
Abstract
Leptin, one of the most important adipokines, is not only an energy regulator but also a regulator of innate immunity. Inflammation plays a key role in the tissue damage after intracerebral hemorrhage (ICH), and we sought to investigate whether leptin has a detrimental effect on ICH. After the injection of a high replacement dose (0.04 mg/kg) and two pharmacologic doses (4 and 8 mg/kg) of leptin, brain water contents increased significantly compared with that of control mice (
INTRODUCTION
Intracerebral hemorrhage (ICH), which accounts for ~15% of all strokes, has a higher incidence in the Hispanic, black, and Asian populations. 1 It is the most devastating and least treatable subtype of stroke, causing severe disability and a high rate of acute mortality. Intracerebral hemorrhage volume is itself a key factor in brain tissue damage. 2 However, secondary brain injury related to hemorrhage is also important,3,4 which is reflected by the edema surrounding the hematoma, and has been a critical therapeutic target for ICH.
Leptin, one of the most important adipokines, has been established as a key regulator of energy balance and body weight. 5 Clinical observations have indicated that hyperleptinemia is associated with risk of cardiovascular diseases6,7 and is more closely related to hemorrhagic stroke than ischemic stroke. 7 Recent clinical observations showed that endogenous leptin levels increased after ICH in humans, and that elevated leptin levels were associated with poor outcomes after ICH.8,9 However, experimental evidence for this relationship is lacking, and the effects of leptin after ICH have never been elucidated. Leptin is not only an energy regulator but also a regulator of innate immunity. 10 Leptin promotes phagocytosis by monocytes/macrophages and their secretion of proinflammatory cytokines.11,12 Inflammation after ICH can cause disruption of the blood–brain barrier, 13 elicit brain edema, 14 and contribute to cell death after hemorrhage via cytotoxic mediators.15,16 In this context, we have focused on leptin and inflammation after ICH, and brain edema is used as an important marker of brain injury related to inflammation. We sought to investigate whether leptin has a detrimental effect on ICH in terms of post-ICH inflammation. First, to evaluate the direct effects of leptin, exogenous leptin was injected into a mouse ICH model, and brain edema surrounding the hematoma and neurologic deficits were observed. Second, to investigate the effects of leptin deficiency after ICH, we induced ICH in leptin-deficient knockout mice. Finally, we investigated the intracellular messengers of leptin using a specific inhibitor for a candidate messenger.
MATERIALS AND METHODS
Animals and Experimental Groups
The study was performed in two stages. First, to examine if exogenous leptin influences the progression of ICH, male imprinting control regions mice (Koatech, Seoul, Republic of Korea) were divided into a leptin-injected group and a control group. Second, to determine the effects of leptin deficiency on ICH, male C57BL/6J strain
Induction of Intracerebral Hemorrhage and the Injection of Leptin
Experimental ICH was induced by the stereotaxic, intrastriatal administration of bacterial collagenase type IV (Sigma, St Louis, MO, USA). After inhalation anesthesia using 3% isoflurane in 30% oxygen and 70% air, the mice were placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA). A burr hole was made, and a 30-gauge Hamilton syringe needle was inserted into the striatum (location: 0.19 mm left lateral to the midline, 0.06 mm posterior to the bregma, 0.4 mm in depth below the skull). Collagenase type IV (0.23 U in 1 μL saline, Sigma) was administered over a 5-minute period. After infusion, the burr hole was sealed with bone wax, the wound was sutured, and the mice were allowed to recover. Physiological parameters, including mean arterial blood pressure, blood gases, and glucose concentration, were measured during the experiment. During the recovery period, the mice were assessed for forelimb flexion and contralateral circling to confirm the procedures. Seizure events were not observed during the experiments. Rectal temperature was maintained at 37°C ± 0.5°C with the use of a thermistor-controlled heating blanket. Free access to food and water was allowed after recovery from anesthesia.
We administered mouse recombinant leptin with replacement doses (0.01 and 0.04 mg/kg, Sigma) and pharmacologic doses (4 and 8 mg/kg) or phosphate-buffered saline (PBS) intraperitoneally just before the induction of ICH. The doses of leptin were determined on the basis of a previous experiment in a rodent stroke model 17 and a leptin replacement study in leptin-deficient adults. 18
Analysis of Brain Water Content
We analyzed brain water content 72 hours after ICH, when brain injury is maximal.
19
The brain was divided into two hemispheres along the midline, and the cerebellum and brainstem were removed. The brain samples were immediately weighed on an electronic analytical balance to obtain the wet weight and were dried in a gravity oven at 100°C for 24 hours to obtain the dry weight. Water content was expressed as a percentage of wet weight using the following formula: (wet weight – dry weight)/(wet weight) × 100 (Song
Measurement of Hemorrhage Volume
Seventy-two hours after ICH, the brains were cut coronally through the needle entry site (identifiable on the brain surface), and serial slices (1-mm thickness) both anterior and posterior to the needle entry site were obtained. Digital photographs of the serial slices were taken, and hemorrhage volume was measured using an image analysis program, Image J (National Institutes of Health, Bethesda, MD, USA). The total hemorrhage volume (mm3) was calculated by summing the clot area in each section and multiplying by the distance between sections.20,21
Behavior Testing
Behavior testing (
Enzyme-Linked Immunosorbent Assay for Measuring Leptin Levels in Serum and Brain
Using a commercially available enzyme-linked immunosorbent assay kit for measuring leptin levels (ALPCO Diagnostics, Salem, NH, USA), we checked leptin levels of serum and brain after ICH without meal. Before induction of ICH, baseline leptin levels in fasting states were measured, and after induction of ICH, leptin levels were checked at 10 minutes and 1 day in fasting states without meal (Figure 1A).
