
Abstract
Central nervous system injury causes a marked increase in the expression of cell cycle-related proteins. In this study, we show that cell cycle activation (CCA) is detected in mature neurons at 24 hours after rat lateral fluid percussion (LFP)-induced traumatic brain injury (TBI), as reflected by increased expression of cyclin G1, phosphorylated retinoblastoma (phospho-Rb), E2F1 and proliferating cell nuclear antigen (PCNA). These changes were associated with progressive cortical, hippocampal, and thalamic neuronal loss and microglial and astrocyte activation. Notably, we detected 5-bromo-2′-deoxyuridine (BrdU)-positive neurons, microglia, and astrocytes at 7 days, but not at 24 hours, suggesting that cell cycle reaches the S phase in these cell types at the latter time point. A delayed systemic post-LFP administration at 3 hours of CR8—a potent second-generation cyclin-dependent kinase (CDK) inhibitor—reduced CCA; cortical, hippocampal, and thalamic neuronal loss; and cortical microglial and astrocyte activation. Furthermore, CR8 treatment attenuated sensorimotor and cognitive deficits, alleviated depressive-like symptoms, and decreased lesion volume. These findings underscore the contribution of CCA to progressive neurodegeneration and chronic neuroinflammation following TBI, and demonstrate the neuroprotective potential of cell cycle inhibition in a clinically relevant experimental TBI model.
Keywords
INTRODUCTION
Traumatic brain injury (TBI) is a major public health problem, with more than 1.7 million new cases reported annually in the United States and accounts for half of all trauma-related deaths. 1 Traumatic brain injury causes direct physical tissue disruption (primary injury), followed by delayed secondary injury that begins within seconds after the insult and may be responsible for a significant component of neurodegeneration and neurologic impairment that potentially continues for years.2, 3
Studies have demonstrated that TBI induces cell cycle activation (CCA) that may result in apoptosis of postmitotic cells (mature oligodendroglia and neurons) and activation of mitotic cells (microglia and astrocytes).4, 5, 6, 7, 8, 9 Microglial and astrocyte activation is associated with the release of proinflammatory molecules that can cause neurotoxicity. 5 Therefore, TBI-induced CCA may initiate multiple secondary injury mechanisms that directly and indirectly contribute to neuronal apoptosis and delayed neurotoxicity, respectively.8, 9, 10, 11 In order to better understand the pathobiology of TBI, it is important to elucidate the progression of the resulting neurodegeneration, inflammatory mechanisms, and cellular proliferative responses. In this study, we present a comprehensive quantitative assessment of neuronal cell loss, and microglial and astrocyte activation in the brain after rat lateral fluid percussion (LFP) injury—a well-established, clinically relevant TBI model. 12
We have previously investigated the neuroprotective potential of a relatively selective cyclin-dependent kinase (CDK) inhibitor, roscovitine, in multiple models of experimental TBI.6, 8 Recently, we demonstrated greater therapeutic efficacy and potency of CR8, an N6-biaryl-substituted derivative of roscovitine, for attenuating CCA and neurodegeneration, and improving functional outcomes in a mouse focal controlled cortical impact model. In this study, we extend the characterization of CCA in the rat LFP model by focusing on the involvement of specific cell cycle phases and using unbiased quantitative stereological methods to evaluate the cellular responses. We also use a more extended therapeutic window as well as a clinically relevant systemic route of administration of CR8 to assess the neuroprotective potential of cell cycle inhibition against progressive neurodegeneration, chronic neuroinflammation, and neurologic dysfunction after LFP.
MATERIALS AND METHODS
Lateral Fluid Percussion Brain Injury
All surgical procedures and experiments were carried out in accordance with protocols and guidelines approved by the Institutional Animal Care and Use Committee at the University of Maryland (Association for Assessment and Accreditation for Laboratory Animal Care-approved). Adult (10 to 12 weeks old) male Sprague–Dawley rats (310 to 330 g) were anesthetized using isoflurane (induction: 4%, maintenance: 2% to 2.5%, supply gas: 70% compressed air+30% oxygen). A sharp dissection was made followed by craniotomy (4 mm) located midway between lambda and bregma sutures over the left parietal cortex. A luer-lock adaptor was cemented over the craniotomy site and attached to our custom-built microprocessor-driven LFP device, 12 which when triggered produced a pressure pulse causing a deformation of the underlying brain. The rats were subjected to moderate LFP brain injury (1.9 to 2.2 atmospheres) or sham operation following approved experimental guidelines, as previously described. 12
Drug Administration
Groups of rats received an intraperitoneal injection of CR8 (5 mg/Kg, n=11), or equal volume vehicle (physiologic saline, n=10) 3 hours after injury. Sham-operated rats (n=6) received an intraperitoneal injection of vehicle 3 hours after surgery. These rats underwent behavioral testing for different functional outcomes and were killed for histology at 28 days. The dose of CR8 was selected based on our previous in vitro and in vivo data showing neuroprotection of CR8 using mouse controlled cortical impact model. 10
Groups of rats (n= 4 to 5 per group) were subjected to the same magnitude of injury followed by intraperitoneal treatment of CR8 or vehicle at 3 hours and killed for histology after 24 hours or 7 days. Groups of rats killed at 24 hours and 7 days were injected with 5-bromo-2′-deoxyuridine (BrdU, 300 mg/Kg, intraperitoneally; Sigma-Aldrich, St Louis, MO, USA) at 8 hours after LFP and 2 hours before killing. 13
Immunocytochemistry
At 24 hours or 7 days after injury, rats (n=4 to 5 per group) were anesthetized and transcardially perfused with saline and 4% buffered paraformaldehyde solution (Fisher Scientific, Pittsburg, PA, USA). The brains were removed, postfixed in paraformaldehyde for 24 hours, and protected in 30% sucrose. Frozen brain sections (60 μm and 20 μm) were cut on cryostat and mounted onto glass slides. Selected slides (20 μm) were stained with Fluoro-Jade B (Chemicon, Temecula, CA, USA), following the manufacturer's protocol. For immunocytochemistry, sections were stained and processed, as previously described,9, 14, 15 using one or more antibodies recognizing the following markers of CCA: PCNA (ab18197, Abcam, Cambridge, MA, USA), Cyclin G1 (7865, Santa Cruz Biotechnology., Santa Cruz, CA, USA), E2F1 (554213, BD Pharmingen, San Diego, CA, USA), phospho-Rb S780 (ab47763, Abcam, Cambridge, MA), neuronal marker: NeuN (MAB377, Millipore, Billerica, MA, USA), microglial activation marker: Iba-1 (#019–19741, Wako Chemicals, Richmond, VA, USA), astrocyte activation marker: glial fibrillary acidic protein (GFAP) (Z0334, Dako, Carpinteria, CA, USA) and S-phase/DNA synthesis marker: BrdU (555627, BD Biosciences, San Jose, CA, USA 347580, BD, Franklin Lakes, NJ; ab6326, Abcam, Cambridge, MA, USA). Imaging based on fluorescence microscopy was carried out using Leica TCS SP5 II Tunable Spectral Confocal microscope (Leica Microsystems, Bannockburn, IL, USA). For acquisition of immunofluorescent images, we used ‘Tile Scan’ function of the Leica Application Suite Advanced Fluorescence software (Leica LAS AF version 2.4.1) to scan multiple partial images of injured brain hemisphere. The scan field was selected to cover an entire hemisphere. The ‘Merge Images' command was activated to automatically join partial images into a complete image after the scan. The acquisition parameters were the same across all specimens: XY format 1024 × 1024; image size 775 um × 775 um; zoom 1; speed 600 Hz; pinhole airy 1; bidirectional X; same gain; autofocus-best focus noise-based method; tile scan auto-stitching and smooth; sequential scan between frames. High magnification (× 20 or × 63) images were cropped from specific locations around the lesion site in merged image. Automatic semi-quantitative counting of Fluoro-Jade B and cyclin G1-positive cells in multitile fluorescent images was performed using the ImageJ software, version 1.47 (NIH, Bethesda, MD, USA), as previously described. 14 The numbers (per representative field of 3 to 5 sections) represent the average number of cells per image (% sham). Semi-quantitative assessment of BrdU/NeuN, BrdU/Iba-1, and BrdU/GFAP double-positive cells was performed in 14 to 16 random fields in lesioned cortex of injured animals. Confocal Z-stacks were acquired using the Leica SP5 II microscope and BrdU single-positive as well as of BrdU/NeuN, BrdU/Iba-1, and BrdU/GFAP double-positive cells were counted using the cell counter plug-in function of ImageJ software (NIH). The numbers of BrdU/NeuN, BrdU/Iba-1, and BrdU/GFAP double-positive cells were expressed as percentages of total number of BrdU-positive cells to estimate the relative distribution of S-phase induction amongst these cell types.
