
Abstract
Hyperammonemia causes glutamine accumulation and astrocyte swelling. Inhibition of glutamine synthesis reduces ammonia-induced edema formation and watery swelling in astrocyte processes. Ordinarily, astrocytes tightly control extracellular K+ activity [K+]e. We tested the hypothesis that acute hyperammonemia interferes with this tight regulation such that [K+]e increases and that inhibition of glutamine synthetase reduces this increase in [K+]e. Ion-sensitive microelectrodes were used to measure [K+]e in parietal cortex continuously over a 6-h period in anesthetized rats. After i.v. sodium acetate infusion in eight control rats, plasma ammonia concentration was 33 ± 26 μmol/L (± SD) and [K+]e remained stable at 4.3 ± 1.6 mmol/L. During ammonium acetate infusion in nine rats, plasma ammonia increased to 594 ± 124 μmol/L at 2 h and to 628 ± 135 μmol/L at 6 h. There was a gradual increase in [K+]e from 3.9 ± 0.7 to 6.8 ± 2.7 mmol/L at 2 h and 11.8 ± 6.7 mmol/L at 6 h. In eight rats, L-methionine-D,L-sulfoximine (150 mg/kg) was infused 3 h before ammonium acetate infusion to inhibit glutamine synthetase. At 2 and 6 h of ammonium acetate infusion, plasma ammonia concentration was 727 ± 228 and 845 ± 326 μmol/L, and [K+]e was 4.5 ± 1.9 and 6.1 ± 3.8 mmol/L, respectively. The [K+]e value at 6 h was significantly less than that obtained with ammonium acetate infusion alone but was not different from that obtained with sodium acetate infusion. We conclude that acute hyperammonemia impairs astrocytic control of [K+]e and that this impairment is linked to glutamine accumulation rather than ammonium ions per se.
Various forms of liver disease and congenital urea cycle disorders cause selective swelling of astrocytes (Bruton et al., 1970; Martinez, 1968; Zimmerman et al., 1981). Experimental hyperammonemia also causes selective astrocyte swelling (Diemer, 1978; Gibson et al., 1974; Voorhies et al., 1983). Plasma-labelled ammonia is rapidly taken up by brain (Cooper et al., 1979, 1985) and used in the amidation of glutamate by glutamine synthetase, an enzyme enriched in astrocytes (Norenberg and Martinez-Hernandez, 1979). With acute hyperammonemia, tissue glutamine concentration increases over a period of several hours (Bosman et al., 1990; Fitzpatrick et al., 1989), and watery astrocyte processes are observed at 6 h (Willard-Mack et al., 1996). We found that inhibition of glutamine synthetase with L-methionine-
Glutamine-dependent astrocyte swelling may lead to astrocyte dysfunction. A major function of astrocytes is tight regulation of extracellular potassium activity (Henn et al., 1972). Swelling may interfere with astrocyte K+ uptake capacity and with the ability to dissipate excess K+ through gap-junction networks (Robinson et al., 1993). In hippocampal slices exposed to ammonia, increases in extracellular K+ activity ([K+]e) were observed that may have passively contributed to alterations in postsynaptic potentials (Alger and Nicoll, 1983). Injection of ammonium acetate directly into the carotid artery of a cat was shown to increase [K+]e (Gronczewski and Leniger-Follert, 1984). However, [K+]e has not been well studied in vivo during hyperammonemia.
In the present study, we continuously measured [K+]e during 6 h of i.v. ammonium acetate infusion in anesthetized rats. We tested the hypothesis that increasing plasma ammonia concentration to approximately 600 to 700 μmol/L causes increased [K+]e, and that increases in [K+]e are attenuated by pretreatment with MSO.
MATERIALS AND METHODS
Surgical preparation
All procedures were approved by the institutional animal care and use committee. Twenty-five male Wistar rats ranging in weight from 350 to 450 g were anesthetized with an intraperitoneal dose of 65 mg/kg of sodium pentobarbital. Anesthesia was maintained by reptitive injections 15 mg/kg sodium pentobarbital every 1.5 h for the duration of the experiment. This dosing regimen prevents spontaneous limb movements without abolishing electroencephalographic activity. Rats were mechanically ventilated via a tracheotomy with approximately 30% O2. Catheters were inserted into the femoral artery for monitoring blood pressure and taking blood samples and into a femoral vein for infusion of salt solutions. Rectal temperature was maintained at about 37°C with a water-circulating blanket. After catheterization, pancronium bromide (0.1 mg/kg, i.v.) was administered.