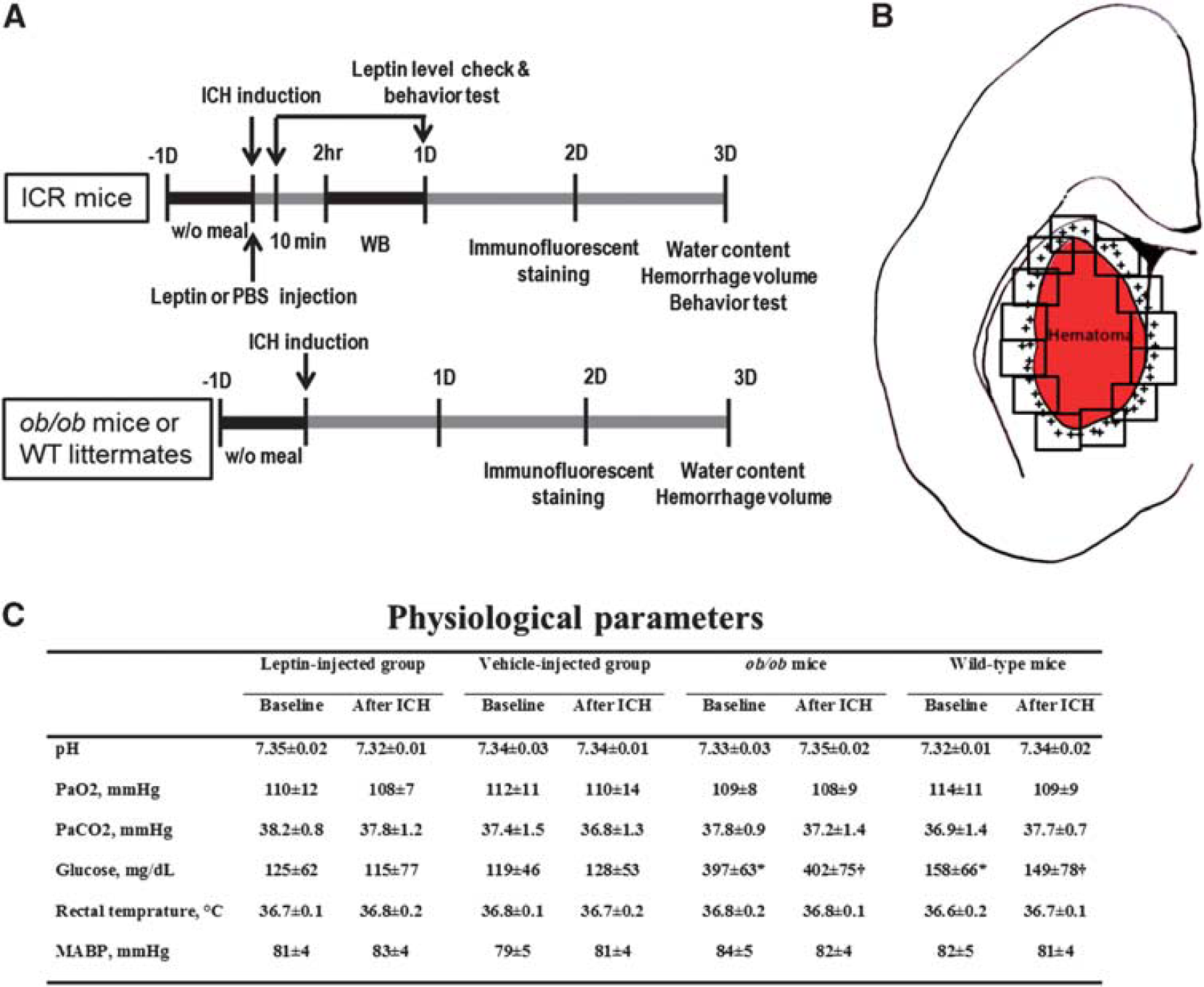
Schematic diagram of the study protocols and regions of cell quantification. (
Immunofluorescent Staining and Cell Quantification
Immunofluorescent staining of brain tissue was performed using cryopreserved 40-μm coronal sections. Each section was incubated with 0.5% bovine serum albumin/0.3% Triton-X followed by 10% normal serum in PBS for 1 hour for blocking. Sections with a primary antibody were placed at 4°C for 16 hours. After washing, each section was subsequently incubated for 2 hours at room temperature with the fluorophore-conjugated secondary antibody. The following primary antibodies were used: monoclonal antibodies against MHC class II Ia (Ox6; Santa Cruz Biotech., Santa Cruz, CA, USA) to label activated microglia/macrophages; MPO (myeloperoxidase; DAKO, Carpinteria, CA, USA) to stain neutrophils; NeuN (neuronal nuclei antigen; Chemicon, Temecula, CA, USA) to label mature neurons; GFAP (glial fibrillary acidic protein; Chemicon) to label astrocytes; and phosphorylated signal transduction and activator of transcription 3 (pSTAT3; Cell Signaling Tech., Danvers, MA, USA). Cell nuclei were visualized with 4,6-diaminodino-2-phenylindole staining. Stained cells were then examined under a confocal laser scanning biological microscope (LSM 410 META; Carl Zeiss, Jena, Germany). To analyze the expression of leptin in the brain, we performed immunohistochemistry assay using anti-leptin antibody (Abcam, Cambridge, UK) in hemorrhagic brain with or without leptin injection, or in nonhemorrhagic brain.