Behavioral Assessments for Functional Recovery
Composite neurologic scoring
Chronic sensorimotor recovery was assessed using the composite neurologic scoring paradigm (n=10 to 11 per group), as previously described.4, 6, 12 The composite neuroscore reflects a combination of certain individually scored sensitive tests, each scored using an ordinal scale ranging from zero (severe impairment) to five (normal function). The total composite neurologic score (0 to 35) is obtained by combining the scores of the following tests: (i) lateral pulsion (left and right), (ii) forelimb flexion (left and right), and (iii) inclined plane (two vertical and two horizontal positions for assessing maximum angle at which the animal can stand for 5 seconds; scoring: >50°=5, 45 to 50°=4, 40 to 45°=3, 35 to 40°=2, 30 to 35°=1, and <29°=0).5, 12, 14, 16 Rats were tested on postinjury days (PIDs) 1, 7, 14, 21, and 28 days.
Morris water maze
Spatial learning and working memory following LFP was assessed using the Morris water maze (MWM) acquisition paradigm (n=10 to 11 per group) on PIDs 14, 15, 16, and 17, as previously described.6, 12 Computer-based AnyMaze video tracking system (Stoelting, Wood Dale, IL, USA) was used to determine the latency (seconds) to locate submerged hidden platform with a 90-second limit per trial. Reference spatial memory was assessed by probe trial, with a 60-second limit, on PID 18. A visual cue test was subsequently performed on PID 18 using a flagged platform in one of the quadrants (with a 90-second limit per trial) and latency (seconds) to locate the platform was recorded.
Novel object recognition
Retention or intact memory was assessed by the novel object recognition (NOR) test (n=10 to 11 per group) on PID 21. The apparatus consists of an open field (40 cm × 20 cm × 60 cm) with two adjacently located imaginary circular zones, as previously designed. 17 The zones are equally spaced from the sides in the center of the square and designated as ‘old object’ and ‘novel object’ zones using the AnyMaze video tracking system. The old object used was square-shaped, whereas the novel object was L-shaped, assembled by building blocks from Lego toys and were clearly distinct in shape and appearance. On PID 20, all animals were placed in the open field without objects for 5 minutes each for habituation. Two 5-minute trials were performed on PID 21: the first (training) trial with two old objects in both zones and the second (testing) phase with one old object and one novel object present in the respective zones of the open field, with an inter-trial interval of 60 minutes. The time spent in novel and old object zones was assessed manually as well as by using AnyMaze software. Retention memory was determined by ‘discrimination index’ (DI) for the second trial, which was calculated as follows9, 10, 17: % DI=time spent in novel object zone × 100 (time spent in old object zone+time spent in novel object zone)
Passive avoidance
The passive avoidance test (n=10 to 11 per group) was used to evaluate nonspatial fear-based amygdala-dependent contextual and emotional memory on PIDs 24 to 25, as previously described using one-trial step-through PACS-30 passive avoidance apparatus (Columbus Instruments, Columbus, OH, USA) adjusted to an electric foot shock setting of 0.5 mA for 3 seconds with a 24-hour lag between training and testing. 18 The cognitive impairment (% sham) was calculated using the following formula: % cognitive impairment=latency to the dark compartment in testing phase × 100 mean latency to the dark compartment in testing phase for sham group
Forced swim
The forced swim test (n=10 to 11 per group) was used to evaluate depressive-like behavior. 19 On PID 26, rats were individually forced to swim inside a vertical rectangular container, containing 30 cm of water maintained at 24°C to 25°C for a time period of 6 minutes. The total duration of immobility versus struggle was recorded manually using a timer.
Histology
Rats were perfused and brains were collected and sectioned, as described under ‘Immunocytochemistry’. Out of the total ninety-six 60 μm sections, every fourth 60 μm section was processed for immunohistochemical analysis and every eighth section was used for lesion volume assessment, beginning from a random start point.
Lesion volume estimation
Sections (60 μm) from brains harvested at 28 days were stained with Cresyl Violet (FD NeuroTechnologies, Baltimore, MD, USA), dehydrated, and mounted for analysis (n=6 to 7 per group). Lesion volume was quantified based on Cavalieri method of stereology using Stereo Investigator software (MBF Biosciences, Williston, VT, USA) by outlining missing tissue on the injured hemisphere using the Cavalieri estimator with grid spacing of 150 μm.
Neuronal cell loss assessment
Stereo Investigator software was used to count the total number of surviving neurons in the cornu ammonis (CA) 1, CA3, and dentate gyrus (DG) sub-regions of the hippocampus, cortex, and thalamus at 24 hours, 7 days, and 28 days using the optical fractionator method of stereology (n=4 to 7 per group). Cresyl violet staining enables discrimination of morphologic features of neurons and glial cells. 20 Every fourth 60 μm section between −1.22 mm and −2.54 mm from bregma was analyzed beginning from a random start point. Sections were analyzed using a Leica DM4000B microscope. The optical dissector had a size of 50 μm by 50 μm in the x and y-axis, respectively with a dissector height of 10 μm and guard zone height of 4 μm from the top of the section. The sampled region for each hippocampal subfield was demarcated in the injured hemisphere and Cresyl violet neuronal cell bodies were counted. For CA1 and CA3 sub-regions, a grid spacing of 300 μm in the x-axis and 100 μm in the y-axis was used. For DG sub-region, a grid spacing of 300 μm in the x-axis and 150 μm in the y-axis was used. For the cortex and thalamus, grid spacing of 1500 μm in the x-axis and 500 μm in the y-axis was used. The volume of the hippocampal subfield was measured using the Cavalieri estimator method with grid spacing of 100 μm. The volume of the cortex and thalamus was quantified with grid spacing of 150 μm. The estimated number of surviving neurons in each field was divided by the volume of the region of interest to obtain the cellular density (counts per mm3).
Microglial and astrocyte morphologic assessment
Microglia or astrocytes were immunostained with anti-Iba-1 or anti-GFAP (1:1000) overnight, respectively, washed in phosphate-buffered saline, incubated with biotinylated anti-rabbit immunoglobulin G antibody (Vector Laboratories, Burlingame, CA, USA) and processed further as previously described.8, 9 The stained sections were counterstained with Cresyl violet and mounted for analysis.
