
Abstract
The presenilin-associated rhomboid-like (PARL) protein and high temperature requirement factor A2 (HtrA2) are key regulators of mitochondrial integrity and play pivotal roles in apoptosis. However, their roles after cerebral ischemia have not been thoroughly elucidated. To clarify these roles, mice were subjected to transient global cerebral ischemia, and striatal neuronal injury was assessed. Western blot and coimmunoprecipitation analyses revealed that PARL and processed HtrA2 localized to mitochondria, and that PARL was bound to HtrA2 in sham animals. Expression of PARL and processed HtrA2 in mitochondria significantly decreased 6 to 72 hours after ischemia, and the binding of PARL to HtrA2 disappeared after ischemia. In contrast, expression of processed HtrA2 increased 24 hours after ischemia in the cytosol, where HtrA2 was bound to X chromosome-linked inhibitor-of-apoptosis protein (XIAP). Administration of PARL small interfering RNA inhibited HtrA2 processing and worsened ischemic neuronal injury. Our results show that downregulation of PARL after ischemia is a key step in ischemic neuronal injury, and that it decreases HtrA2 processing and increases neuronal vulnerability. In addition, processed HtrA2 released into the cytosol after ischemia contributes to neuronal injury via inhibition of XIAP.
Keywords
INTRODUCTION
The integrity of mitochondria is fundamental to cell life.1,2 Disruption of this integrity induced by cellular stress causes an abatement in adenosine triphosphate production, oxidative damage, and release of multiple mitochondrial proapoptotic proteins, such as cytochrome c, second mitochondria-derived activator of caspases/direct inhibitor-of-apoptosis binding protein with low pI, and apoptosis-inducing factor, all of which are involved in the pathogenesis of numerous disorders.3,4 Mitochondrial dysfunction also plays a pivotal role in ischemic neuronal injury, and maintaining mitochondrial integrity against cerebral ischemia is a very important cellular response.5-7
The presenilin-associated rhomboid-like (PARL) protein is a member of the rhomboid family and is located in the inner mitochondrial membrane. As with other rhomboids, PARL is an intramembrane serine protease. 8 Previous studies in both animal models and humans have suggested that PARL is a key regulator of mitochondrial integrity and function and that it plays an important role in apoptosis.8-10 Because cells lacking PARL were more susceptible to apoptosis, PARL is regarded as an antiapoptotic protein. 11 In addition, recent studies revealed that PARL participates in the processing of high temperature requirement factor A2 (HtrA2) in mitochondria.12,13 The HtrA2, also known as Omi, is a mammalian homolog of the bacterial heat-inducible serine protease known as HtrA14,15 and is synthesized in the nucleus as a precursor containing a mitochondrial target sequence. When imported into mitochondria, it is processed to a mature form and resides in the intermembrane space. Originally described as a proapoptotic protein, 16 recent studies have shown that HtrA2 in neurons exerts either proapoptotic or antiapoptotic activities, depending on its subcellular localization; within mitochondria, processed HtrA2 maintains mitochondrial integrity against cellular stress.17,18 When released into the cytosol after cell death stimuli, however, the processed form interacts with the X chromosome-linked inhibitor-of-apoptosis protein (XIAP) through its processed N-terminal and antagonizes XIAP, resulting in the promotion of caspase activation and apoptosis.15,16,19
Because both the mitochondrial proteases, PARL and HtrA2, are deeply involved with mitochondrial function and cell death, they may play important roles in cerebral ischemia. In fact, several studies have shown the role of HtrA2 in cerebral ischemia;20-22 however, these studies were focused on the role of HtrA2 released into the cytosol and did not investigate its role in mitochondria. With regard to PARL, its role in cerebral ischemia remains unknown. This study was designed to clarify the roles of PARL and HtrA2 in ischemic striatal neuronal injury using a mouse model of transient global cerebral ischemia (tGCI).
MATERIALS AND METHODS
Global Cerebral Ischemia
All animals were treated in accordance with the Stanford University guidelines and the animal protocols were approved by Stanford University's Administrative Panel on Laboratory Animal Care. Male C57BL/6J mice (8 to 12 weeks) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Anesthesia was induced with inhalation of 4% isoflurane and intraperitoneal injection of xylazine (4 mg/kg) and maintained with 1.5% isoflurane in 70% nitrous oxide and 30% oxygen via a face mask. Rectal temperatures were maintained at 37 ± 0.5 °C with a heating blanket (Harvard Apparatus, Holliston, MA, USA) and a heating lamp. Regional cerebral blood flow (rCBF) was monitored by laser Doppler flowmetry (Laserflo BMP2; Vasamedics, Eden Prairie, MN, USA). The probe was fixed on the skull, 4 mm lateral to the bregma. Transient global cerebral ischemia was induced by applying microaneurysm clips to both common carotid arteries. Changes in rCBF after ischemia were expressed as a percentage of the preischemic value. Mice whose rCBF decreased to <13% of the preischemic value were used. Sham-operated animals were treated as described above but without clamping the carotid arteries. After reperfusion, the skin incision was sutured and the animals were wrapped with a heating blanket to maintain rectal temperatures above 36.0 °C for 24 hours after surgery. Acetate Ringer's solution (0.5 ml) was administered subcutaneously in all animals 30 minutes and 24 hours after reperfusion. Using this method, striatal neurons were consistently injured after 22 minutes of tGCI,23,24 after which striata were used as samples for western blotting, coimmunoprecipitation, and immunohistochemistry. For assessment of the effects of the treatments on ischemic neuronal injury, we used 17-minute and 22-minute tGCI models as mild and severe ischemia, respectively.
