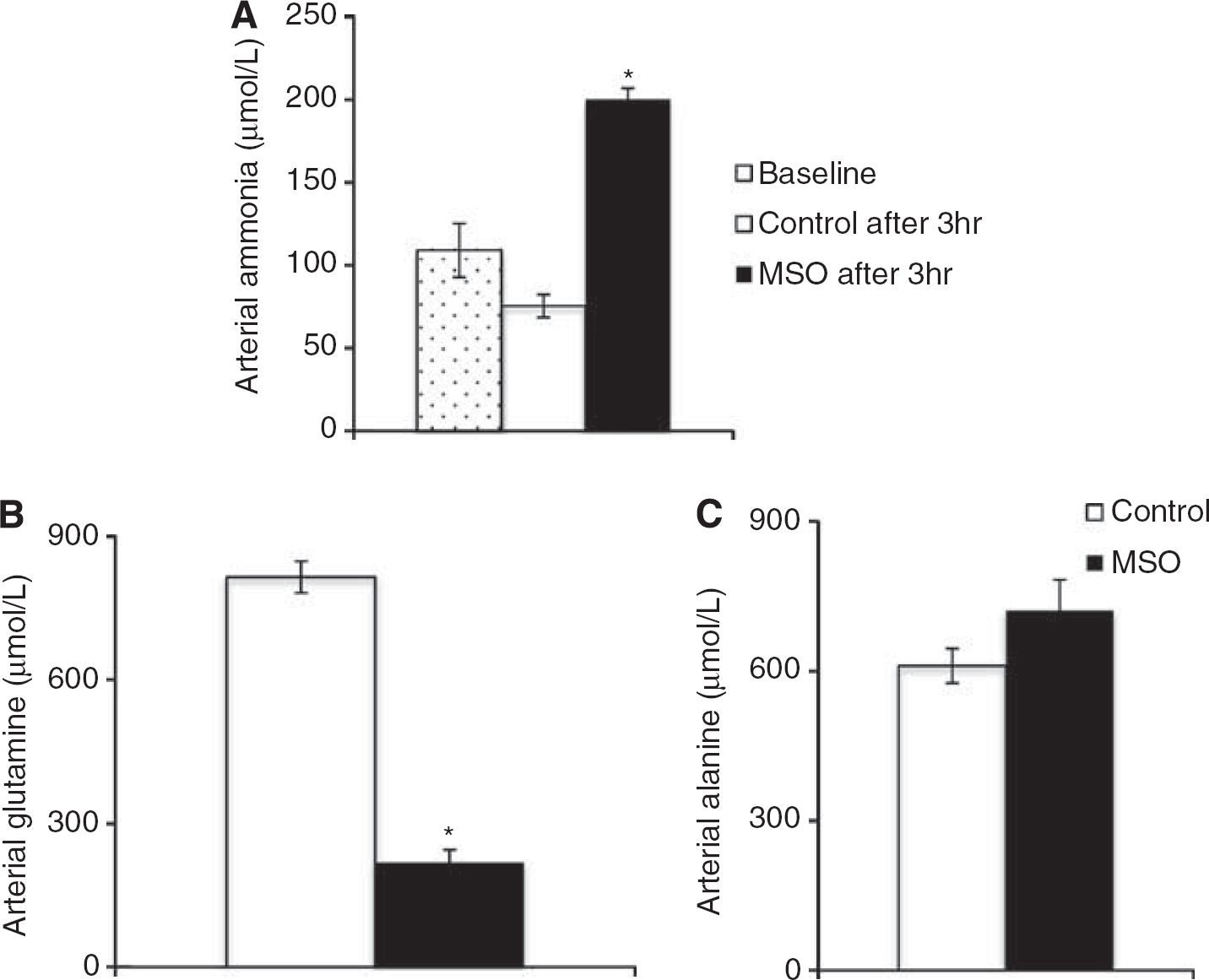
Arterial ammonia concentrations before and 3 hours after treatment with MSO or saline but before the 15NH4+ infusion show that MSO treatment doubled arterial ammonia whereas saline treatment did not (Figure 4A). After 15NH+4 infusion, the arterial glutamine concentration was decreased in rats pretreated with MSO (Figure 4B), while that of alanine was unchanged (Figure 4C).
Cerebral 15N-labeling (%) in (A) glutamine and (B) alanine from 15NH4+ in saline-treated rats (white bars) and methionine sulfoximine (MSO)-treated rats (black bars). Results are shown as mean ± s.e.m. of 7 to 8 rats. The asterisk indicates a statistically significant difference between the two groups of animals (P < 0.05).