Characterization of cellular morphologic phenotypes: Stereo Investigator Software was used to count and classify the number of cortical microglia in each of the three microglial morphologic phenotypes (ramified, hypertrophic, and bushy), and cortical astrocytes in each of the two astroglial morphologic phenotypes (resting and reactive) at 24 hours, 7 days, and 28 days using the optical fractionator method of unbiased stereology (n=4 to 7 per group).8, 9 The sampled region was the ipsilateral cortex between −1.22 mm and −2.54 mm from bregma, and dorsal to a depth of 2.0 mm from surface. The optical dissector had a size of 50 μm by 50 μm in the x and y-axis, respectively with a dissector height of 10 μm and guard zone height of 4 μm from the top of the section. Grid spacing of 400 μm in the x-axis and 400 μm in the y-axis was used. Microglial8, 9, 11, 21 and astrocyte phenotypic22, 23 classification was based on length and thickness of projections, number of branches, and size of cell body. The volume of the region of interest was measured using the Cavalieri estimator method with grid spacing of 150 μm for the cortex. The estimated number of microglia or astrocytes in each phenotypic class was divided by volume of the cortex to obtain cellular density. The live image setting tool of the Neurolucida software (MBF Biosciences) was used to create reconstructions of microglia and astrocytes at different stages of activation by tracing the cell bodies (contour tool) and processes (dendrite line tool), as previously described.8, 9
Statistical Analysis
The number of animals per group for each assessment was based on our prior studies using the LFP model5, 6, 12 and satisfied power requirements. Lesion volume, functional data, and stereological analysis were performed by an investigator masked to groups. Quantitative data were expressed as mean±s.e.m. Functional data for composite neuroscore and MWM acquisition phase, respectively were analyzed by repeated measures one-way analysis of variance (ANOVA) followed by Student–Newman–Keuls post hoc test. The data for MWM probe trial, NOR and forced swim tests, and stereological assessments were analyzed by one-way ANOVA followed by Student–Newman–Keuls test. The data for cognitive impairment in passive avoidance test were analyzed by Mann–Whitney U- and t-test. Lesion volume, Fluoro-Jade B and Cyclin G1-positive cell assessments were analyzed by one-tailed unpaired Student's t-test. The data for distribution of S-phase induction was statistically analyzed by Kruskal–Wallis ANOVA followed by Dunn's post hoc test. The data were analyzed using SigmaPlot 12 (Systat Software, San Jose, CA, USA) or GraphPad Prism Version 4.0 (GraphPad Software, San Diego, CA, USA). A P<0.05 was considered statistically significant.
RESULTS
Systemic Administration of CR8 Inhibits Lateral Fluid Percussion-Induced Neuronal Cell Cycle Activation
In order to evaluate the role of CCA in pathophysiology of TBI and to explore the neuroprotective potential of CR8, we examined four cell cycle markers: cyclin G1, phospho-Rb, PCNA, and E2F1 at 24 hours post injury (Figure 1). Staining with NeuN confirmed the colocalization of cell cycle markers with neurons. Immunocytochemical analysis revealed that at 24 hours, there was increased expression of cyclin G1 (Figures 1A, 1B) and PCNA (Figure 1C) in neurons within the central area of lesion site and their colocalization in cells with neuronal morphology. This elevated expression of cyclin G1 and PCNA was reduced in LFP rats treated with CR8 (Figures 1A–1C). Semi-quantitative assessment revealed a significant decrease in the number of cyclin G1-positive cells after CR8 treatment (P=0.026, versus vehicle, Figure 1B). Lateral fluid percussion also increased expression and colocalization of phospho-Rb and E2F1, which was attenuated by CR8 treatment (Figure 1D).
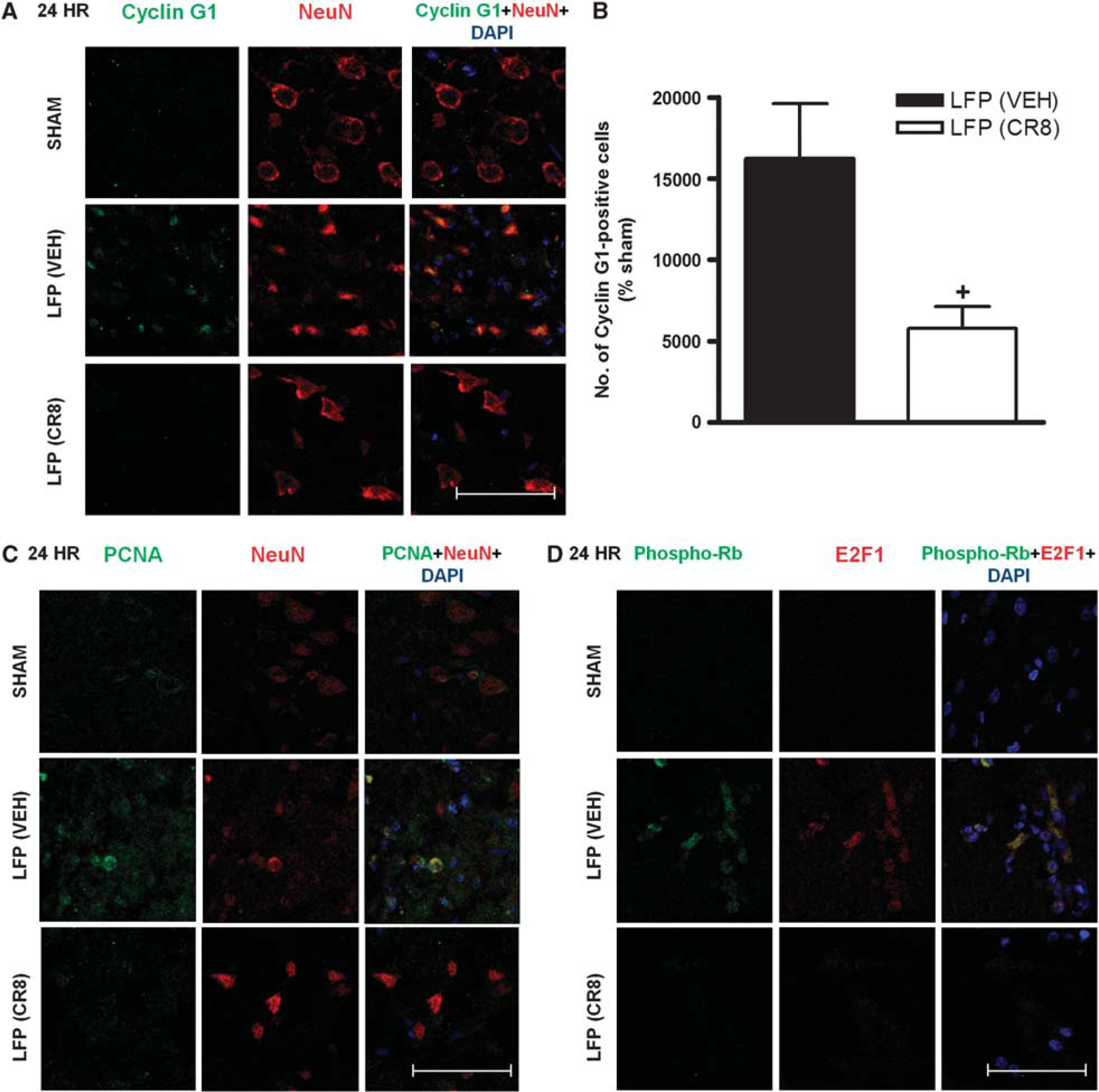
Systemic administration of CR8 inhibits lateral fluid percussion (LFP)-induced neuronal cell cycle activation (CCA). Immunocytochemistry for assessment of CCA at 24 hours after LFP. (
LFP-induced Cell Cycle Activation Reaches DNA Replication Phase in Neurons and Microglia
To assess the progression of LFP injury-induced CCA and characterize DNA replication, we used BrdU labeling, a marker of S phase at 24 hours and 7 days post injury. Samples were immunostained for BrdU in combination with NeuN, Iba-1, and GFAP to label neurons, microglia, and astrocytes, respectively. We observed no colocalization of BrdU with NeuN, Iba-1, or GFAP at 24 hours (data not shown). In contrast, we observed BrdU/NeuN- (Figure 2A), BrdU/Iba-1- (Figure 2B), and BrdU/GFAP double-positive cells (Figure 2C) at 7 days. Semi-quantitative assessment revealed that microglia (39.10±1.69%) constituted larger component of BrdU-positive cells than neurons (19.03±1.80%) and astrocytes (21.62±1.32%; ∗∗∗P<0.001, versus microglia, Figure 2D). The three cell types altogether constituted close to 80% of the total cellular population undergoing the S phase of cell cycle.