Quantitative analysis of the positively stained cells was performed in the perihematomal regions by two independent investigators (W-SR, BJK) who were masked to the group allocation. To count activated microglia/macrophages and neutrophils, 16 high-power fields were taken from the section staining through the center of the ICH lesion (Figure 1B) 48 hours after ICH, when the intensity of inflammation is maximal. 23 Total counts in the measured sections were converted into cell densities for comparison between the ICH groups according to our established protocol. 24 In addition, to locate the pSTAT3 site of action, we performed double-immunofluorescence staining with antibodies for pSTAT3 and cell type-specific markers (Ox6, NeuN, and GFAP).
Western Blot Analysis
The mice were killed via decapitation, and the brains were immediately extracted 2 or 24 hours after the induction of ICH. After the centrifugation of hemisphere homogenates, 50 μg of protein was separated on a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel and transferred to nitrocellulose membranes. These membranes were incubated in blocking buffer (5% skim milk in 50 mmol/L Tris pH 7.5, 0.15 mmol/L NaCl, 0.05% Tween-20), and the blots were probed with antibodies recognizing STAT3, pSTAT3 (Cell Signaling Tech.), extracellular signal-regulated kinase (ERK; Santa Cruz Biotech.), pERK (Cell Signaling Tech.), Akt (Santa Cruz Biotech.), pAkt (Cell Signaling Tech.), cyclooxygenase-2 (COX-2; BD Biosciences, Franklin Lakes, NJ, USA), and inducible nitric oxide synthase (iNOS; NOVUS Biologicals, Littleton, CO, USA). Immunoreactivity was visualized by enhanced chemiluminescence, and the relative optical densities were determined by comparison of the measured values with the mean values of the control group.
Inhibition of Phosphorylated Signal Transduction and Activator of Transcription 3