For continuous recording during a 6-h period of sodium or ammonium acetate infusion, ion-sensitive electrodes were stabilized by securing them in a closed cranial window. A craniotomy was made over the right parietal cortex (diameter 2 × 3 mm). The hole was enclosed in a plastic ring (diameter 10 mm, depth 3 mm) that was fixed to the skull with acrylic and filled with warm (38°C) artificial CSF. A small slit was made in the dura. A coverslip was glued to the ring to seal the fluid-filled window. Using a microheat cautery, two small holes were made on the plastic cover slip. Through these holes, the K+ and reference microelectrodes were inserted 0.5 to 1.0 mm below the surface of cerebral cortex with micromanipulators. The coverslip holes were filled with glue to seal the window and thereby to minimize movement caused by cardiovascular and ventilatory pulsation and brain edema. The tip of the reference electrode was within 100 μm of the tip of the K+ electrode.
Extracellular K+ and direct potential measurements
K+-selective single barreled electrodes were fabricated using K+-selective resin (World Precision Instruments, Sarasota, FL, U.S.A.), backfilled with 500 mmol/L KC1 and connected to a preamplifier via a Ag-AgCl junction. Direct current (DC) measurements were made using single-barreled electrodes filled with 0.9% sodium chloride. Changes in potential were referred to a Ag-AgCl ground placed in the neck muscle. The K+-selective electrode and DC electrode were amplified (World Precision Instruments, model FD223 electrometer, Sarasota, FL, U.S.A.), sampled at 1 Hz, and digitally stored. [K+]e was calculated from the difference between the K+ electrode and the DC electrode potential. The electrodes were calibrated before and after each experiment using standard solutions containing 3.13, 6.25, 12.5, 25, 50, and 100 mmol/L KCl in 100 mmol/L sodium chloride.
Experimental protocol
Three groups were studied. Nonhyperammonemic controls (n = 8) received 200 mmol/L sodium acetate in water for 30 min followed by 200 mmol/L sodium acetate dissolved in 200 mmol/L hydrochloric acid (to reduce metabolic alkalemia) for 330 min. The infusion rate was regulated to deliver 50 μmol/min/kg for 360 min. In a hyperammonemic group (n = 9), rats were infused with 200 mmol/L ammonium acetate in water at a rate of 50 μmol/min/kg for 360 min. In a third group (n = 8), rats were pretreated with MSO (150 mg/kg in 5 ml saline; 0.83 mmol/kg; Sigma Chemical Co., St. Louis, MO, U.S.A.) as an i.v. infusion from 3 h to 2 h before starting a 6 h infusion of ammonium acetate (50 μmol/min/kg). This dose of MSO was found to inhibit glutamine synthetase activity measured in cortical homogenates by 64% and to prevent increases in cortical glutamine concentration during the 6-h infusion of ammonium acetate (Takahashi et al., 1991).
Arterial blood samples (1 ml) were drawn at baseline and at 2 h and 6 h of salt infusion. Ammonia concentration was measured in duplicate on 50 μl aliquots of plasma by a cation exchange-visible spectrophotometric technique previously described (Brusilow, 1991). Arterial blood gases and pH were measured with an ABL3 electrode system (Radiometer, Copenhage, Denmark). Arterial hemoglobin concentration was measured with a Radiometer OSM3 hemoximeter. Arterial glucose concentration was measured with a Yellow Springs Glucose Analyzer (model 2300A; Yellow Springs, OH, U.S.A.).
Data analysis
Values are expressed as mean ± SD. Data were subjected to two-way analysis of variance (ANOVA) among the three groups, with measurements repeated over time as a within-subject factor and treatment as a between subject factor. At individual time points, mean values were contrasted among groups by the Newman–Keuls multiple range test at the 0.05 significance level. Because the SD increased with the mean value for [K+]e, these data were logarithmically transformed.