Western Blot Analysis
Both sides of the striatum were removed 3,6,24, or 72 hours after 22 minutes of tGCI. Protein extraction of the cytosolic and mitochondrial fractions was performed using a multiple centrifugation method as described previously. 25 Protein concentrations were determined by comparison with a known concentration of bovine serum albumin using a kit (Pierce, Rockford, IL, USA). Equal amounts of samples were loaded per lane. Sodium dodecyl sulfatepolyacrylamide gel electrophoresis was performed on a 10% NuPAGE Bis-Tris gel (Invitrogen, Carlsbad, CA, USA) and then immunoblotted. Anti-PARL (1:500, ARP44851; Aviva Systems Biology, San Diego, CA, USA), anti-HtrA2 (1:2,000, AF1458; R&D Systems, Minneapolis, MN, USA), anti-cytochrome c (1:1,000, 556433; BD Biosciences, San Jose, CA, USA), anti-spectrin (1:5,000, MAB1622; Millipore, Billerica, MA, USA), anti-β-actin (1:100,000, A5441; Sigma-Aldrich, St Louis, MO, USA), or anti-cytochrome oxidase subunit IV (COX-IV) (1:2,000, A21348; Invitrogen) primary antibodies were used. After incubation with an appropriate horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology, Beverly, MA, USA), the bound antibodies were detected by a chemiluminescence system (Pierce). Images were scanned and the results were quantified using Multi-Analyst software (Bio-Rad Laboratories, Hercules, CA, USA).
Immunohistochemistry
Anesthetized animals were perfused with 10 U/mL heparin saline and subsequently with 4% paraformaldehyde in phosphate-buffered saline (PBS) 6, 24, or 72 hours after 22 minutes of tGCI. Brains were removed, postfixed for 24 hours, and cut on a vibratome into slices 30 μm thick at the level of the striatum (from bregma + 1.1 mm to 0.1 mm).
Double immunostaining was performed with immunofluorescence. Brain sections were reacted with a primary antibody, then incubated with appropriate Alexa 488- or 594-conjugated immunoglobulin G (IgG) antibodies (Invitrogen). Negative controls were treated with similar procedures, except that the primary antibody was omitted. The sections were covered with VECTASHIELD mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA) and examined under an Axioplan 2 microscope (Carl Zeiss, Thornwood, NY, USA). We used the following as primary antibodies: anti-PARL (1:50), anti-HtrA2 (1:25, sc-15467; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti-neuronal nuclei (NeuN, a neuronal marker) (1:200, MAB377; Millipore).
For double immunofluorescent staining of PARL and COX-IV, the sections were reacted with a rabbit antibody against PARL (1:50), followed by an Alexa 594-conjugated anti-rabbit IgG antibody (Invitrogen). After blocking the unbound secondary antibody with 20% rabbit normal serum, the sections were reacted with an Alexa 488-conjugated anti-rabbit COX-IV antibody (1:50, 4853; Cell Signaling Technology).
We performed double staining of PARL and terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL) using a fluorescent method to clarify the spatial relationship between PARL expression and DNA fragmentation. At 24 hours after 22 minutes of tGCI, the brain sections were reacted with the PARL antibody as described above, then incubated with Alexa 594-conjugated anti-rabbit IgG (Invitrogen). Subsequently, we performed TUNEL staining with a commercial kit (11684817910; Roche Molecular Biochemicals, Indianapolis, IN, USA), following the manufacturer's protocol. The slides were covered with VECTASHIELD mounting medium with DAPI.
Coimmunoprecipitation
Both sides of the striatum were removed 24 hours after 22 minutes of tGCI (n = 3). Tissue was homogenized and sonicated in ice-cold 1 x lysis buffer (Cell Signaling Technology) with 1% protease inhibitor mixture (Sigma-Aldrich). The homogenate was centrifuged (900 g for 10 minutes at 4 °C), and the resulting supernatant was used for quantification. Protein concentrations were determined by comparison with a known concentration of bovine serum albumin using a kit (Pierce). Coimmunoprecipitation experiments were performed using Dynabeads magnetic beads covalently coupled with Protein G (Invitrogen). The Dynabeads were incubated with 1 μg of rabbit polyclonal anti-HtrA2 antibody (AF1458; R&D Systems) or an equivalent amount of normal rabbit IgG. After washing three times with 200 μL of PBS containing 0.01% Tween-20 (PBST), the beads were incubated with 60 μg of protein samples in 200 μL of PBST for 2 hours at 4 °C with gentle agitation. After washing three times with PBS, 22 μL of lithium dodecyl sulfate sample buffer were added to the beads, and the samples were boiled. Then, 20 mL of the supernatant was immunoblotted with an anti-XIAP antibody (1:1,000,610762; BD Biosciences), the anti-PARL antibody, or anti-HtrA2 antibody as described above in the section on western blot analysis.
Small Interfering RNA Treatment
All small interfering RNA (siRNA) treatments were administered with 10 mM of jetSI (403-05; Polyplus Transfection, New York, NY, USA) into the right striatum (bregma: 2.5 mm lateral, 0.5 mm anterior, 3.0 mm deep) as described previously. 26 JetSI was used with 20 mmol/L of dioleoylphosphatidylethanolamine (P1223; Sigma-Aldrich) in a 1:2 molar ratio. JetSI and dioleoylphosphatidylethanolamine were used in a nitrogen/phosphate ratio of 1.8. Transfection mixes containing 0.2 μg/μL siRNA (for 1 μg siRNA injection) were prepared according to the manufacturer's protocol. We used siSTABLE non-targeting control siRNA and siSTABLE PARL-siRNA (Dharmacon, Lafayette, CO, USA). Nontargeting control siRNA (D-001700-01-05; Dharmacon) was used as an off-site siRNA control. siSTABLE PARL-siRNA was used to inhibit PARL expression, and is a siRNA targeting the PARL sequence of 50-AUUAACAAGUGGUGGAAUA-30 with in vivo siSTABLE option, which chemically modifies the enhancing stability of siRNAs even under non-RNase-free conditions. For titration, control-siRNA and siSTABLE PARL-siRNA were injected into the right striatum (0.5, 1, and 2 μg). Animals were killed 48 hours after injection and the right striata were removed for the western blotting samples. For inhibition of PARL after tGCI, 2 μg of control siRNA or siSTABLE PARL-siRNA were injected into the right striatum 48 hours before tGCI.
Histologic Analysis of Striatal Injury
Injury to the right striatum was histologically evaluated 72 hours after tGCI by cresyl violet and TUNEL staining. For TUNEL staining, we used the commercial kit described above, and this staining was visualized with diaminobenzidine. Nuclei were counterstained with hematoxylin solution.
Five subregions (central, dorsomedial, dorsolateral, ventromedial, and ventrolateral) were assigned for quantification of TUNEL staining, each consisting of a rectangle of 250 μm x 174 μm. The TUNEL-positive cells in each subregion of the right striatum were counted by two masked counters.