The pSTAT3 inhibitor NSC74859 (Merck, Darmstadt, Germany), dissolved in 50% dimethyl sulfoxide and 50% PBS in a 0.2-mL volume, was administered intraperitoneally after the injection of leptin in the leptin-injected group. To investigate the effect of NSC74859 in the absence of exogenous leptin, NSC74859 was also administered to the control group at the same time. The pSTAT3-inhibiting effect of NSC74859 was analyzed using Western blot analysis.
Statistical Analysis
The values are presented as the mean ± s.d. Data were analyzed with the unpaired Student's
RESULTS
Physiological Parameters and Leptin Levels After Intracerebral Hemorrhage
The physiological parameters, including mean arterial blood pressure, blood gases, serum glucose, and body temperature, were similar between the leptin-injected and control groups (Figure 1C). The glucose level and body weight were higher in
Compared with leptin levels in serum and brain in fasting state, leptin increased significantly 1 day after ICH in serum and brain without meal (208 ± 38 versus 32 ± 16 pg/mL in serum, and 167 ± 7 versus 116 ± 6 pg/mL in brain,
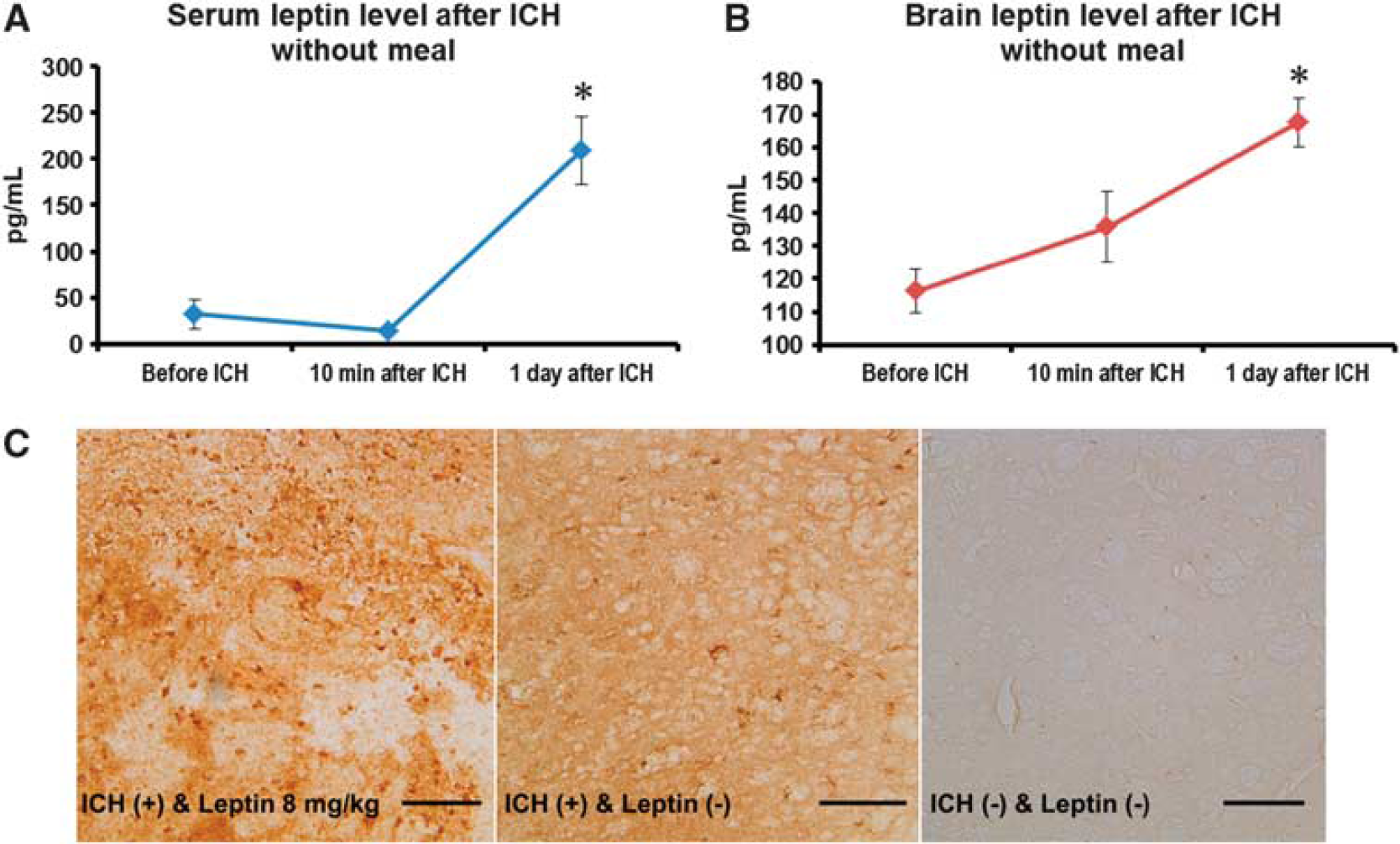
Leptin levels after intracerebral hemorrhage (ICH). (
Leptin Increases Brain Water Content and Neurological Deficits after Intracerebral Hemorrhage
To investigate the dose-dependent effects of leptin, we compared the results of water content at replacement doses (0.01 and 0.04 mg/kg) and pharmacologic doses of leptin (4 and 8 mg/kg), and then determined an optimal dose for this study protocol. We replicated the same procedures in leptin-deficient
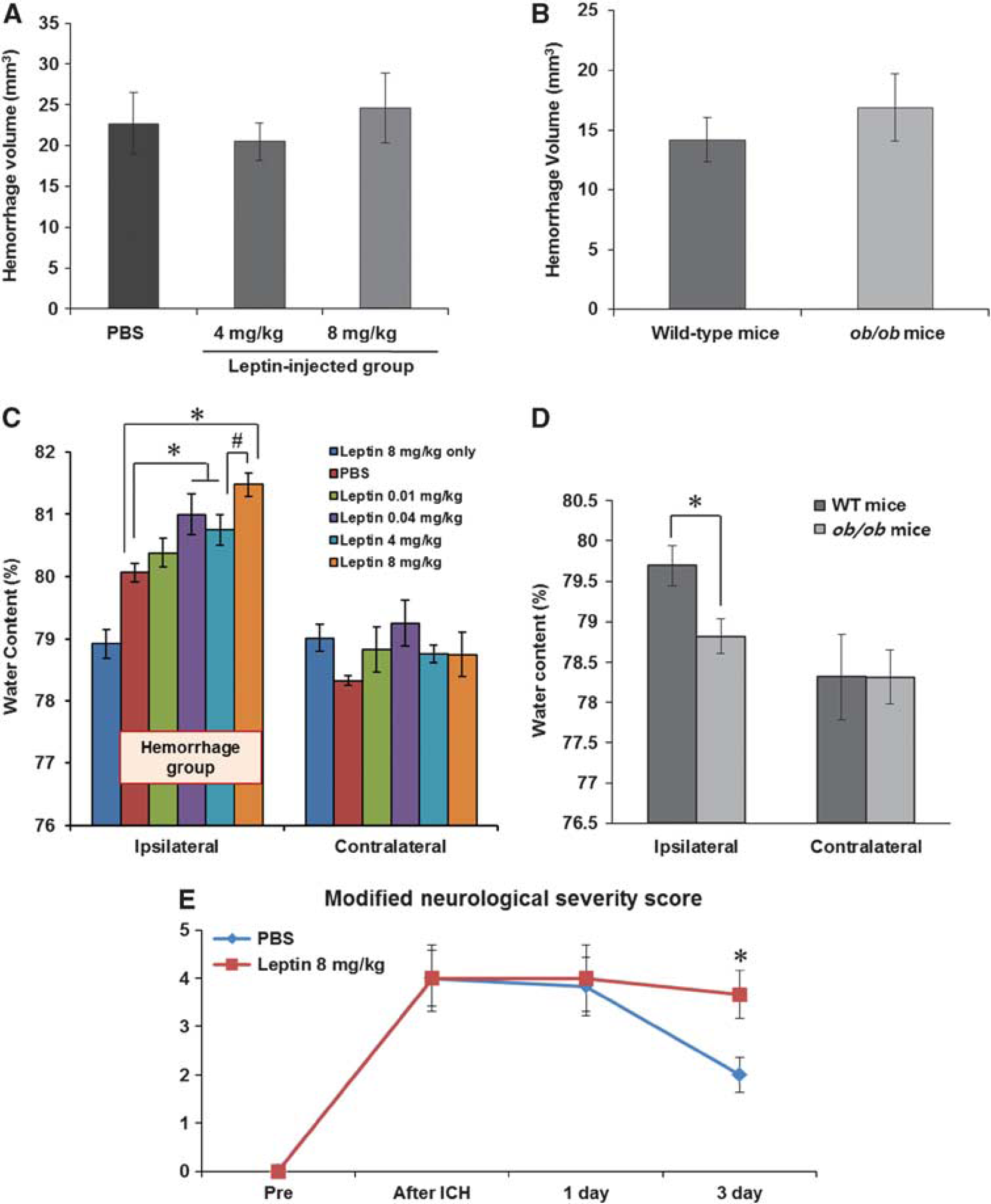
Measurements of brain water content, hemorrhage volume, and neurologic deficits. (
Leptin injection aggravated neurologic deficits in ICH injury (Figure 3E). In all mice, the mNSS was 0 before the ICH, indicating normal neurologic function. In the control group, the mNSS was peaked at 4 ± 1.4 at 30 minutes after ICH and decreased to 2 ± 0.9 on day 3. In the leptin-injected group, the mNSS was not reduced on days 1 and 3 compared with 30 minutes after ICH (4 ± 1.7 at 30 minutes, 4 ± 1.7 on 1 day, and 3.7 ± 1.2 on day 3), and was significantly higher than that of control group on day 3 after ICH (