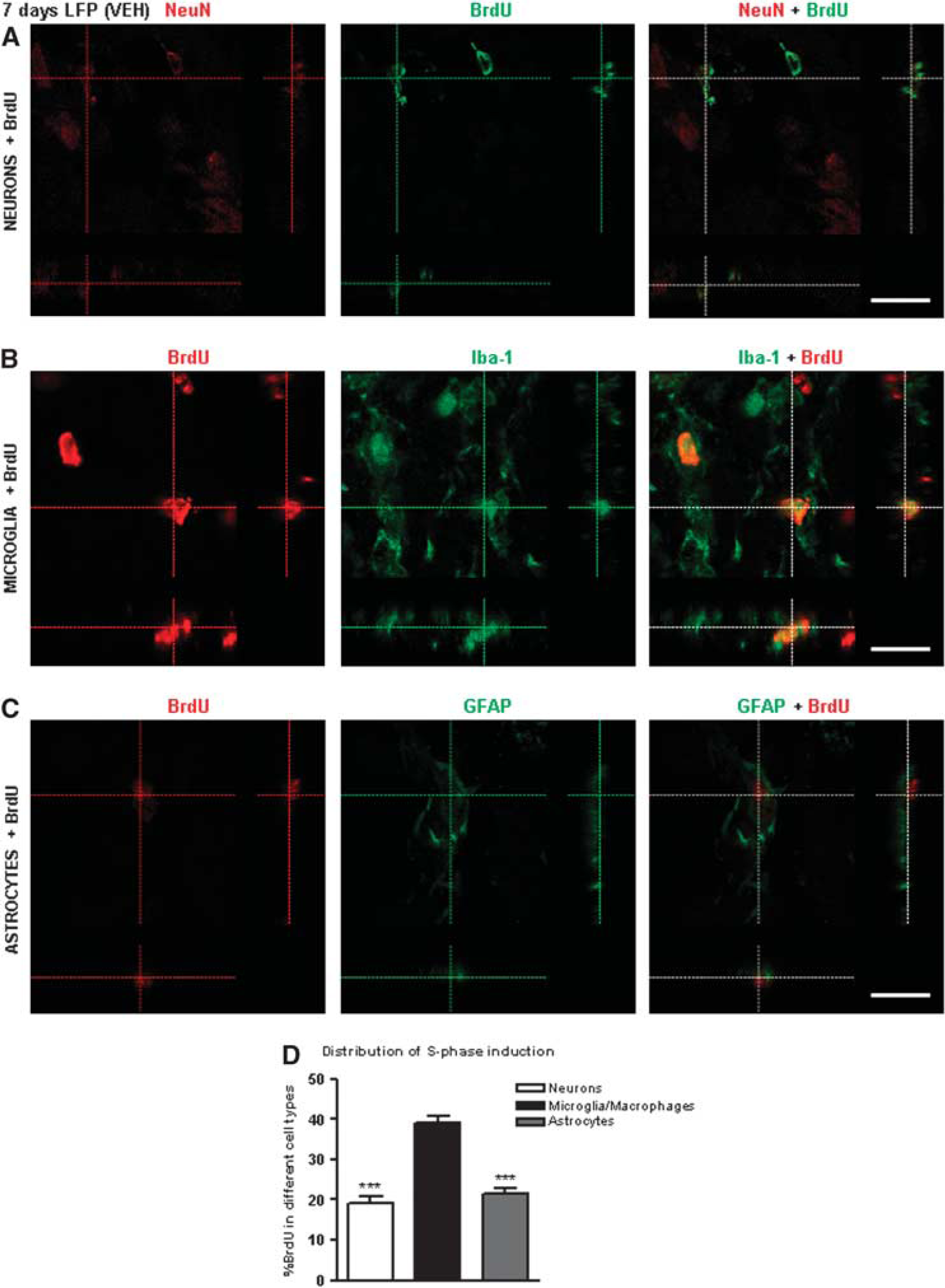
Lateral fluid percussion-induced cell cycle activation reaches DNA replication phase in neurons, microglia, and astrocytes. Immunocytochemistry for qualitative assessment of S-phase induction in neurons, microglia, and astrocytes at 7 days after LFP. (
CR8 Treatment Reduces Lateral Fluid Percussion injury-Induced Lesion Volume and Progressive Neuronal Cell Loss in the Hippocampus, Cortex and Thalamus
Histological assessment at 28 days showed that vehicle-treated rats developed a large lesion following LFP injury (12.11±1.21 mm3, Figures 3A, 3B), and that systemic treatment of CR8 resulted in a significant reduction in lesion size (P<0.001, versus vehicle; 4.24±0.83 mm3).
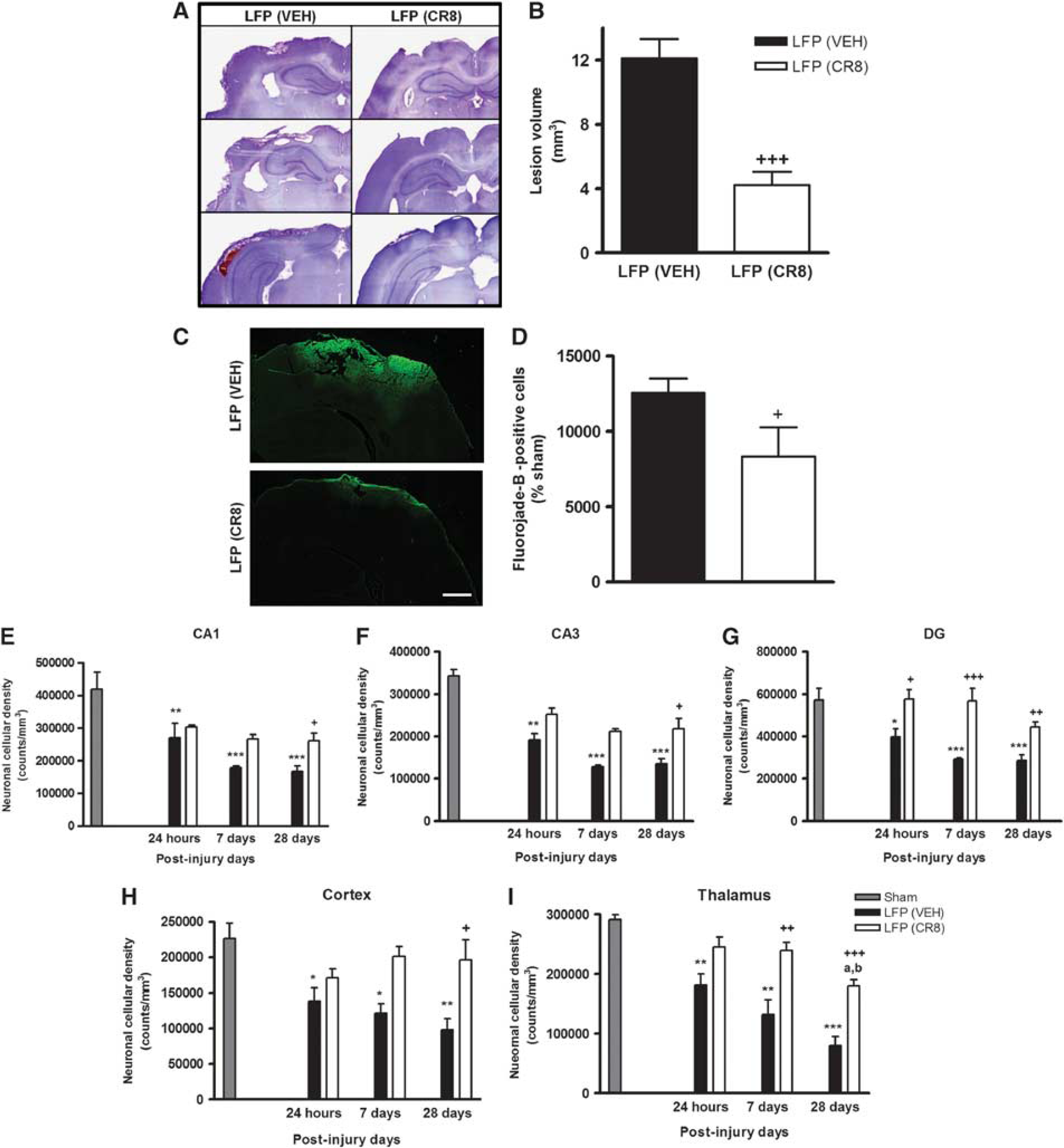
CR8 treatment reduces lateral fluid percussion (LFP)-induced lesion volume and progressive neuronal cell loss in the hippocampus, cortex, and thalamus. (
To determine specific effects of CR8 treatment on LFP injury-induced neurodegeneration, we performed Fluoro-Jade B staining at 24 hours post injury (Figure 3C). Immunocytochemical analysis revealed that LFP injury-induced neurodegeneration in perilesional cortical regions, indicated by higher number of Fluoro-Jade B-positive cells, was significantly attenuated by CR8 treatment (P=0.003, versus vehicle, Figure 3D).