Cell Death Assay
For quantification of apoptosis-related DNA fragmentation in the right striatum, we used a commercial enzyme immunoassay (11774425001; Roche) to determine cytoplasmic histone-associated DNA fragments, which detect apoptotic but not necrotic cell death. The right striata were removed 72 hours after tGCI and samples were prepared as described for the western blotting method. A cytosolic volume containing 20 μg of protein was used for the enzyme-linked immunosorbent assay, according to the manufacturer's protocol.
Statistical Analysis
Statistical analyses were performed by analysis of variance with Dunnett's multiple comparison post hoc test (SigmaStat; Systat Software, Chicago, IL, USA). Comparisons between two groups were achieved with a Student's unpaired t-test. Data are expressed as mean ± s.d. and significance was accepted with P<0.05.
RESULTS
Expression of Presenilin-Associated Rhomboid-Like Protein Was Localized in Neuronal Mitochondria and Decreased After Transient Global Cerebral Ischemia
Western blot analysis showed that PARL immunoreactivity was evident with a molecular mass of 36 kDa, and its expression was localized in mitochondria (Figure 1A). In the mitochondrial fraction, there was a gradual decrease in the expression of PARL after tGCI. A quantitative analysis showed that mitochondrial PARL expression was decreased significantly 6 to 72 hours after tGCI (P<0.05; Figure 1B). Expression of PARL in the cytosolic fraction was not detected even after tGCI.
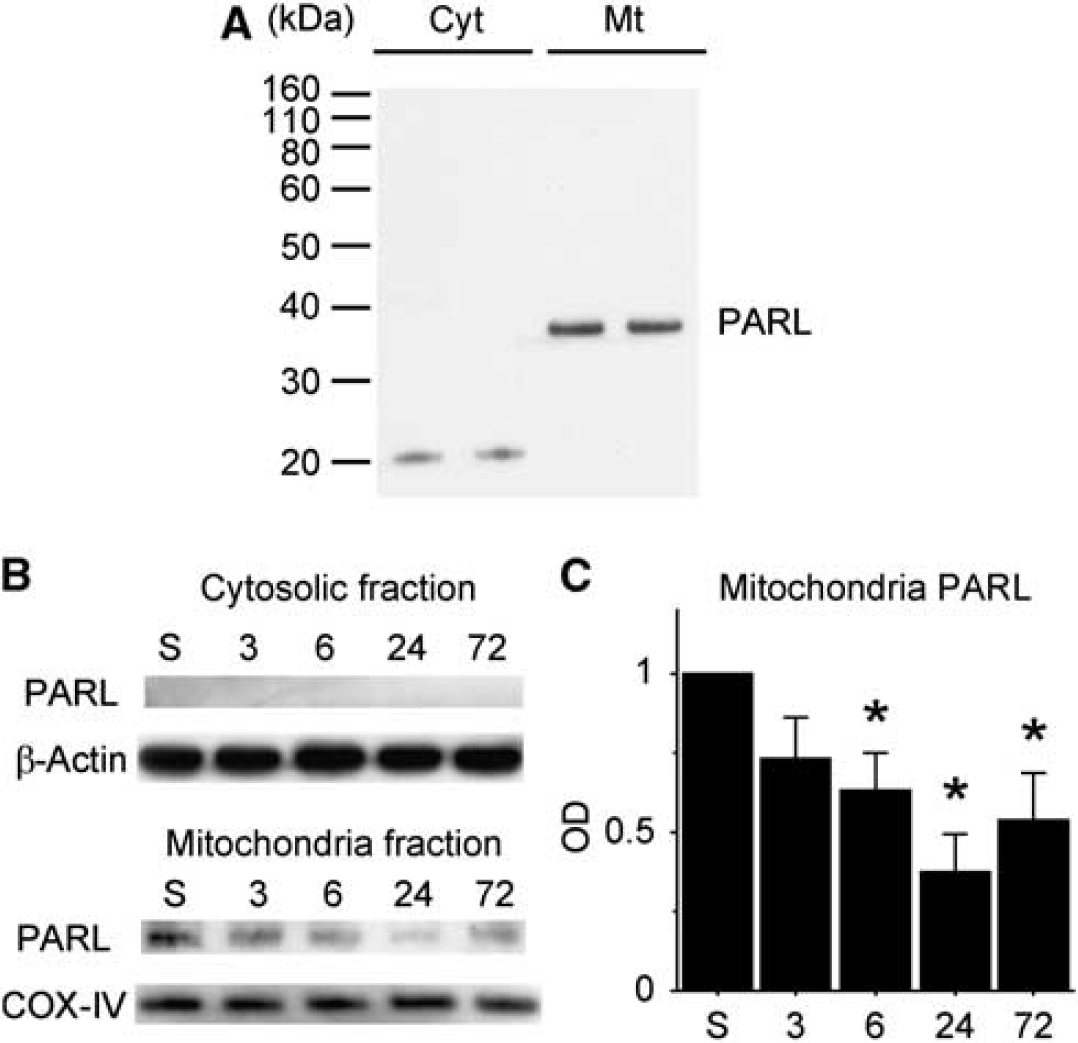
Western blot analysis of presenilin-associated rhomboid-like (PARL) protein in the striatum. (
Immunohistochemistry revealed that expression of PARL was decreased after ischemia, which was consistent with the results of the western blot analysis (Figure 2A). Double immunofluorescence showed PARL-positive cells colocalized with NeuN-positive cells and COX-IV-positive cells (Figures 2B and 2C). These results reveal that PARL is expressed in neuronal mitochondria in the striatum and that PARL expression decreases after tGCI.