Leptin Increases the Number of Inflammatory Cells in the Perihematomal Area
Ox6-stained microglia/macrophages were frequently found in the periphery around the lesion, and the density in the leptin-injected group was higher than that in the control group (Figure 4A). In quantitative analysis, the leptin-injected group exhibited a higher number of Ox6-positive cells (128.9 ± 13.1 versus 88.0 ± 20.7 cells/mm2,
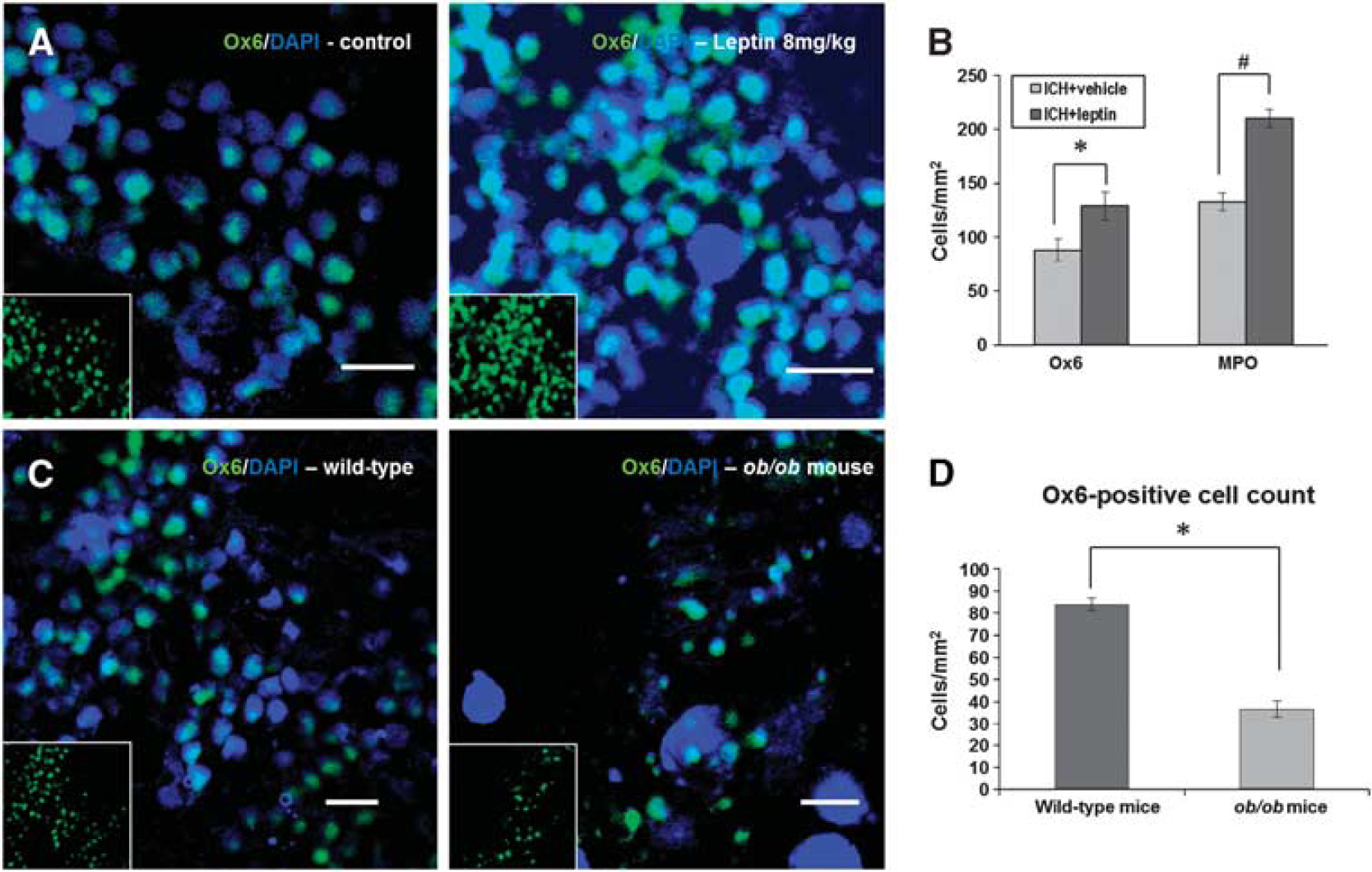
Immunofluorescent staining of inflammatory cells. (
The Signal Transduction and Activator of Transcription 3 Signaling Pathway Plays a Critical Role in Inflammatory Cells
Phosphorylated STAT3 was minimally detected 2 hours after ICH, but at 24 hours, a 1.8-fold increase in pSTAT3 density was detected in the leptin-injected group compared with the control group, and the density in the hemorrhagic hemisphere was also higher than that in the nonhemorrhagic hemisphere (Figure 5B;
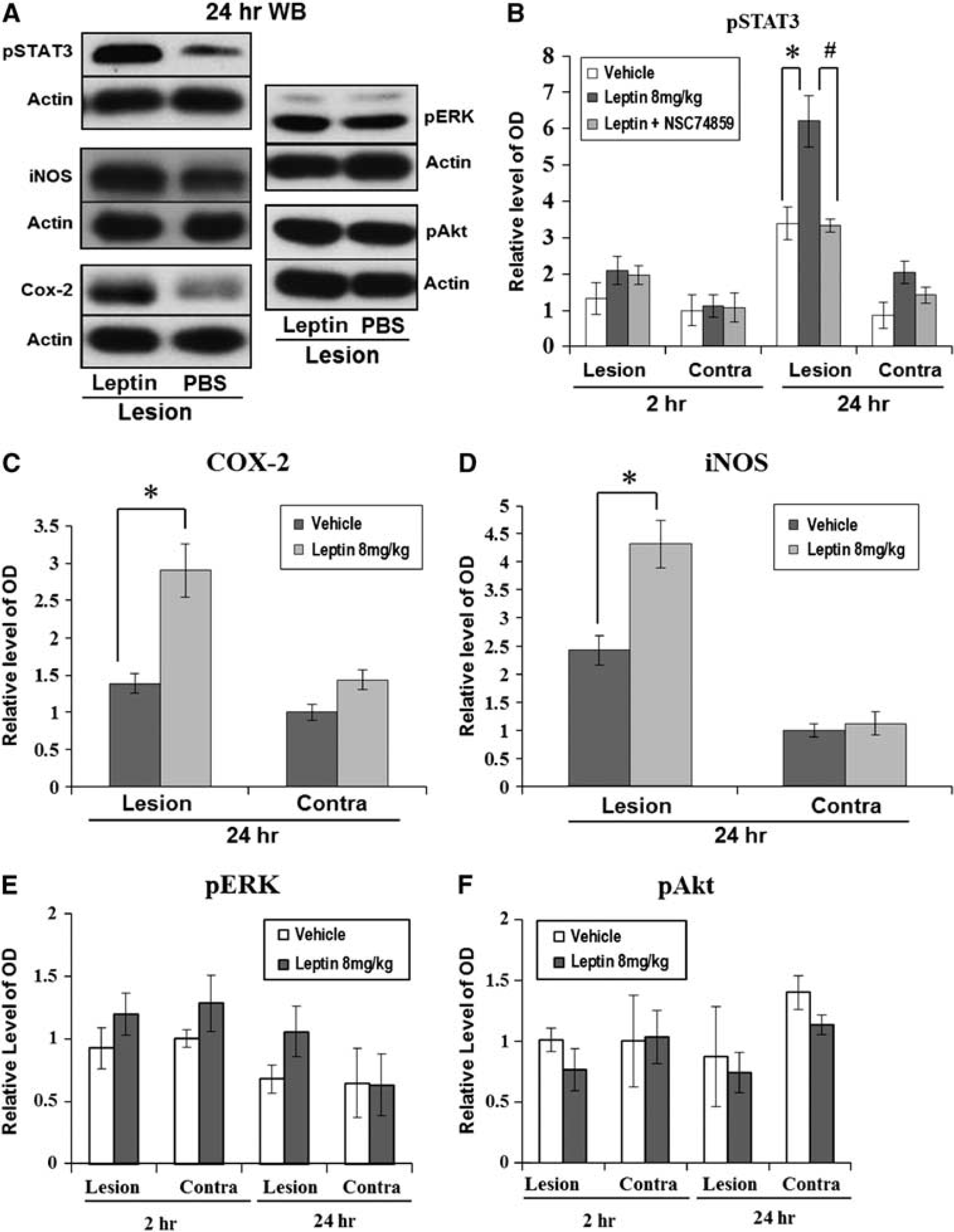
Western blot analyses for signal transduction and activator of transcription 3 (STAT3), extracellular signal-regulated kinase (ERK), Akt, and cyclooxygenase-2 (COX-2). (
After the inhibition of pSTAT3 using NSC74859 in the leptin-injected group, the brain water content decreased to the level of the control group (Figure 6A; leptin + NSC74859: 79.8% ± 0.8% versus leptin-only group: 81.5% ± 0.5%,
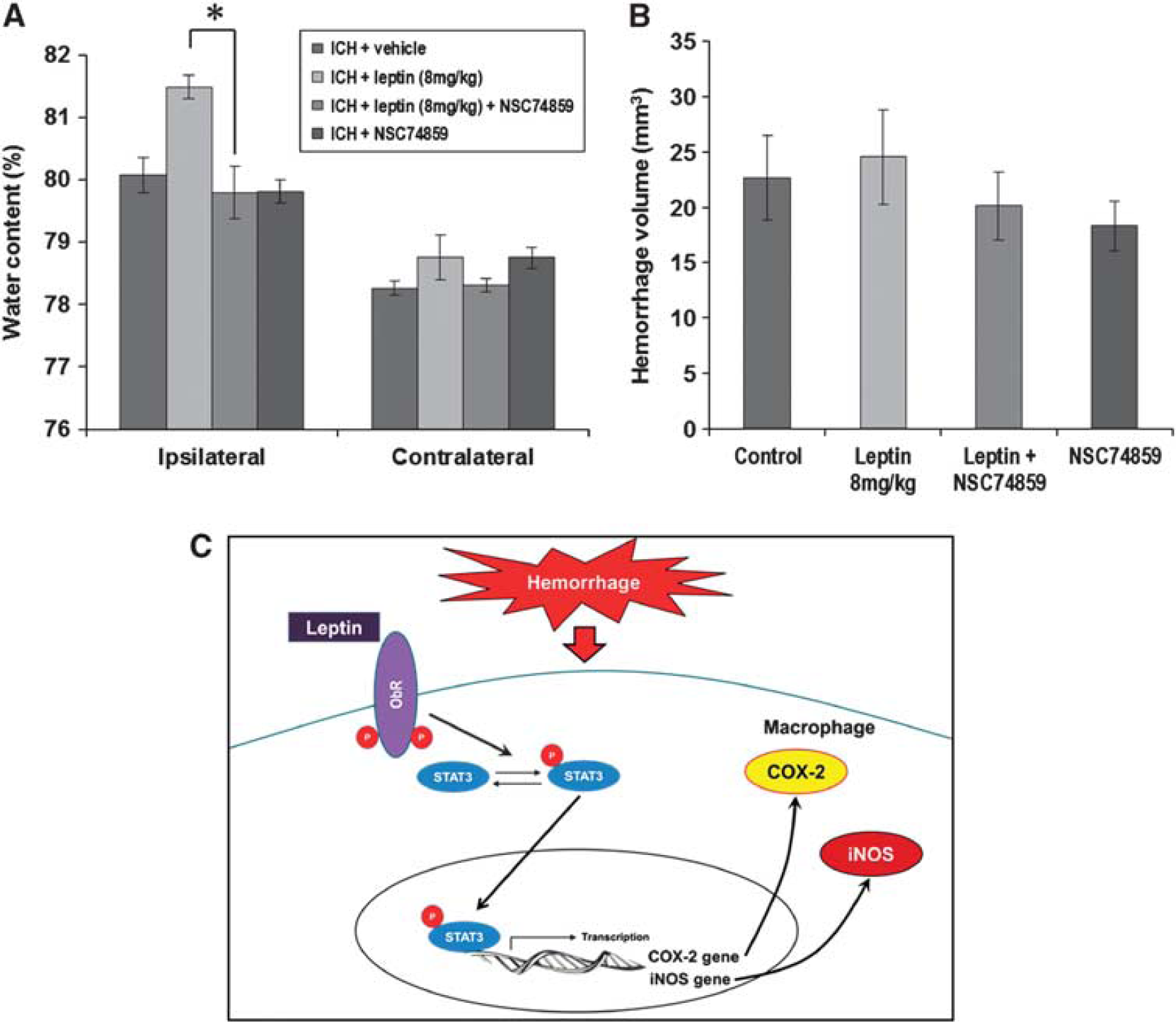
Changes in brain water content and hemorrhage volume after the injection of a phosphorylated signal transduction and activator of transcription (pSTAT) inhibitor (NSC74859). (