RESULTS
Ammonium acetate infusion increased plasma ammonia concentration to a similar extent with and without MSO pretreatment (Table 1). Other than an increase in arterial pH at 6 h of sodium acetate plus HC1 infusion, there were no differences among groups in arterial blood measurements. Mean arterial blood pressure decreased from baseline during ammonium acetate infusion, but there were no differences from values in the sodium acetate group at any time point (Table 2). With MSO pretreatment, arterial pressure was lower than values in the other two groups during the first 3 h of salt infusion.
Arterial blood analysis
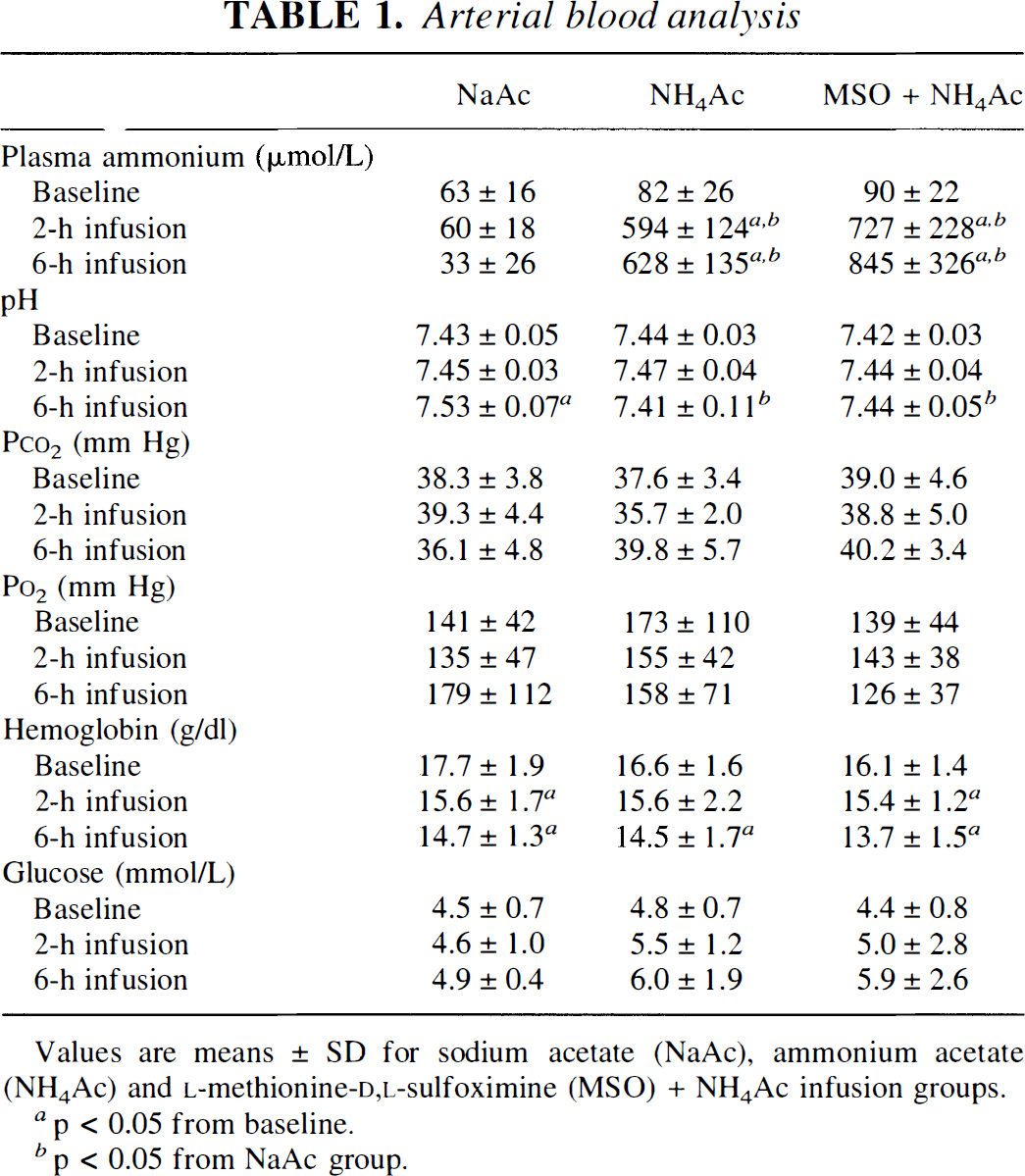
Values are means ± SD for sodium acetate (NaAc), ammonium acetate (NH4Ac) and L-methionine-D,L-sulfoximine (MSO) + NH4Ac infusion groups.
p < 0.05 from baseline.
p < 0.05 from NaAc group.