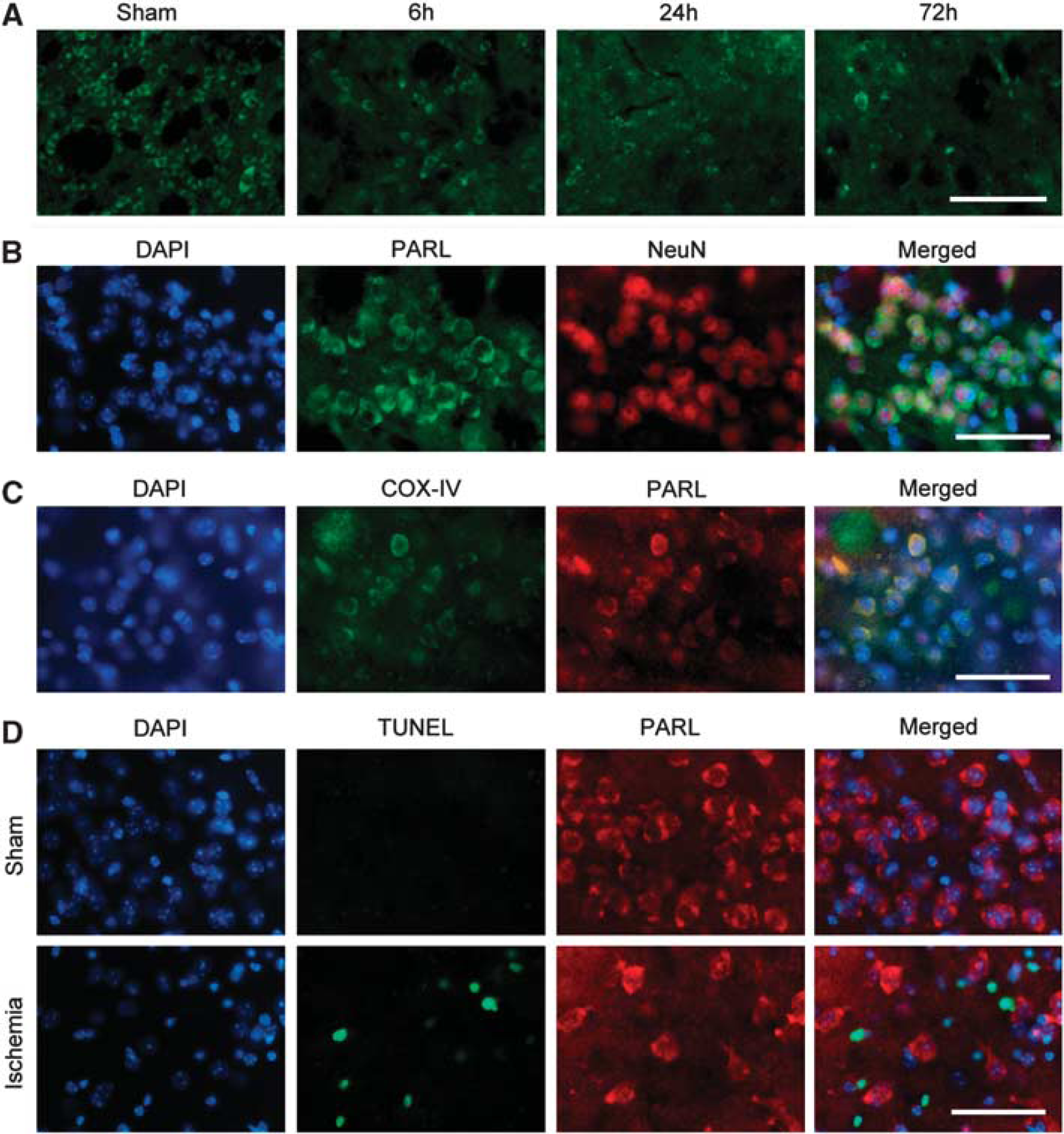
Representative photomicrographs of presenilin-associated rhomboid-like (PARL) protein immunohistochemistry. (
To investigate the relationship between PARL expression and apoptosis, we performed double immunofluorescence for PARL and TUNEL. The TUNEL-positive cells were observed in the striatum 24 hours after tGCI. There were few PARL-positive cells at this time point, and these cells did not colocalize with the TUNEL-positive cells (Figure 2D). These results suggest that the cellular population of PARL is different from that of DNA fragmentation after tGCI.
Expression of High Temperature Requirement Factor A2 in the Striatum After Transient Global Cerebral Ischemia
Western blot analysis of HtrA2 showed unprocessed forms at 50 kDa and processed forms at 38 kDa (Figure 3A). Under normal conditions, both unprocessed and processed forms were observed in the mitochondrial fraction, and only the unprocessed forms were observed in the cytosolic fraction. After tGCI, expression of processed HtrA2 in the mitochondrial fraction showed a gradual decrease in a time-dependent manner. A quantitative analysis showed that processed HtrA2 in mitochondria was decreased significantly 6 to 72 hours after tGCI (P<0.05). Unprocessed forms in mitochondria tended to decrease after tGCI, but this decrease was not statistically significant. In the cytosolic fraction, expression of processed HtrA2 increased after tGCI, with a significant difference at 24 hours (P<0.05). Expression of unprocessed forms in the cytosolic fraction did not change significantly (Figure 3B).