Stereological assessment of surviving neurons was performed in CA1, CA3, and DG hippocampal sub-regions as well as the cortex and thalamus at 24 hours, 7 days, and 28 days post injury. Lateral fluid percussion injury resulted in significant and progressive neuronal cell loss in all hippocampal sub-regions (P<0.001, <0.01, and <0.05 versus sham, Figures 3E–G). Systemic administration of CR8 at 3 hours resulted in improved neuronal survival in CA1 (261,232.3±22,863.14 versus167,496.6±16,185.99 counts/mm3, P<0.05, versus vehicle, Figure 3E) and CA3 sub-regions at 28 days (217,877.4±25,109.72 versus 135,241.7±11,956.99 counts/mm3, P<0.05, versus vehicle, Figure 3F). In addition, CR8 treatment consistently improved neuronal survival in DG at 24 hours (576,697.5±45,226.49 versus 398,071.6±37,623.83 counts/mm3, P<0.05, versus vehicle, Figure 3G), 7 days (568,087.6±59,417.56 versus 291,401.3±7,867.87 counts/mm3, P<0.001, versus vehicle, Figure 3G), and 28 days (444,541.9±23,241.86 versus 285,591.3±27,108.77 counts/mm3, P<0.01, versus vehicle, Figure 3G), when compared with vehicle-treated samples.
Lateral fluid percussion injury also induced significant and progressive neuronal cell loss in the cortex and thalamus (P<0.001, <0.01, and <0.05 versus sham, Figures 3H, 3I). CR8 treatment resulted in significantly improved neuronal survival in the cortex at 28 days (196,625.8±28,429.34 versus 97,755.35±16,336.02 counts/mm3, P<0.05, versus vehicle, Figure 3H). In addition, systemic administration of CR8 significantly attenuated neuronal loss in thalamus at 7 days (239,311.8±13,623.13 versus 132,047.0±24,642.91 counts/mm3, P<0.01, versus vehicle, Figure 3I) and 28 days (179,901.2±10,673.82 versus 80,012.36±15,450.92 counts/mm3, P<0.001, versus vehicle, Figure 3I) when compared with vehicle-treated samples.
CR8 Treatment Modulates Lateral Fluid Percussion-Induced Cortical Microglial and Astrocyte Activation
Quantitative assessment of microglial and astrocyte cell number and activation phenotype was performed in the cortex at 24 hours, 7 and 28 days using stereological methods (Figures 4, 5). There was a significant decrease in ramified microglia in vehicle-treated samples at 28 days when compared with sham controls (P<0.05, versus sham, Figure 4B). Lateral fluid percussion injury resulted in a significant increase in hypertrophic microglia that peaked at 7 days followed by a return toward control levels by 28 days (P<0.001, versus