Mean arterial blood pressure
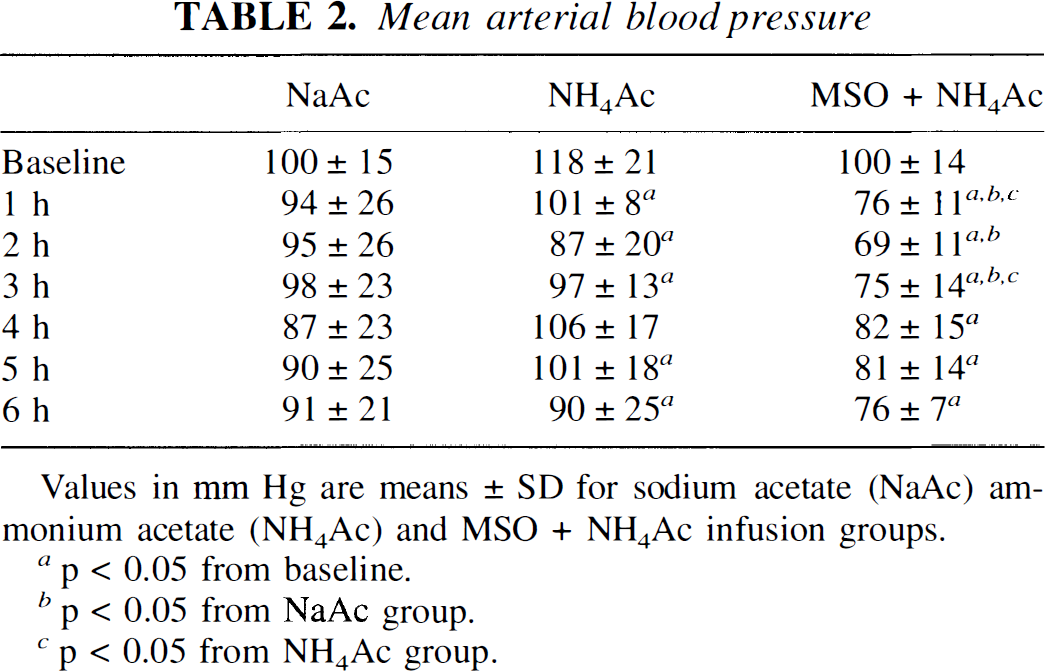
Values in mm Hg are means ± SD for sodium acetate (NaAc) ammonium acetate (NH4Ac) and MSO + NH4Ac infusion groups.
p < 0.05 from baseline.
p < 0.05 from NaAc group.
p < 0.05 from NH4Ac group.
With sodium acetate infusion, recordings of [K+]e remained reasonably stable over a 6-h period (Fig. 1). With ammonium acetate infusion, [K+]e increased in a gradual fashion. In some rats, [K+]e began to increase within the first hour, as shown in Fig. 1. In others, obvious increases were delayed until the second or third hour of infusion. None of the rats showed abrupt increases typical of anoxic depolarization. With MSO treatment, there was little change in [K+]e during the 6-h ammonium acetate infusion (Fig. 1).