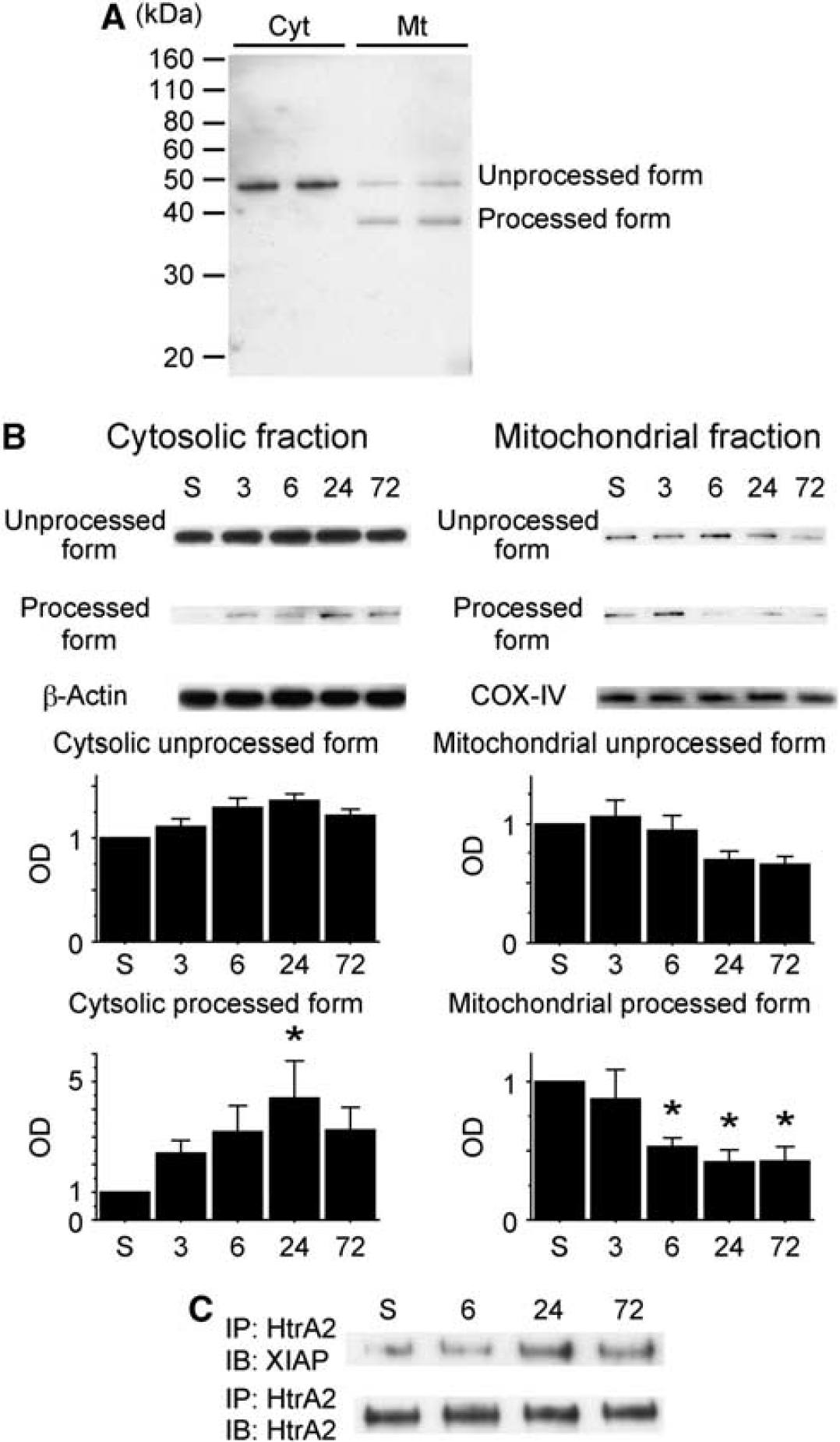
Western blot analysis of high temperature requirement factor A2 (HtrA2) in the striatum. (
Interaction Between X Chromosome-Linked Inhibitor-of-Apoptosis Protein and High Temperature Requirement Factor A2 To investigate the interaction between XIAP and HtrA2, we performed coimmunoprecipitation. The expression of XIAP precipitated by HtrA2 showed a gradual increase in a time-dependent manner after tGCI (Figure 3C). This result indicates that HtrA2 interacts with XIAP after tGCI.
Interaction Between Presenilin-Associated Rhomboid-Like Protein and High Temperature Requirement Factor A2
To investigate the interaction between PARL and HtrA2, we performed coimmunoprecipitation and double immunofluorescence. The immunoreactivity of PARL precipitated by HtrA2 was observed in the sham animals; however, it disappeared 24 hours after tGCI (Figure 4A). Double immunofluorescence showed that PARL colocalized with HtrA2 in the striatum of the sham animals (Figure 4B). These results suggest that PARL interacts with HtrA2 under nonischemic conditions and that this interaction decreases after tGCI.
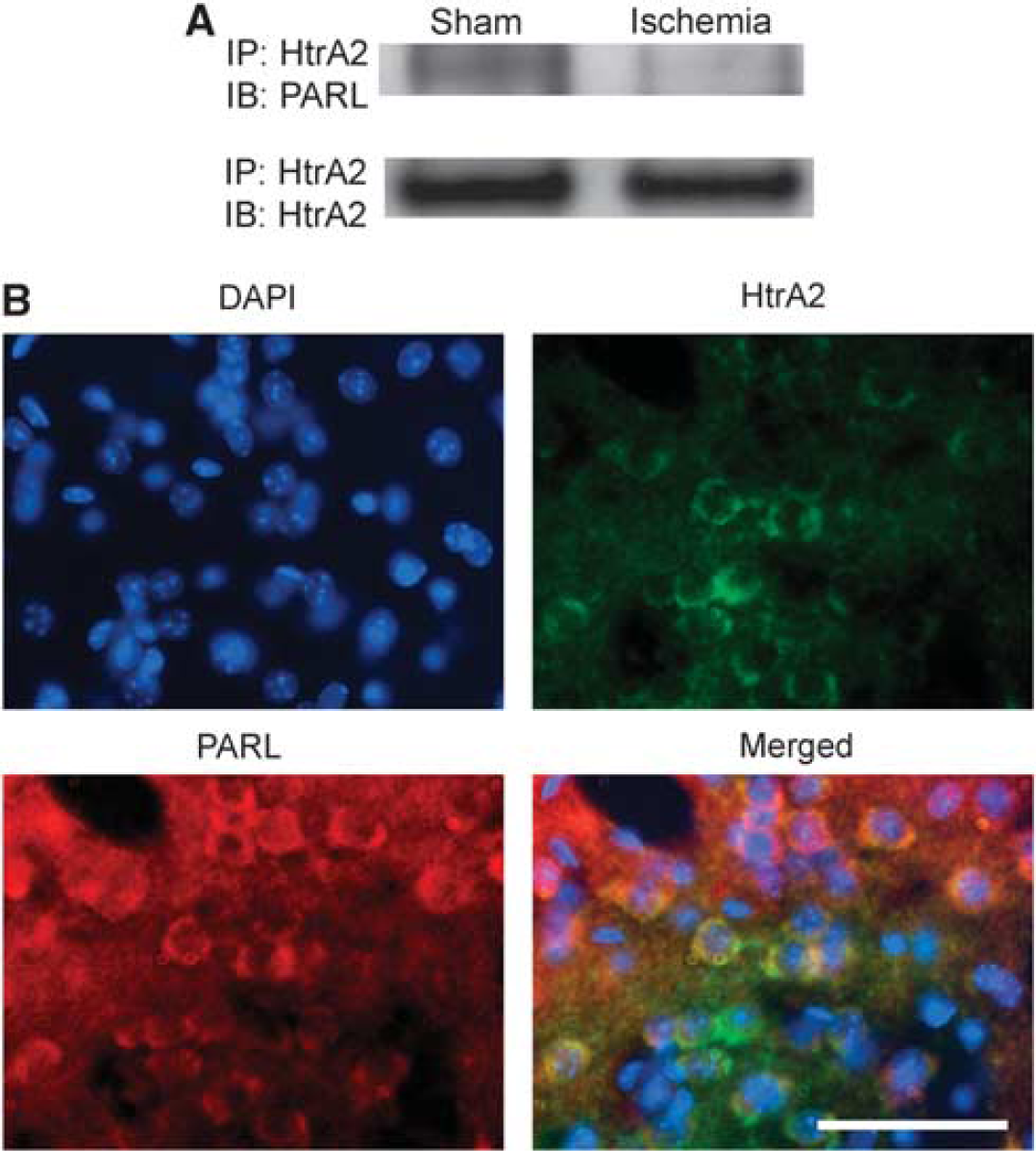
Interaction between presenilin-associated rhomboid-like (PARL) protein and high temperature requirement factor A2 (HtrA2). (
Presenilin-Associated Rhomboid-Like Protein-Small Interfering RNA Reduced Expression of Processed High Temperature Requirement Factor A2