sham, Figure 4C). CR8 treatment resulted in a significant attenuation in numbers of hypertrophic microglia at 7 days (12,031.26±1,776.52 versus 16,607.21±1,721.43 counts/mm3, P<0.05, versus vehicle). Lateral fluid percussion injury also resulted in a significant increase in the numbers of bushy microglia, and although a peak was reached at 7 days (P<0.001, versus sham, Figure 4D), the levels remained elevated when compared with sham-operated samples through 28 days (P<0.05, versus sham). CR8 treatment significantly reduced the number of bushy microglia at both 7 (10,520.89±1,236.78 versus 39,606.09±1,126.82 counts/mm3, P<0.001, versus vehicle) and 28 days (7,955.89±807.01 versus 19,146.77±296.19 counts/mm3, P<0.001, versus vehicle), when compared with vehicle-treated samples.
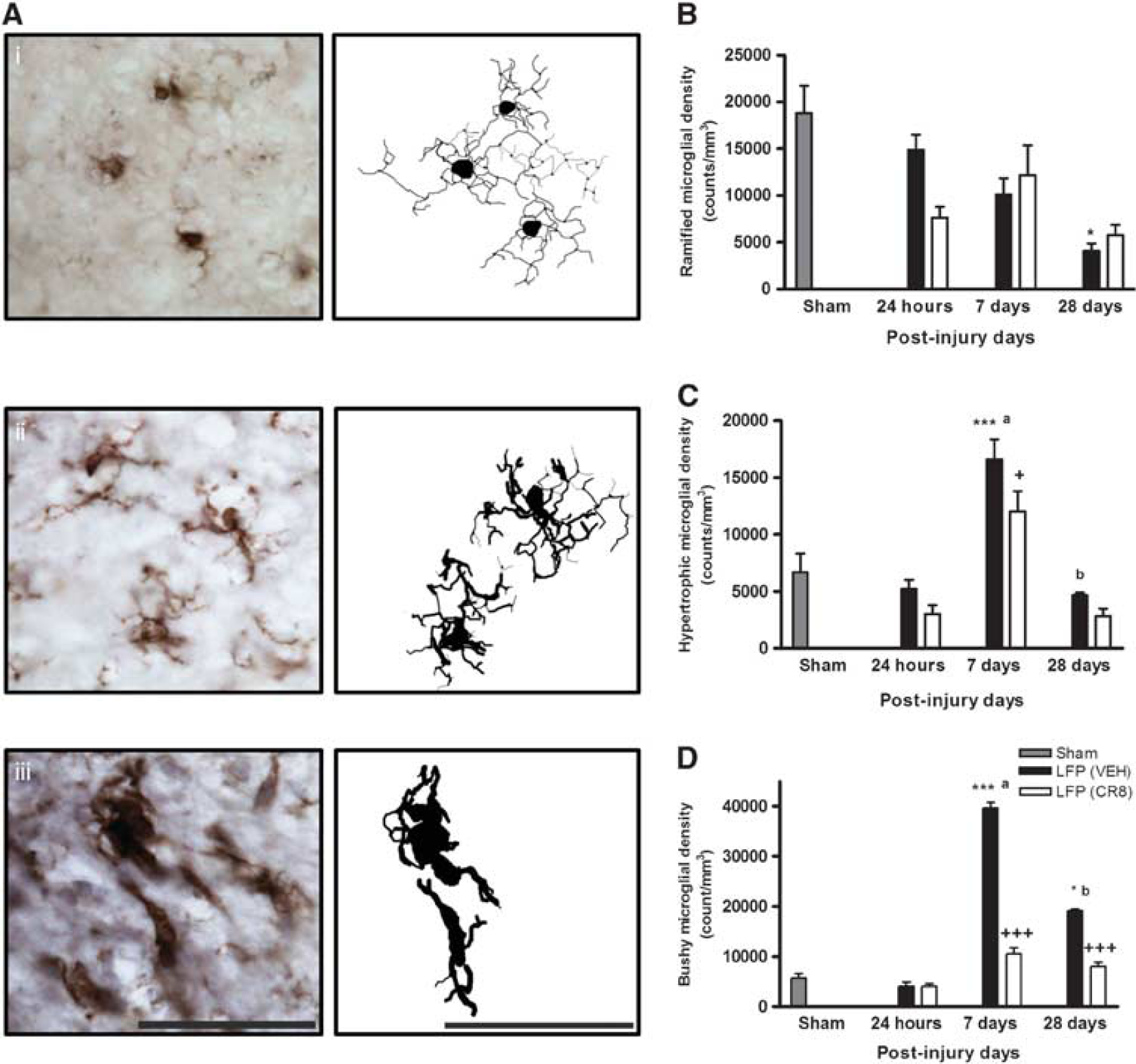
CR8 treatment modulates lateral fluid percussion (LFP)-induced cortical microglial activation. (
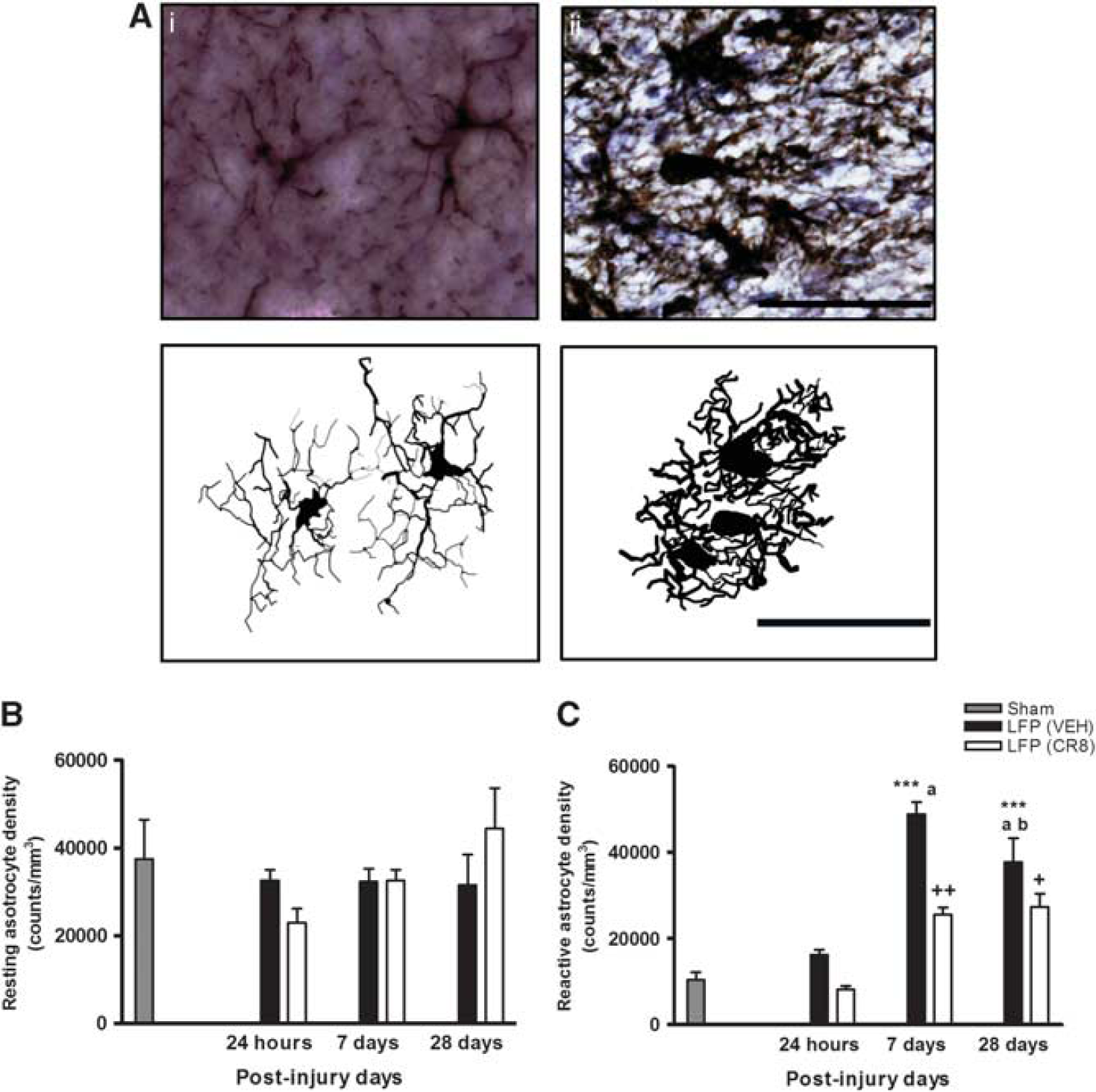
CR8 treatment attenuates lateral fluid percussion (LFP)-induced cortical astrocyte activation. (
Furthermore, LFP injury resulted in a significant increase in the number of reactive astrocytes, which peaked at 7 days followed by a slow decline, and the levels of reactive astrocytes remained elevated after LFP at 28 days when compared with sham group (P<0.001, versus sham, Figure 5C). CR8 treatment significantly reduced reactive astrocytes at both 7 (25,505.14±1,762.86 versus 48,815.17±2,884.76 counts/mm3, P<0.01, versus vehicle) and 28 days (27,393.22±3,040.16 versus 37,705.55±5,569.20 counts/mm3, P<0.05 versus vehicle).
CR8 Treatment Limits Lateral Fluid Percussion-Induced Sensorimotor Impairment
Functional assessment of fine sensorimotor function was performed using the composite neurologic scoring paradigm6, 12 and results were statistically analyzed by one-way (groups) repeated measures (scores/time) ANOVA followed by Student–Newman–Keuls test. The interaction of ‘PIDs X groups' (F(8,115)=0.611, P=0.767) was not significant. However, the factors of ‘PIDs' (F(4,115)=4.297, P=0.003) and ‘groups' (F(2,115)=109.370, P<0.001) were statistically significant. Lateral fluid percussion injury-induced significant sensorimotor impairments when compared with sham rats (P<0.001, versus sham, Figure 6A). CR8-treated LFP rats exhibited significant sensorimotor improvement at 14 (P=0.026, versus vehicle) and 28 days (P=0.027, versus vehicle) when compared with vehicle-treated rats.