Ox6-positive microglia/macrophages were stained by pSTAT3 in nucleus or cytosol (Figures 7A and 7B), and we confirmed the identity of double-stained cells with three-dimensional analysis (Figures 7C and 7D). However, the cells labeled by pSTAT3 were not stained by NeuN or GFAP in the periphery around a hemorrhagic lesion (Figures 7E and 7F).
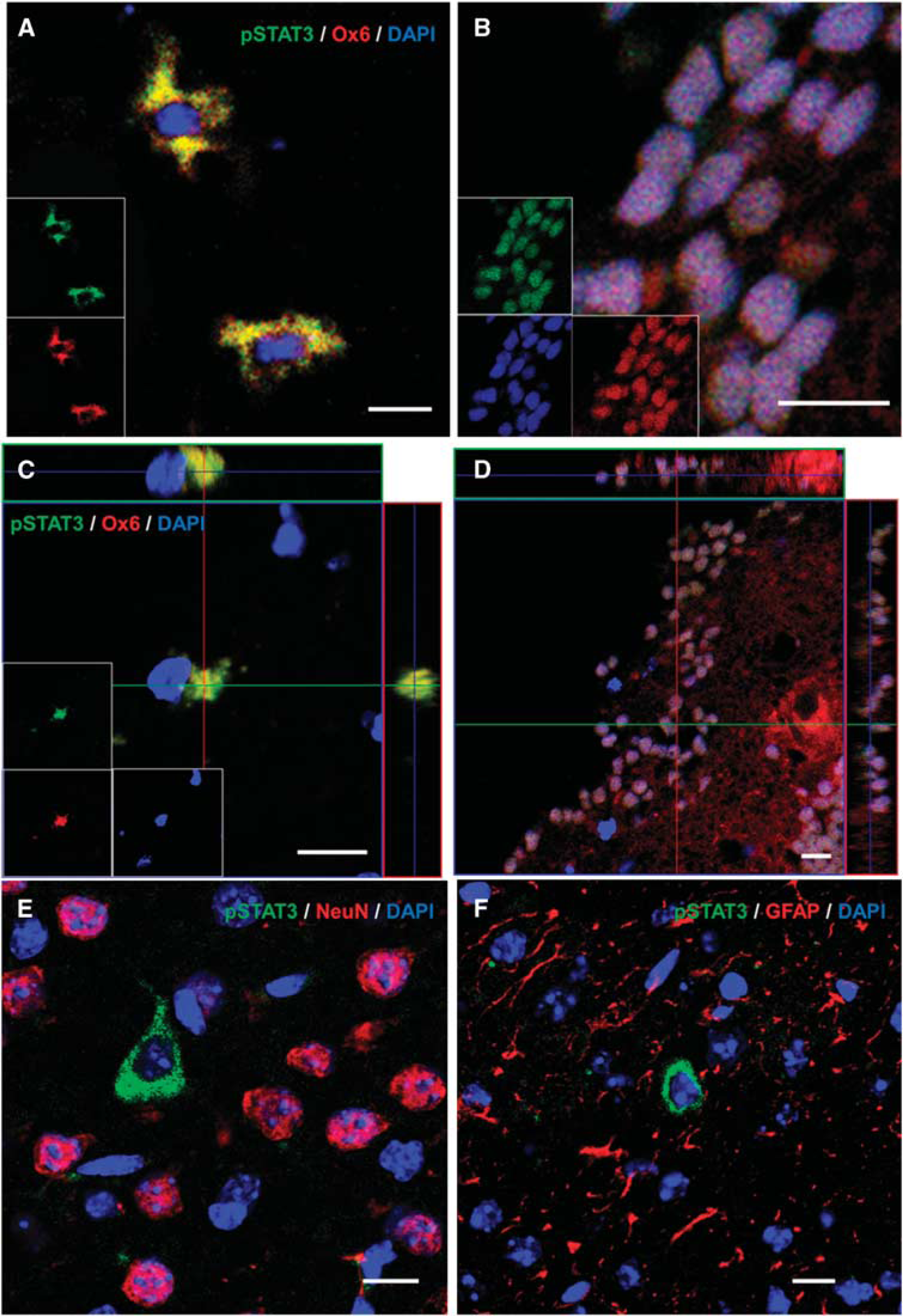
Double staining of phosphorylated signal transduction and activator of transcription 3 (pSTAT3) and cell-specific markers (
DISCUSSION
In this study, leptin increased the degree of brain edema, neurologic deficits, recruitment of inflammatory cells, and production of inflammatory end products after ICH, and these results were confirmed in leptin-deficient
The early phase of secondary brain injury after ICH is mainly mediated by clot-derived thrombin, which is associated with local injury such as direct cellular toxicity, blood–brain barrier breakdown, and inflammatory cell infiltration. 13 After the early phase, subsequent inflammatory reaction plays an important role in tissue damage that occurs after ICH. As one of the critical mediators of the immune system, leptin promotes the secretion of proinflammatory cytokines such as tumor necrosis factor-α, interleukin-6, and interferon-γ. 11 Functional receptors for leptin are expressed not only in the hypothalamus, where it regulates energy homeostasis, but also in various cell types related to immunity. Leptin binding to the functional receptor recruits Janus tyrosine kinase, which then serves as a docking site for cytoplasmic adaptors such as STAT3, ERK, and Akt, candidate signal transducers in this study. 25 In this study, leptin increases inflammation after ICH via the STAT3 pathway, and the target gene related to inflammation of STAT3 might be COX-2 and iNOS, which were highly expressed after injection of leptin in hemorrhagic hemisphere. 26 According to previous studies about the role of STAT3 in inflammatory processes, STAT3 is involved in the expression of iNOS and is partly dependent on Ser727 phosphorylation, which is also necessary for nuclear translocation and DNA binding, 27 and STAT3 upregulated COX-2 for late preconditioning in cardioprotection. 28