To examine the effects of PARL inhibition on the processing of HtrA2, we used PARL-siRNA. Western blot analysis showed that the expression of PARL in the mitochondrial fraction was significantly reduced in the animals treated with 2 μg of PARL-siRNA compared with the non-treated mice (P<0.05). In addition, expression of processed HtrA2 in the mitochondrial fraction was significantly decreased with 2 μg of PARL-siRNA treatment (P<0.05), although expression of unprocessed HtrA2 did not change significantly (Figure 5). In contrast, PARL and processed HtrA2 were not detected in the cytosolic fraction of either control or PARL-siRNA-treated animals because they were in the nontreated animals. Expression of unprocessed HtrA2 in the cytosolic fraction was not changed with siRNA treatment.
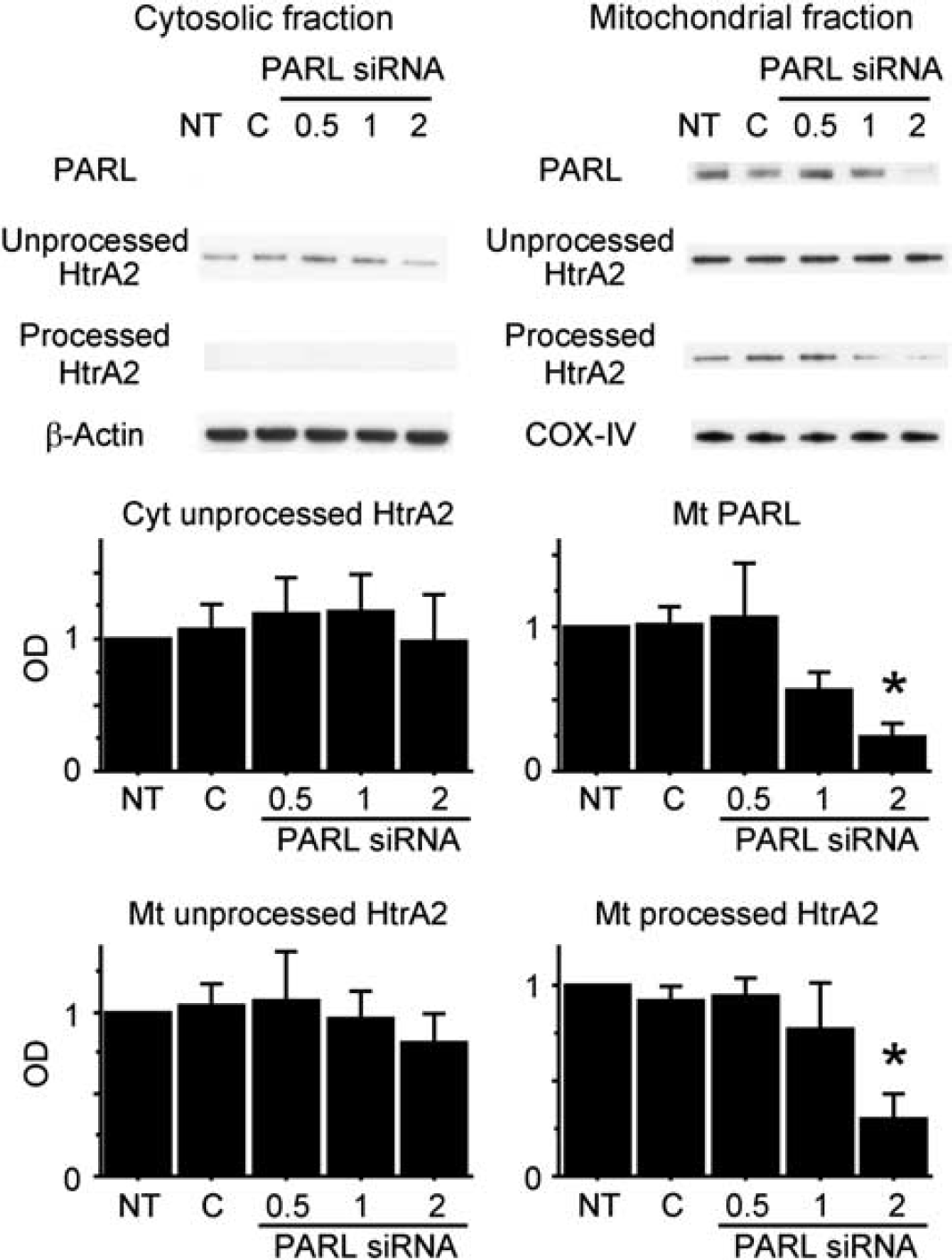
The effects of presenilin-associated rhomboid-like protein-small interfering RNA (PARL-siRNA) on the expression of pArL and high temperature requirement factor A2 (HtrA2). Control and PARL-siRNA (0.5, 1, or 2 μg) were administered into the right striatum. The animals were killed 48 hours after the siRNA injection. Western blot analysis showed that expression of PARL and processed HtrA2 in the mitochondrial fraction (Mt) was significantly decreased in the animals treated with 2 μg PARL-siRNA compared with the non-treated animals (n = 4, ∗P<0.05). In the cytosolic fraction (Cyt), expression of HtrA2 and PARL was not changed with siRNA treatment. As internal controls, β-actin and cytochrome oxidase subunit IV (COX-IV) were used for the cytosolic and mitochondrial fractions, respectively. C, control; NT, nontreated; OD, optical density.