Cerebral content (nmol/mg protein) of (A) glutamine, (B) alanine, (C) glutamate, and (D) aspartate after 15NH4+ infusion in control (white bars) and methionine sulfoximine (MSO) rats (black bars). Results are mean ± s.e.m. of 7 to 8 rats.
Arterial ammonia concentration (μM) (A) before 15NH4+ infusion at baseline t = 0 hours (white dotted bars), in saline-treated rats t = 3 hours (white bars) and methionine sulfoximine (MSO)-treated rats t = 3 hours (black bars) as well as (B) glutamine and (C) alanine after 15NH4+ infusion in saline-treated rats (white bars) and MSO-treated rats (black bars). Results are shown as mean ± s.e.m. of 7 to 8 rats. The asterisk indicates a statistically significant difference between baseline in (A) or control animals in (B-C) (P < 0.05).
In Vitro Effect of MSO on Metabolism of 15NH4+ in Co-Cultures of Neurons And Astrocytes
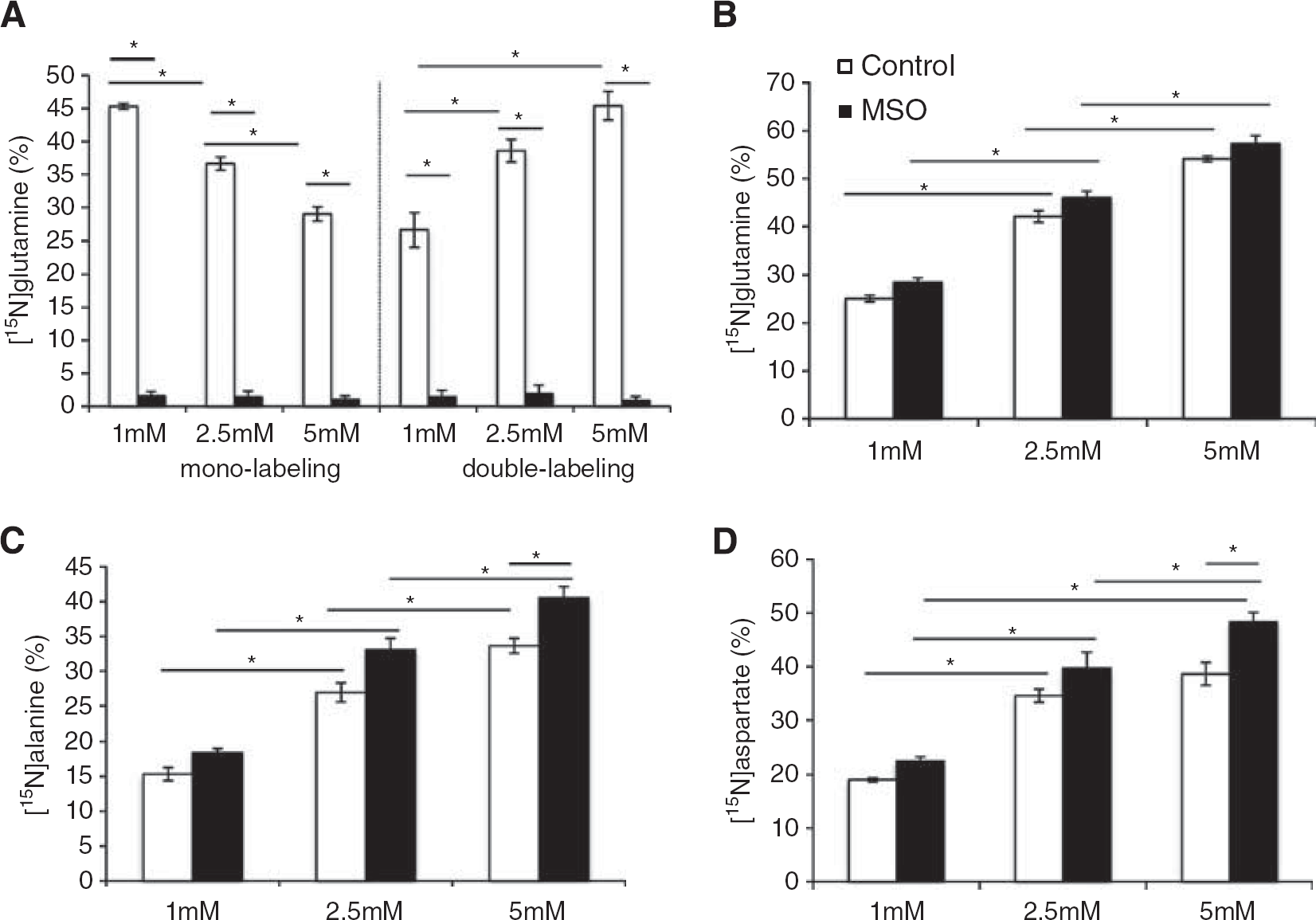
The 15N-labeling of intracellular glutamate, glutamine, aspartate, and alanine after incubation of the co-cultures with 15NH4+ (1 mM, 2.5 mM, or 5 mM) in the presence or absence of MSO (10 mM) is shown in Figures 5A–5D. Increasing concentrations of 15NH4+ led to decreased mono-labeling of glutamine with a simultaneous increase in double labeling of glutamine (Figure 5A), showing that an elevated ammonia concentration results in ammonia being detoxified not only through GS activity but also through an elevated extent of reductive amination catalyzed by GDH. In line with this, increasing 15NH4+ concentrations resulted in increased 15N labeling in glutamate (Figure 5B). However, the presence of MSO did not lead to a significant increase in 15N labeling of glutamate. Exposure to MSO essentially blocked both mono- and double-labeling in glutamine (Figure 5A). Alanine labeling was significantly increased in cells exposed to 5 mM 15NH4+ in the presence of MSO (Figure 5C), thus demonstrating ammonia being detoxified by the concerted action of GDH and ALAT when GS is inhibited. Lower ammonia concentrations did not lead to significantly increased [15N]alanine labeling in the presence of MSO, however, a clear dose-dependent correlation was observed between the 15N-labeling