In the current study, leptin-deficient
Janus tyrosine kinase 2-STAT3 signaling is an important pathway mediating the effects of leptin on immune cells. 35 It is well established that leptin stimulation leads to the tyrosine phosphorylation of STAT3 and its translocation to the nucleus in human or murine macrophages. 36 This pathway has not been observed in stroke models, but we found that the detrimental effect of leptin on ICH disappeared after the injection of a pSTAT inhibitor (NSC74859) and that, after ICH, pSTAT3 was located in microglia/macrophages and not in neurons or astrocytes. The STAT3 activation was higher at 24 hours than at 2 hours after ICH in our study; this delayed activation is highly correlated with the time course of inflammation after ICH because the infiltration of inflammatory cells begins at 12 hours and is maximal 48 hours after ICH. 23 In this context, we postulate that STAT3 is a key molecule of the leptin-induced detrimental effect on ICH. However, the inhibitor of STAT3, NSC74859, only works in the presence of the pharmacologic dose of leptin (8 mg/kg), and it did not reduce brain edema in mice administered ICH + inhibitor without injection of leptin. This result shows that the protective effects of reducing activation of STAT3 without hyperleptinemia against perihematomal edema and inflammation after ICH are still elusive.
Leptin is an adipokine with physiologic function, and increased leptin in stressful conditions associated with various diseases is not always harmful. After focal ischemia in the brain or heart, increased leptin showed protective effects via the antiapoptotic pathway, but a possible proinflammatory effect of hyperleptinemia was not evaluated in these studies.17,37 As demonstrated in the current study, increased leptin in inflammatory