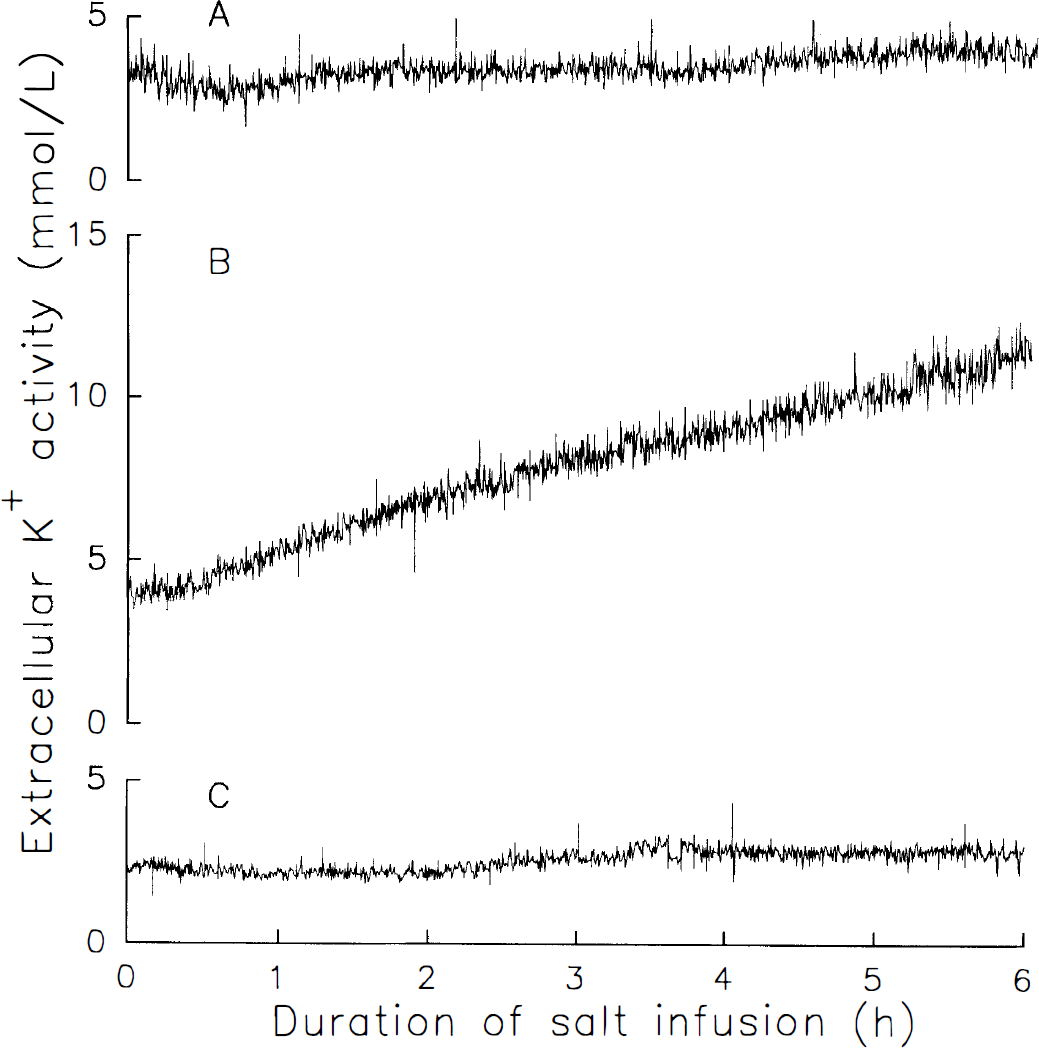
Representative recordings of extracellular K+ activity in parietal cortex in rats during 6 h i.v. infusion of sodium acetate (
Group data are shown in Fig. 2. Two-way ANOVA indicated a significant interaction of treatment with time. Within group analysis indicated a significant increase in [K+]e by 1 h of ammonium acetate infusion, whereas there was no effect of time in the sodium acetate group or in the group pretreated with MSO. Between group analysis indicated that [K+]e in the ammonium acetate group exceeded [K+]e in the sodium acetate group by 3 h, and exceeded [K+]e in the MSO plus ammonium acetate group by 5 h. Three of eight rats in the MSO group showed a moderate increase in [K+]e, but group data were not significantly different from sodium acetate controls. The values of [K+]e at six hours were 4.3 ± 1.6, 11.8 ± 6.7 and 6.1 ± 3.8 mmol/L in the sodium acetate, ammonium acetate, and MSO plus ammonium acetate groups, respectively.