Presenilin-Associated Rhomboid-Like Protein-Small Interfering RNA Worsened Ischemic Injury in the Striatum After Transient Global Cerebral Ischemia
We then investigated the effects of PARL-siRNA on neuronal injury after tGCI. A total of 2 μg PARL-siRNA was injected into the right striatum 48 hours before surgery. In the cresyl violet-stained sections, ischemic change 3 days after 17 minutes of tGCI was mild in the control siRNA treatment group; however, severe ischemic change was observed in the PARL-siRNA treatment group. In the 22-minute tGCI group, severe damage was observed in both the control and PARL-siRNA treatment groups 3 days after ischemia (Figure 6A). These observations were confirmed by the results of TUNEL staining. A cell counting study showed that the number of TUNEL-positive cells in the PARL-siRNA treatment group was significantly higher than in the control-siRNA treatment group 3 days after 17 minutes of tGCI (P<0.05). In the 22-minute tGCI group, ischemic damage was severe in both the control and PARL-siRNA treatment groups, and there were no significant differences between them (Figure 6B). An apoptotic DNA fragmentation quantitative assay and investigation of cleaved spectrin expression supported these results. The DNA fragmentation 3 days after 17 minutes of tGCI in the PARL-siRNA treatment group was significantly higher than in the control-siRNA group (P< 0.05; Figure 6C). The spectrin antibody is known to recognize both caspase-cleaved (150 and 120 kDa) and calpain-cleaved (150 kDa) products and is used to assess brain injury after ischemia. 27 Cleaved spectrin products (both 150 and 120 kDa) were significantly higher in the PARL-siRNA group than in the control-siRNA group 24 hours after 17 minutes of tGCI (P<0.05; Figure 6D). Finally, we investigated the effects of PARL-siRNA on the mitochondria-dependent apoptotic pathway. Treatment with PARL-siRNA significantly increased cytochrome c release into the cytosol 24 hours after 17 minutes of tGCI (P<0.05) (Figure 6D).
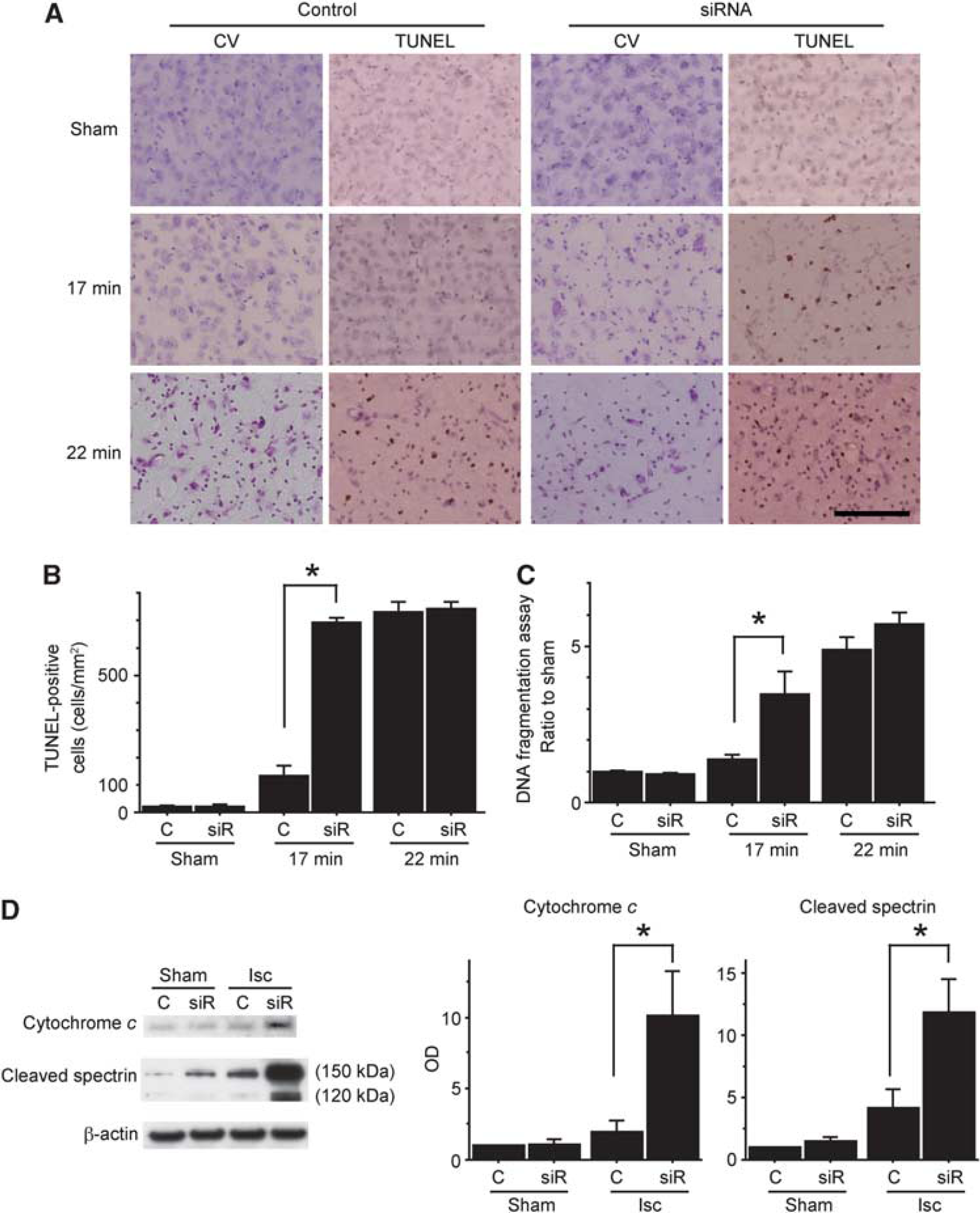
Treatment with 2 mg presenilin-associated rhomboid-like protein-small interfering RNA (PARL-siRNA) worsened ischemic injury in the striatum after transient global cerebral ischemia (tGCI). Small interfering RNA was injected into the right striatum 48 hours before surgery. (
DISCUSSION
Maintaining mitochondria is a pivotal cellular response against injurious stress, including ischemia. In this study we investigated the roles of PARL and HtrA2 (which are believed to control mitochondrial integrity) in ischemic neuronal injury. The major findings of our study are: (1) PARL resides in mitochondria of striatal neurons under physiologic conditions, and its expression is reduced after ischemia; (2) processed HtrA2 in mitochondria is decreased after ischemia at a time course similar to that of PARL, and is released into the cytosol after ischemia where it binds to XIAP;(3) PARL interacts with HtrA2 under physiologic conditions and this interaction decreases after ischemia; and (4) administration of PARL-siRNA reduces expression of processed HtrA2 in mitochondria and worsens ischemic neuronal injury.