diseases was harmful, and in an experimental ICH model, leptin showed a detrimental effect through the enhancement of inflammation. The protective or harmful effect of leptin after various diseases is not seen uniformly, and it may depend on the specific disease conditions, action timing, or doses of leptin.
According to glucose homeostasis, leptin acts on peripheral leptin receptors expressing various tissues. The direct action of leptin on the pancreas inhibits insulin and glucagon secretion, and leptin suppresses insulin signaling and action in adipocytes and liver. The centrally mediated action of leptin is that autonomic efferents increase direct leptin action on the pancreas and insulin-sensitive tissues via activation of the hypothalamus. 38 From these reasons, in the leptin-deficient state such as in ob/ob mice, hyperglycemia is induced with combined overnutrition, obesity, and hyperinsulinemia.
Taken together, we have demonstrated that leptin is a novel mediator of inflammation after ICH and plays a critical role in secondary brain injury around the hematoma via the STAT3 signaling pathway in microglia/macrophages. Specific medical or surgical treatment for ICH patients does not exist: the benefit of surgical treatment has not been proven, 39 and furthermore, hemostatic medical treatment using the early introduction of recombinant factor VIIa to inhibit the expansion of hematoma failed in a large clinical trial. 40 As the incidence of ICH is very high worldwide and increases up to 30–50% in Asian countries, specific treatment to improve the outcomes of ICH patients must be discovered and immediately applied to clinical practice. However, whether there is a key molecule that modulates the ICH-induced inflammation is still controversial. We believe that the perihematomal inflammation mediated by leptin can be a new therapeutic target for ICH treatment.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.