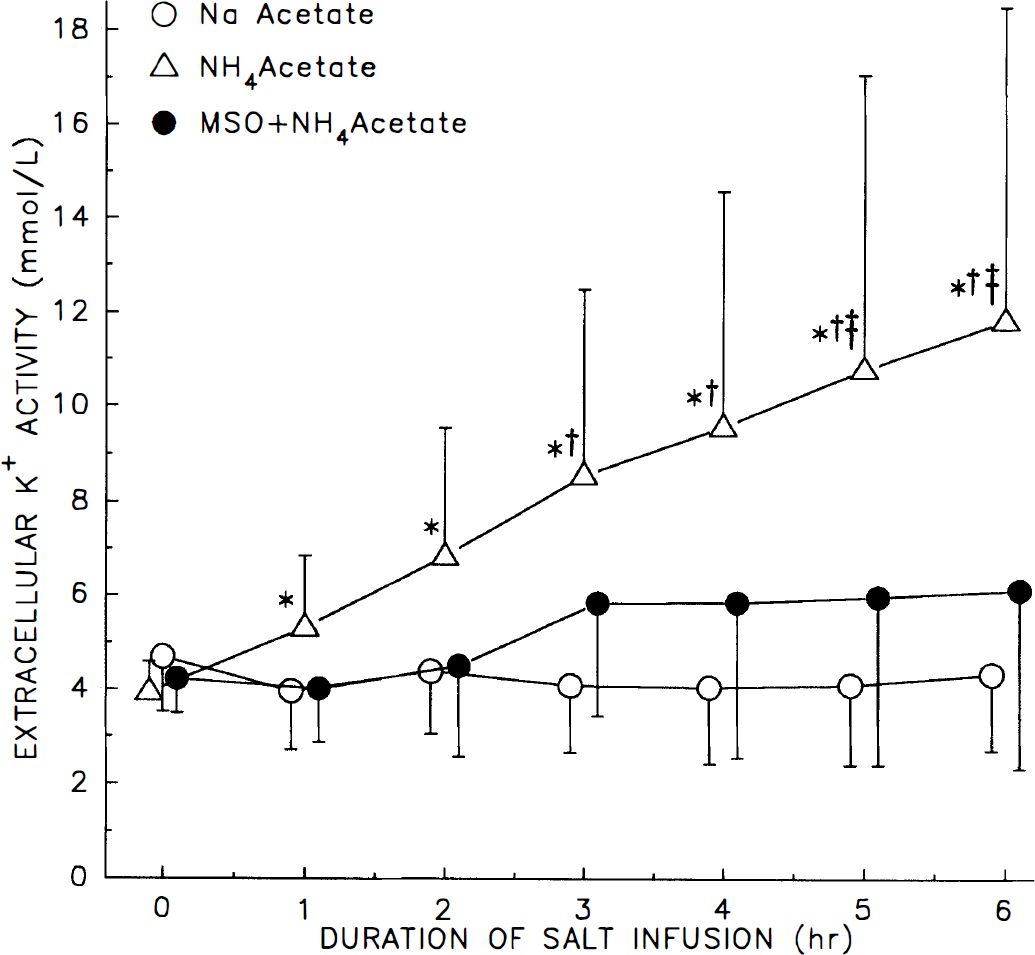
Cortical extracellular K+ activity (± SD) in groups of rats during intravenous infusion of sodium acetate (n = 8), ammonium acetate (n = 9), and ammonium acetate after MSO pretreatment (n = 8). *, p < 0.05 from baseline value within group; †, p < 0.05 from sodium acetate group at that time; ‡, p < 0.05 from MSO + ammonium acetate group at that time.
DISCUSSION
The major findings of this study are that acute increases in plasma ammonia to approximately 600 to 700 μmol/L results in progressive increases in [K+]e over a 6-h period in anesthetized rats, and that inhibition of glutamine synthetase with MSO largely attenuates this increase in [K+]e.
There were no differences in arterial PO2, hemoglobin concentration, or arterial blood pressure among the groups that could account for differences in [K+]e. Hyperammonemia does not substantially decrease cerebral blood flow in this model (Hirata et al., 1996; Takahashi et al., 1992). Although hyperammonemia produced a moderate decrease in arterial pressure in the present study, we previously showed that cerebral blood flow was well-maintained down to arterial pressures of 48 mmHg (Hirata et al., 1995). Thus, the increase in [K+]e during hyperammonemia is unlikely to be attributable to anoxia.