in alanine with increasing [15N]ammonia concentrations in the presence and absence of MSO (Figure 5C). Similarly, an augmented 15N-labeling in aspartate was observed with increasing 15NH4+ concentrations, while exposure to MSO only led to increased [15N]aspartate in the presence of 5 mM ammonia (Figure 5D). The extent of 15N-labeling in the extracellular pool of glutamine and alanine, shown in Figures 6A and 6B, was similar to that observed intracellularly (Figures 5A and 5C). The labeling intensity for extracellular glutamine (Figure 6A) reflected the intracellular values, while alanine displayed lower amount (P < 0.05) of extracellular 15N-labeling (Figure 6B) compared with that in the intracellular pool (Figure 5C).
Intracellular 15N-labeling (%) in co-cultures of neurons and astrocytes in (A) glutamine, (B) glutamate, (C) alanine, and (D) aspartate from 15NH4+ (1–5 mM) in the absence (white bars) or presence (black bars) of MSO. Results are shown as mean ± s.e.m. of 7 to 9 cell cultures originating from different batches. The asterisk indicates a statistically significant difference between the two conditions (P < 0.05).
Extracellular 15N-labeling (%) in co-cultures of neurons and astrocytes in (A) glutamine and (B) alanine from 15NH4+ (1-5 mM) in the absence (white bars) or presence (black bars) of methionine sulfoximine (MSO). Results are shown as mean ± s.e.m. of 5 to 9 cell cultures originating from different batches. The asterisk indicates a statistically significant difference between the two conditions (P < 0.05).
Exposure to MSO decreased intracellular glutamine content by 80% (Figure 7A), while the content of glutamate, alanine, and aspartate were unaffected by MSO (Figures 7B–7D).
Intracellular content (nmol/mg protein) in co-cultures of neurons and astrocytes of (A) glutamine, (B) glutamate, (C) alanine, and (D) aspartate in the presence of 15NH4+ (1 to 5 mM) in the absence (white bars) or presence (black bars) of methionine sulfoximine) (MSO). Results are shown as means ± s.e.m. of 5 to 9 cell cultures originating from different batches. The asterisk indicates a statistically significant difference between the two conditions (P < 0.05).