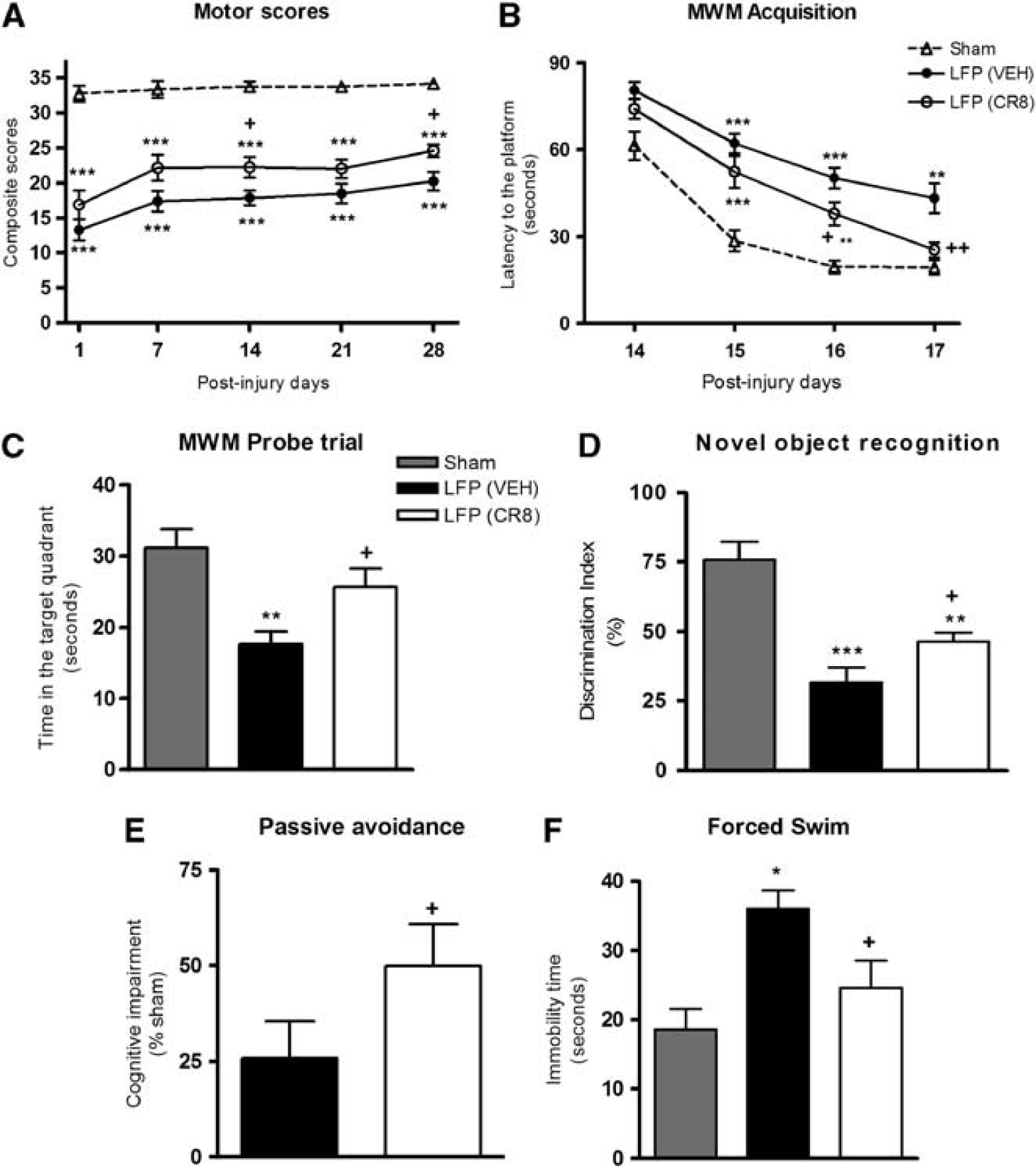
CR8 treatment attenuates lateral fluid percussion (LFP)-induced sensorimotor impairment, cognitive dysfunction, and depressive-like symptoms. (
CR8 Treatment Attenuates Lateral Fluid Percussion-Induced Cognitive Dysfunction
Spatial learning was tested using acquisition phase of the MWM6, 12 and results for latency to find the platform were statistically analyzed by one-way (groups) repeated measures (trials/time) ANOVA followed by Student–Newman–Keuls test. The interaction of ‘PIDs X groups' (F(6,92)=1.037, P=0.406) was not significant. The factors of ‘PIDs' (F(3,92)=55.491, P<0.001) and ‘groups' (F(2,92)=33.378, P<0.001) were found to be statistically significant. Lateral fluid percussion injury resulted in learning impairments on PIDs 15, 16, and 17 (P<0.001 or 0.01, versus sham, Figure 6B). CR8-treated LFP rats showed cognitive improvements with significantly reduced latency to find the platform on PIDs 16 (P=0.022, versus vehicle) and 17 (P=0.01, versus vehicle) when compared with vehicle-treated rats. Reference memory was assessed using MWM probe trial.6, 12 Lateral fluid percussion injury resulted in significant cognitive impairments when compared with sham rats (P<0.01, versus sham, Figure 6C). CR8-treated LFP rats exhibited significant cognitive improvements in probe trial (P<0.05, versus vehicle), when compared with vehicle-treated rats. All rats performed well in visual cue test and swim speeds did not differ across groups (data not shown). Retention or intact memory was evaluated using the NOR test. 17 Lateral fluid percussion injury resulted in significant cognitive impairments when compared with sham rats (P<0.001 or <0.01, versus sham, Figure 6D). CR8 treatment resulted in significant improvements in retention memory performance (P<0.05, versus vehicle). Nonspatial contextual/emotional memory was assessed using passive avoidance test. 18 CR8 treatment resulted in a significant attenuation of LFP-induced cognitive deficits (P<0.05, versus vehicle, Figure 6E).
CR8 Treatment Decreases Lateral Fluid Percussion-Induced Depression
Depressive-like behavior was evaluated using forced swim test. 19 Lateral fluid percussion-induced depressive-like symptoms (P<0.05, versus sham, Figure 6F) were significantly alleviated by CR8 treatment (P<0.05, versus vehicle).
DISCUSSION
Lateral fluid percussion injury is an experimental TBI model that simulates many pathobiological features of human contusive head trauma.12, 24, 25, 26 The LFP model produces both focal and diffuse injury with vascular disruption, neuronal cell death, and glial proliferation.12, 24, 25, 26 The involvement of CCA as a key pathophysiological mechanism for neuronal cell death in central nervous system injury is well established.4, 5, 6, 8, 9, 10, 27 CR8, an N6-biaryl-substituted derivative of the selective CDK inhibitor, roscovitine, was synthesized to generate a second-generation analog with greater therapeutic potential. 28 CR8 not only exhibits higher solubility and 50-fold greater potency in vitro, 28 but also 10 or 20 times higher potency in vivo. 10 In order to simulate a more clinically relevant treatment paradigm, we administered CR8 systemically at 3 hours post injury and investigated its long-term neuroprotective effects on LFP injury-induced neurologic deficits, neurodegeneration, and neuroinflammation. Physiologic monitoring of LFP injury revealed a drop in oxygen saturation levels (to 71.6±7.96%) at the onset of TBI, which recovered to 98.28±0.32% within 1 minute (data not shown). There were no significant changes in breathing and heart rates after LFP (data not shown), and systemic administration of CR8 did not affect any of the physiologic parameters (data not shown).
We used BrdU labeling, a thymidine analog and a marker of DNA synthesis to establish the presence and role of DNA replication, a key component of the S phase of cell cycle in posttraumatic responses in three types of cellular population: neurons, microglia/macrophages, and astrocytes. 29 The dose of BrdU used in this study was previously shown to be required for optimal identification of DNA synthesis. 13 Thus, when administered within the final 24 hours before killing, it serves to identify active DNA replication processes at that time. At 24 hours after LFP, a time of intense CCA and neuronal degeneration,4, 5, 6, 8, 9, 10 we failed to detect colocalization of BrdU with NeuN, Iba-1, and GFAP. These data suggest that CCA in mature neurons, microglia/macrophages, and astrocytes fails to progress to the S phase of DNA replication at 24 hours. The ability of BrdU to label cells engaged in DNA synthesis was demonstrated by the presence of BrdU-positive cells in the SVZ (subventricular zone) and DG (data not shown). Interestingly, we observed a striking colocalization between BrdU and NeuN at 7 days, particularly at the lesion site, and the morphology of these cells indicated degenerating neurons. In addition, higher numbers of BrdU/Iba-1 and BrdU/GFAP double-positive cells were observed at 7 days, indicating the involvement of proliferation in microglial and astrocyte activation following LFP. Previous studies have reported that microglia/macrophages, astrocytes, and neurons show signs of proliferation at 14 days after TBI.30, 31 Here we demonstrate that at least until 7 days after LFP injury, BrdU-positive cellular population evidently comprises of neurons, microglia, and astrocytes. The three cell types altogether constituted close to 80% of the total cellular population undergoing the S phase of cell cycle. Interestingly, we observed that the largest component of BrdU-positive cells is represented by microglia. Previous studies have correlated the proliferating cell differentiation in an injured brain with the degree of cellular repair and restoration.30, 31 Notably, in our study, based on the morphologic appearance of degenerating neurons and activated astrocytes/microglia, CCA/proliferation appears to be occurring before or in parallel with neurodegeneration and neuroinflammation, respectively.