Using the same doses of MSO and ammonium acetate in this model, we previously showed that increased cortical glutamine and water content (Takahashi et al., 1991), increased intracranial pressure (Takahashi et al., 1992) and watery swelling of astrocyte processes (Willard-Mack et al., 1996) at 6 h of hyperammonemia were inhibited by MSO. One explanation of these results is that the 13 mmol/kg increase in tissue glutamine concentration seen in this model (Hirata et al., 1995; Takahashi et al., 1991) constitutes a significant osmotic load if the increase is concentrated in the astrocyte compartment. Astrocytes have a high K+ conductance and have long been thought to be important in controlling [K+]e (Henn et al., 1972). Our results suggest that glutamine-dependent swelling impairs the ability of the astrocyte to control [K+]e.
The present data do not distinguish whether the increase in [K+]e is attributable to a net shift of K+ out of astrocytes or neurons. Benjamin et al. (1978) observed a decrease in K+ content of neocortical slices after a 1-h exposure to ammonium chloride, leading Szerb and Butterworth (1992) to speculate that ammonium ions decrease K+ content in neurons. Potassium channels are permeable to ammonium ions (Hille, 1992); however, permeability to ammonium ions is approximately an order of magnitude less than K+ permeability when both ions are present in similar concentrations (Blatz and Magleby, 1984; Hille, 1973). Hindfelt (1975) estimated intracellular [NH4+] to be about twice that of plasma [NH4+]. At a plasma [NH4+] of 628 μmol/L in the present study, intracellular NH4+ is predicted to be about 1.3 mmol/L, a value that is lower than intracellular [K+] by at least two orders of magnitude and thus is expected to exert a negligible amount of competition with intracellular [K+] for K+ efflux through K+ channels. The control value of extracellular [NH4+] is reported to be 116 μmol/L (Kraig and Cooper, 1987), which is probably intermediate between plasma and intracellular concentrations (Hindfelt, 1972, 1974). Assuming that extracellular [NH4+] remains intermediate during hyperammonemia (0.6–1.3 mmol/L), then extracellular [NH4+] may be less than an order of magnitude lower than extracellular [K+] and could compete with extracellular [K+] for K+ influx through K+ channels. Selective inhibition of K+ influx by NH4+, although probably not large as a percent of total flux, could conceivably cause accumulation of K+ over a period of hours, particularly considering that the volume of the interstitial fluid space is nearly an order of magnitude less than the intracellular fluid volume. Thus, ammonium ions may act to impede K+ influx more than K+ efflux and lead to a gradual increase in [K+]e. Inhibition of K+ influx may be more prominent in neurons than in astrocytes. Temporary inhibition of a small fraction of K+ channels by NH4+ may not significantly reduce K+ influx in astrocytes because total K+ conductance is high in astrocytes.
An increase in [K+]e ordinarily produces a large increase in astrocyte K+ content. For example, increasing [K+]e in astrocyte culture medium from 3 to 12 mmol/L (i.e., values comparable to that currently obtained during hyperammonemia) caused a doubling of intracellular K+ content without a counterbalancing loss of Na+ (Walz and Hertz, 1983). If a similar K+ influx occurs in astrocytes during hyperammonemia, the normal dissipative K+ routes through the purported network of astrocyte gap junctions (Robinson et al., 1993) could become overloaded, thereby exacerbating glutamine-dependent astrocyte swelling. Thus, as [K+]e begins to increase during hyperammonemia, a significant amount of K+ may have already been translocated from neurons to astrocytes, and further increases in [K+]e may begin to saturate the K+ buffering capacity of astrocytes. When glutamine synthesis is inhibited, the K+ buffering capacity apparently does not become saturated, possibly because K+ can be effectively dissipated through astrocyte gap junctions and exported back to neurons or into blood when astrocytes are not swollen by glutamine.