DISCUSSION
The principal route for metabolism of ammonia in brain is through glutamine formation and at least 85% of the blood-derived ammonia is metabolized via GS into the amide position of glutamine in the astrocytes.26–28 Accordingly, during hyperammonemia, astrocytic glutamine levels are increased several fold in patients with HE.5,6 However, in rat models of fulminant hepatic failure and in patients with fulminant hepatic failure, it has been shown by microdialysis that also brain extracellular alanine levels are augmented,29,30 indicating that alanine might have a role in cerebral ammonia metabolism. In support of this, HE patients with fulminant hepatic failure and patients with acute exacerbations of chronic liver disease displayed significant net cerebral effluxes of glutamine and alanine.31 Hence, cellular alanine synthesis and release from brain may constitute an additional ammonia detoxification mechanism resembling the role of glutamine synthesis. In agreement with this, the present study shows that rats infused with 15NH4+ had increased fixation of 15N into cerebral alanine when glutamine synthesis was inhibited with MSO, demonstrating that, in vivo, GS inhibition may enhance the alanine-synthesizing pathway consistent with what has been shown in cell culture systems.14,15 In support of the in vivo findings, a dose dependent increase of 15N-labeling in alanine from 15NH4+ together with an increased [15N]glutamate labeling was observed in the co-culture experiment clearly demonstrating that ammonia is detoxified by the concerted action of GDH and ALAT. Exposure to 5 mM but not 1 and 2.5 mM, 15NH4+ resulted in a significant increase of [15N]alanine in the presence of MSO implying that the enhancement of the alanine-synthesizing pathway during inhibition of GS is most relevant in acute hyperammonemic conditions. Furthermore, augmented extracellular levels of [15N]alanine in MSO-treated co-cultures support the findings of increased cerebral efflux of alanine in HE patients.29–31
Although no 15N-labeling was observed in glutamate and aspartate in the rat brain, small pools of these amino acids must be 15N labeled since 15N-labeling of alanine as well as aspartate exclusively occurs directly from [15N]glutamate, which is also clear from the data obtained in the co-culture experiments. Thus, [15N]alanine is formed from a small pool of 15N-labeled glutamate that is camouflaged by a large non-labeled pool of glutamate. Most likely, the high extent of [15N]alanine observed in vivo originates from the small astrocytic pool of glutamate that is not in equilibrium with the large neuronal glutamate pool. Alternatively, a small compartmentalized 15N-enriched pool of neuronal glutamate equilibrates with a large pool of alanine. Incorporation of ammonia into alanine requires that GDH catalyzes the amination of α-ketoglutarate. In support of the suggestion that this could have occurred in the neuronal compartment, particularly during acute hyperammonemic conditions, is a study using GABAergic neurons in culture showing prominent removal of 15NH4+ by reductive amination at 3 mM concentrations but not at 0.3 mM.32 This study by Yudkoff et al.32 showed up to 15% 15N enrichment of glutamate in neurons, however, studies performed using co-cultures of astrocytes and neurons displayed 50% 15N enrichment in glutamate after exposure to 5 mM NH4+ and up to 45% [15N]glutamate in astrocyte cultures exposed to 1 mM NH4+.14,33 Hence, it is most likely that the fixation of 15NH4f by GDH and synthesis of [15N]alanine primarily occurs in astrocytes. The elevated double labeling in glutamine observed with increasing 15NH4+ concentrations when GS is not inhibited confirms an augmented activity of GDH in the amination direction in the co-cultures. In agreement with this, in vivo studies in rats have shown that an increased amount of [13N]ammonia derived from blood could be found in glutamate when GS is inhibited.26 As expected, we found 15N-labeling in aspartate to be elevated in parallel with glutamate, which reflects the equilibrium of the aspartate aminotransferase reaction. For both aspartate and alanine intracellular 15N-labeling is 40% to 50% in the co-cultures demonstrating the relevance of both transamination reactions. Even though this is in contrast to findings of relatively low activity of ALAT compared with AAT in homogenates of rat brain,34 a cell culture study found that ALAT activity is cell-type specific with a three-fold higher enzyme activity in astrocytes compared with neurons.35 Since aspartate is not released from healthy astrocytes owing to glutamate transporters generating a large inward gradient of glutamate as well as aspartate,36 this amino acid cannot be considered an alternative ammonia scavenger. Moreover, increased alanine and lactate synthesis has been demonstrated to correlate with the severity of encephalopathy in acute liver failure rats, signifying the importance of alanine synthesis in hyperammonemia.16,37
Although removal of ammonia through alanine synthesis and release from cells may have a less significant role under modest hyperammonemic conditions, the results of the present study support the notion that this alternative mechanism could have therapeutic interest for acute hyperammonemia. Although blocking glutamine synthesis significantly elevated arterial ammonia levels 3 hours after administration of MSO, the data obtained in the co-culture experiments suggest that the increased ammonia level is caused by the inhibition of GS, which dose dependently will lead to increased incorporation of ammonia into alanine in the brain. Indeed, the importance of inhibiting brain glutamine synthesis in acute hyperammonemic rats has been demonstrated in studies showing amelioration of ammonia-induced astrocyte swelling and cerebral edema when GS is blocked.10–12 Since diminshed glutamine synthesis may completely abolish ammonia-induced astrocyte swelling, enhancement of alanine synthesis cannot be considered having a similar swelling effect.38 Furthermore, inhibition of this enzyme has recently been shown to increase survival and decrease cytokine response in a mouse model of acute liver failure.39
In summary, we find that inhibition of GS during an acute ammonia load in rats enhances incorporation of 15N into alanine mediated through the concerted action of GDH and ALAT. The increased ammonia fixation in alanine was in co-culture experiments demonstrated to occur in a dose-dependent manner, which is probably a direct result of the GS inhibition.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
ACKNOWLEDGEMENTS
The expert technical assistances of Mette Simonsen and Heidi Nielsen are cordially acknowledged.