We have previously demonstrated the significant role of CCA as the principal pathophysiological mechanism using multiple models of central nervous system trauma. Both experimental TBI and spinal cord injury led to elevated expression of several important cell cycle markers: CDK1, n-myc phosphorylation at Ser (54) indicating specific CDK1 inhibition, phosphorylated CDK, CDK co-activators such as cyclins, and pro-apoptotic E2F1 transcription factor and phospho-Rb.6, 8, 9, 32, 33 Interestingly, cell cycle inhibition via systemic administration of a selective CDK inhibitor, CR8 downregulated the expression of CDKs and other cell cycle proteins.10, 32, 33 In this study, we validated the elevated expression of certain key cell cycle markers (cyclin G1, phospho-Rb, E2F1, and PCNA) in the injured cortex at 24 hours. We observed predominant colocalization of cyclin G1 and PCNA with NeuN indicating that the majority of cells showing CCA at 24 hours were neurons. Importantly, the administration of CR8 attenuated all these changes. The sequential phosphorylation of Rb family and activation of E2F (E2 promoter binding factor) family of transcription factors has an important role in initiation of the first stage of cell cycle, G1 phase. E2F transcription factors may lead to apoptosis in mature postmitotic neurons via activation of p53 and p73, 34 B- and C-myb genes 35 and upregulation of caspases-3, 9, 8 and Apaf-1. 36 Proliferating cell nuclear antigen is a marker of late G1/early S phase during cell cycle progression, and its role in neuronal death has been suggested in models of neurodegeneration and excitotoxicity.7, 37 The contribution of cyclin G1 expression and Rb phosphorylation in neuronal cell death has been demonstrated in the experimental models of ischemia, neurotrauma, excitotoxicity, and neurodegeneration.6, 38 Moreover, cyclin G1 has a role in G2/M-phase transition to induce apoptosis of mature neurons. 6 Thus, our results indicate that LFP injury strongly induces CCA in neurons, and the ability of CR8 treatment to inhibit CDKs and significantly reduce CCA upregulation in neurons may be an essential part of its neuroprotection.
Lateral fluid percussion injury resulted in widespread neurodegeneration, as indicated by numerous Fluoro-Jade B-positive neurons in the injured hemisphere at 24 hours, and CR8 treatment significantly attenuated these changes. In order to examine temporal profile of LFP injury-induced neurodegeneration, we used stereological techniques to quantify neuronal loss in CA1, CA3, and DG sub-regions of the hippocampus, cortex, and thalamus, as well as lesion volume. We have previously demonstrated that controlled cortical impact causes progressive hippocampal neuronal cell loss, and that treatment with selective CDK inhibitors significantly improves hippocampal neuronal survival.8, 9, 10 This is the first reported detailed stereological assessment of progressive neuronal loss in specific hippocampal sub-regions, cortex, and thalamus following LFP injury. Neuronal loss in the CA1, CA3, and DG regions of hippocampus was observed as early as 24 hours, followed by a progressive albeit slower neurodegeneration at later time points. Notably, CR8 treatment significantly improved neuronal survival in the CA1 and CA3 sub-regions at 28 days post injury. In addition, CR8-treated LFP rats showed significantly improved DG neuronal recovery at all time points. We observed a similar pattern of progressive neurodegeneration continuing through 28 days in the cortex and thalamus. CR8 treatment significantly improved cortical and thalamic neuronal survival at 7 and 28 days and also significantly reduced LFP-induced lesion size.
Lateral fluid percussion injury caused cognitive impairment in spatial learning, as well as reference and retention memory. Studies have suggested that functional improvements in behavioral tasks are correlated to neuronal integrity in specific brain regions.8, 9, 10, 39 Cognitive performance in the MWM in part reflects the hippocampal integrity.8, 9, 10 The parameters assessed in the acquisition phase and probe trial evaluate spatial learning and reference memory, respectively.8, 9, 10 The object exploration and discrimination trials of NOR task assess retention or intact memory.9, 10, 17 CR8 treatment significantly improved each of these cognitive aspects. Furthermore, we assessed nonspatial fear-based retention memory function using the passive avoidance task that may reflect amygdala- and hippocampal-dependent contextual and emotional memory and learning ability. 18 CR8 treatment alleviated LFP-induced impairment in fear-based contextual and emotional memory function in the passive avoidance task. Overall, CR8-induced cognitive improvement can be attributed at least in part to neuronal preservation in the hippocampus.
Lateral fluid percussion injury impaired the performance on the composite neuroscore, which is designed to offer a comprehensive assessment of the motor recovery and includes a combination of individually scored sensitive tests.5, 12, 14, 16 The injury not only impacts the sensorimotor cortex but also compromises more distant regions that have important roles in motor function including the thalamus.6, 26 CR8-treated LFP rats showed markedly improved sensorimotor outcomes in composite neuroscore test compared with the vehicle-treated LFP groups. We also assessed depressive-like symptoms using the forced swim task. 19 LFP-injured rats showed classical signs of depression, which were significantly alleviated by CR8 treatment.
Chronic neuroinflammation following central nervous system trauma may provide a mechanistic connection between early and chronic neurodegeneration.11, 40 Our previous studies have indicated that sustained microglial and astrocyte activation after central nervous system trauma may have a role in the chronic neuronal cell loss and loss of neurologic function.11, 27 To better assess the neuroinflammatory responses, we performed a quantitative assessment based on the morphologic characterization of microglial and astrocyte activation in the injured cortex. This is the first detailed analysis of the temporal profile of cortical microglial and astrocyte activation after LFP injury using unbiased stereological techniques and sensitive morphologic criteria.
Based on morphology, microglia can be classified into three categories corresponding to increasing activation status: ramified (resting), hypertrophic, and bushy.8, 9, 11, 21 Traumatic brain injury causes transformation of ramified microglia into the more active phenotypes, hypertrophic, and bushy8, 9, 11, 21 and the relative proportion of these microglial phenotypes determines the extent of microglial activation and associated neuroinflammation. We observed significant increases in activated hypertrophic and bushy microglial phenotypes at 7 and 28 days, but not at 24 hours, following LFP injury. Traumatic brain injury also causes astrocyte activation from a resting form to a more reactive morphology and this response may contribute to the secondary injury.22, 23 Resting astrocytes are characterized by fine long fibers emanating from cell body with relatively lighter GFAP staining in the cytoplasm whereas the reactive astrocytes exhibit a hypertrophic or fully activated morphology characterized by thicker, more intensely stained fibers or shortened processes with intense GFAP staining, respectively.22, 23 The numbers of reactive astrocytes were significantly elevated at 7 days and 28 days after LFP injury when compared with sham controls. Previous studies suggested that CDK inhibitors reduce glial scar formation and microglial activation after TBI.4, 5, 6, 8, 9 Here we demonstrate that systemic administration of CR8 consistently attenuated microglial and astrocyte activation as indicated by the reduced numbers of activated bushy microglia and reactive astrocytes. The robust effect of CR8 on reactive microglia and astrocytes might reflect an important component of its overall neuroprotection.
In conclusion, our studies underscore the contribution of CCA to the progressive neurodegeneration and chronic neuroinflammation in pathophysiology of TBI using the rat LFP model. Lateral fluid percussion-induced CCA is associated with widespread S-phase induction in neurons, microglia/macrophages and astrocytes, with microglia constituting the largest component of BrdU-positive cells. We demonstrated that a delayed systemic administration of the selective and potent CDK inhibitor, CR8, inhibited LFP-induced CCA and functional deficits and reduced lesion size. In addition, CR8 reduced progressive neuronal cell loss/neurodegeneration in the cortex, thalamus and hippocampus, and attenuated cortical microglial and astrocyte activation/neuroinflammation. The therapeutic effects of CR8 likely reflect its multipotential neuroprotective activities due to direct inhibitory effects on CCA-dependent neurodegeneration, as well as microglial/macrophage- and astrocyte-mediated neuroinflammation. Our study provides further evidence that selective CDK inhibitors show therapeutic promise for treatment of clinical TBI.
Footnotes
The authors declare no conflict of interests.
ACKNOWLEDGMENTS
We thank Rainier Cabatbat, Katherine Cardiff, Kelsey Guanciale, Juliane Faden, and Tess Maseda for expert technical assistance.