An increase in [K+]e may also affect glutamine accumulation. In cultured mouse astrocytes, increasing the pH of the medium increased intracellular pH and glutamine content without substantially accelerating glutamine efflux (Brookes 1992, 1993). The increase in glutamine at pH 7.8 was inhibited by MSO (Brookes, 1992). Increasing K+ in the medium from 3 to 12 mmol/L caused an increase in intracellular pH and glutamine content (Brookes and Turner, 1993). The increase in pH may be secondary to K+-induced depolarization. Moderate depolarization and increases in intracellular pH have been reported to occur in astrocytes in vivo after portacaval shunting (Swain et al., 1991). Therefore, another potential effect of increased [K+]e in hyperammonemia is to depolarize astrocytes partially, increase their intracellular pH, and augment glutamine accumulation.
An increase in [K+]e is likely to affect neighboring neurons by altering excitability. Decreased synaptic transmission by ammonium ions has been well known for many years (Binstock and Lecar, 1969; Lux, 1971; Meyer and Lux, 1974; Raabe and Gumnit, 1975) and has been attributed both to a decreased capacity of neurotransmitter release (Hamberger et al., 1979; Theoret et al., 1985) and to a postsynaptic action (Fan et al., 1990). Postsynaptic effects may be secondary, in part, to a decrease in membrane potential (Alger and Nicoll, 1983; Raabe, 1989; Szerb and Butterworth, 1992); however, most of these studies on decreased neurotransmission used ammonium concentrations in the 1 to 10 mmol/L range. Our results indicate that plasma ammonium concentrations of approximately 600 μmol/L are capable of increasing [K+]e threefold. A threefold increase in [K+]e would be expected to decrease the resting membrane potential in neurons and account, at least in part, for some of the observed inhibition of synaptic transmission. Because the increase in [K+]e was inhibited by MSO, astrocyte dysfunction needs to be considered when interpreting electrophysiological alterations in neurons with clinically relevant ammonium concentrations. With short exposure to higher ammonium concentrations, other electrophysiological properties of ammonium ions may come into play, but the clinical relevance of extremely high ammonium concentrations is uncertain.
In addition to effects on neurons, an increase in [K+]e can affect cerebral vessels. Potassium conductance in perivascular endfeet of astrocytes may be exceptionally high (Newman, 1986), and postulated siphoning of K+ through endfeet processes may alter vascular regulation (Paulson and Newman, 1987). Moderate increases in [K+]e can cause dilation of pial arterioles (Kuschinsky et al., 1972) and may contribute to the increase in pial arteriolar diameter seen in this model of hyperammonemia (Hirata et al., 1996). Perivascular endfeet compression of capillaries and venules may offset dilation of upstream arterioles and account for the lack of increase in blood flow in this model (Takahashi et al., 1992; Hirata et al., 1996). Thus, increased [K+]e may help maintain cerebral blood flow in the face of astrocyte edema.
Glutamine synthetase is often considered to be the major detoxifying pathway for ammonia; however, others have shown that MSO reduces mortality with high doses of ammonium salts (Warren and Schenker, 1964; Hindfelt and Plum, 1975), inhibits the increase in brain uptake of tryptophan, leucine, and phenylalanine seen during hyperammonemia (Jonung et al., 1984, 1985) and attenuates the decrease in cerebral glucose consumption, the increase in tryptophan uptake, and the increase in intracranial pressure seen acutely after portacaval shunting (Blei et al., 1994; Hawkins et al., 1993). These results, together with our previous work showing that MSO prevents edema, intracranial hypertension, astrocyte swelling, and impaired cerebrovascular CO2 reactivity during acute hyperammonemia (Hirata et al., 1996; Takahashi et al., 1991, 1992; Willard-Mack et al., 1996), emphasize the importance of glutamine accumulation in the pathophysiology of hyperammonemia. Therefore, the present results demonstrating that MSO inhibits [K+]e dysregulation during acute hyperammonemia contributes to the mounting evidence that ammonia-induced alterations in glutamine metabolism are responsible for some of the pathophysiological features of hyperammonemic encephalopathy.
Footnotes
Acknowledgment:
We thank Ellen Gordes for technical assistance and Lisa DeLoriers for assistance in preparing the manuscript. This work was supported by a grant from the National Institutes of Health NS25275.