HaussingerDSchliessF. Pathogenetic mechanisms of hepatic encephalopathy. Gut2008; 57: 1156–1165.
3.
NorenbergMDMartinez-HernandezA. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res1979; 161: 303–310.
4.
AlbrechtJZielinskaMNorenbergMD. Glutamine as a mediator of ammonia neurotoxicity: a critical appraisal. Biochem Pharmacol2010; 80: 1303–1308.
5.
LavoieJGiguereJFLayrarguesGPButterworthRF. Amino acid changes in autopsied brain tissue from cirrhotic patients with hepatic encephalopathy. J Neurochem1987; 49: 692–697.
6.
LaubenbergerJHäussingerDBayerSGuflerHHennigJLangerM. Proton magnetic resonance spectroscopy of the brain in symptomatic and asymptomatic patients with liver cirrhosis. Gastroenterology1997; 112: 1610–1616.
7.
AlbrechtJNorenbergMD. Glutamine: a Trojan horse in ammonia neurotoxicity. Hepatology2006; 44: 788–794.
8.
Rama RaoKVNorenbergMD. Glutamine in the pathogenesis of hepatic encephalopathy: The Trojan Horse hypothesis revisited. Neurochem Res; e-pub ahead of print2013 doi:10.1007/s11064–012–0955–2.
9.
ChavarriaLAlonsoJRoviraACordobaJ. Neuroimaging in acute liver failure. Neurochem Int2011; 59: 1175.
10.
TakahashiHKoehlerRCBrusilowSWTraystmanRJ. Inhibition of brain glutamine accumulation prevents cerebral edema in hyperammonemic rats. Am J Physiol1991; 261: H825–H829.
11.
Willard-MackCLKoehlerRCHirataTCorkLCTakahashiHTraystmanRJ. Inhibition of glutamine synthetase reduces ammonia-induced astrocyte swelling in rat. Neuroscience1996; 71: 589–599.
12.
TanigamiHRebelAMartinLJChenTYBrusilowSWTraystmanRJ. Effect of glutamine synthetase inhibition on astrocyte swelling and altered astroglial protein expression during hyperammonemia in rats. Neuroscience2005; 131: 437–449.
13.
MasterSGottsteinJBleiAT. Cerebral blood flow and the development of ammonia-induced brain edema in rats after portacaval anastomosis. Hepatology1999; 30: 876–880.
14.
LekeRBakLKAnkerMMeloTMSorensenMKeidingS. Detoxification of ammonia in mouse cortical GABAergic cell cultures increases neuronal oxidative metabolism and reveals an emerging role for release of glucose-derived alanine. Neurotox Res2011; 19: 496–510.
15.
DadsetanSBakLKSorensenMKeidingSVilstrupHOttP. Inhibition of glutamine synthesis induces glutamate dehydrogenase-dependent ammonia fixation into alanine in co-cultures of astrocytes and neurons. Neurochem Int2011; 59: 482–488.
16.
ZwingmannCChatauretNLeibfritzDButterworthRF. Selective increase of brain lactate synthesis in experimental acute liver failure: results of a [H-C] nuclear magnetic resonance study. Hepatology2003; 37: 420–428.
17.
ZaganasIWaagepetersenHSGeorgopoulosPSonnewaldUPlaitakisASchousboeA. Differential expression of glutamate dehydrogenase in cultured neurons and astrocytes from mouse cerebellum and cerebral cortex. J Neurosci Res2001; 66: 909–913.
18.
SchousboeAWestergaardNWaagepetersenHSLarssonOMBakkenIJSonnewaldU. Trafficking between glia and neurons of TCA cycle intermediates and related metabolites. Glia1997; 21: 99–105.
LowryOHRosebroughNJFarrALRandallRJ. Protein measurement with the Folin phenol reagent. J Biol Chem1951; 193: 265–275.
21.
LekeRBakLKSchousboeAWaagepetersenHS. Demonstration of neuron-glia transfer of precursors for GABA biosynthesis in a co-culture system of dissociated mouse cerebral cortex. Neurochem Res2008; 33: 2629–2635.
22.
HertzLJuurlinkBHJFosmarkHSchousboeA. Astrocytes in primary culture. In: PfeifferSE. (ed) Neuroscience Approached Through Cell Culture. CRC Press: Boca Raton, FL, USA, 1982,pp 175–186.
23.
MawhinneyTPRobinettRSAtalayAMadsonMA. Analysis of amino acids as their tert.-butyldimethylsilyl derivatives by gas-liquid chromatography and mass spectrometry. J Chromatogr1986; 358: 231–242.
24.
BiemannK. Mass spectrometry. In: BiemannK. (ed) Organic Chemistry Applications. McGraw: New York, NY, USA, 1962, pp 223–227.
25.
BerlSTakagakiGClarkeDDWaelschH. Metabolic compartments in vivo. Ammonia and glutamic acid metabolism in brain and liver. J Biol Chem1962; 237: 2562–2569.
26.
CooperAJMcDonaldJMGelbardASGledhillRFDuffyTE. The metabolic fate of 13N-labeled ammonia in rat brain. J Biol Chem1979; 254: 4982–4992.
27.
YudkoffMNissimIPleasureD. Astrocyte metabolism of [15N]glutamine: implications for the glutamine-glutamate cycle. J Neurochem1988; 51: 843–850.
28.
SonnewaldUWestergaardNJonesPTaylorABachelardHSSchousboeA. Metabolism of [U-13C5] glutamine in cultured astrocytes studied by NMR spectroscopy: first evidence of astrocytic pyruvate recycling. J Neurochem1996; 67: 2566–2572.
29.
SwainMButterworthRFBleiAT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology1992; 15: 449–453.
30.
ToftengFHauerbergJHansenBAPedersenCBJorgensenLLarsenFS. Persistent arterial hyperammonemia increases the concentration of glutamine and alanine in the brain and correlates with intracranial pressure in patients with fulminant hepatic failure. J Cereb Blood Flow Metab2006; 26: 21–27.
31.
SchmidtLEToftengFStraussGILarsenFS. Effect of treatment with the Molecular Adsorbents Recirculating System on arterial amino acid levels and cerebral amino acid metabolism in patients with hepatic encephalopathy. Scand J Gastroenterol2004; 39: 974–980.
32.
YudkoffMNissimIHertzL. Precursors of glutamic acid nitrogen in primary neuronal cultures: studies with 15N. Neurochem Res1990; 15: 1191–1196.
33.
SkyttDMKlawonnAMStridhMHPajeckaKQuintana-CabreraRBolanosJP. siRNA knock down of glutamate dehydrogenase in astrocytes affects glutamate metabolism leading to extensive accumulation of the neuroactive amino acids glutamate and aspartate. Neurochem Int2012; 61: 490–497.
34.
BenuckMSternFLajthaA. Transamination of amino acids in homogenates of rat brzain. J Neurochem1971; 18: 1555–1567.
35.
WestergaardNVarmingTPengLSonnewaldUHertzLSchousboeA. Uptake, release, and metabolism of alanine in neurons and astrocytes in primary cultures. J Neurosci Res1993; 35: 540–545.
ChatauretNZwingmannCRoseCLeibfritzDButterworthRF. Effects of hypothermia on brain glucose metabolism in acute liver failure: a H/C-nuclear magnetic resonance study. Gastroenterology2003; 125: 815–824.
38.
NorenbergMDBenderAS. Astrocyte swelling in liver failure: role of glutamine and benzodiazepines. Acta Neurochir Suppl (Wien)1994; 60: 24–27.
39.
JambekarAAPalmaENicolosiLRasolaAPetronilliVChiaraF. Aglutamine synthetase inhibitor increases survival and decreases cytokine response in a mouse model of acute liver failure. Liver Int2011; 31: 1209–1221.