The role of the mitochondrial rhomboid protease PARL in ischemic neuronal injury is shown for the first time in this study. Previous studies reported that PARL maintains mitochondrial integrity and plays an antiapoptotic role against intrinsic stimuli, such as tumor necrosis factor-α, etoposide, staurosporine, and H2O2,8,9,11 and the results of our study are consistent with these previous reports. The expression of PARL was not colocalized with TUNEL, and inhibition of PARL expression by siRNA increased the vulnerability of neurons to ischemic stress. Thus, PARL is neuroprotective against cerebral ischemia, and the downregulation of PARL after ischemia may be a key step for ischemic neuronal injury. That the decrease in PARL expression after ischemia preceded the maturation of ischemic neuronal injury also supports this possibility.
In HtrA2 mutant or knockout mice, the susceptibility of mitochondria to cell death stimuli is increased. Therefore, HtrA2 is believed to maintain mitochondrial health.17,18 Based on this evidence, one of the mechanisms that increases neuronal vulnerability by PARL inhibition could be, at least in part, the decrease in processed HtrA2 in mitochondria. However, to confirm the exact roles of HtrA2 under ischemic conditions, a study of HtrA2 overexpression is needed. Some researchers have reported on the overexpression of HtrA2. Inagaki et al. 28 reported that overexpression of HtrA2 by adenovirus vector suppressed mutant huntingtin-induced cell death in primary neurons. Liu et al. 29 reported the establishment of a transgenic mouse with neuron-specific overexpression of HtrA2 that showed normal development without any sign of apoptotic cell death. In a future study, we will investigate the effects of HtrA2 overexpression on ischemic neuronal injury.
A decrease in HtrA2 processing in mitochondria after ischemia could be induced by the downregulation of PARL, because expression of processed HtrA2 was decreased after ischemia at a time course similar to that of PARL, and HtrA2 processing in mitochondria was reduced by inhibition of PARL, although release into the cytosol would also contribute to the decrease. The other issue to be resolved is the mechanism by which HtrA2 in mitochondria protects neurons. In lymphocytes, the presence of HtrA2 prevents accumulation of activated Bax in the mitochondrial outermembrane, which would initiate cytochrome c release and cell death. 12 Moisoi et al. 30 showed that HtrA2 is necessary for preventing accumulation of unfolded protein and oxidative stress in mitochondria. A recent study by Li et al. 31 reported that HtrA2 is responsible for the maintenance of constitutive autophagy, which can eliminate damaged mitochondria. The exact protective mechanisms of HtrA2, especially under ischemic stress, remain unclear and warrant further investigation.
The results of our study showing that PARL participates in HtrA2 processing are consistent with previous reports.12,13 However, the processing mechanism is obscure. According to the report by Chao et al., 12 HS1-associated protein X-1 (HAX-1) is required for PARL-mediated processing of HtrA2. However, we did not confirm the interaction between HAX-1 and HtrA2 by coimmunoprecipitation in our preliminary study (data not shown), and some researchers have questioned the participation of HAX-1 in HtrA2 processing.8,32 Therefore, the mechanism of HtrA2 processing is another issue to be resolved.
The role of HtrA2 depends on its subcellular localization.17,18 The results of this study indicate that processed HtrA2 released into the cytosol after ischemia would induce apoptosis, in addition to the protective roles in mitochondria. In this study, HtrA2 interacted with XIAP after ischemia, which is consistent with results previously reported. 20 Similar to second mitochondria-derived activator of caspases/direct inhibitor-of-apoptosis binding protein with low pI, processed HtrA2 in the cytosol can bind and degrade XIAP, causing the release and subsequent activation of caspases.15,16,19 Processed HtrA2 in the cytosol also induces cell death in a caspase-independent manner. Some endogenous substrates of HtrA2 in the caspase-independent pathway are amyloid precursor protein, 33 ped/pea-15, 34 WARTS kinase, 35 and HAX-1. 36 However, this pathway remains largely unknown. Because reduction of processed HtrA2 induced by PARL-siRNA worsened ischemic neuronal injury, the major role of HtrA2 under ischemic conditions might be neuroprotective. However, processed HtrA2 released into the cytosol after ischemia can also contribute to the promotion of ischemic neuronal injury.
In addition to HtrA2, optic atrophy protein 1 (OPA-1) is another substrate for PARL. It is a dynamin-like GTPase involved in mitochondrial inner membrane fusion, and it plays a crucial role in the regulation of mitochondrial cristae remodeling. 37 Optic atrophy protein 1 cleaved by PARL generates a soluble intermembrane space form of OPA-1, which maintains mitochondrial cristae morphology and prevents cytochrome c release during apoptosis. 11 Mitochondria from PARL knockout cells are reported to undergo faster apoptotic cristae remodeling and cytochrome c release. 11 Therefore, there is a possibility that this PARL/OPA-1 pathway might have exacerbated ischemic neuronal injury in the mice treated with PARL-siRNA in this study.
There are other important issues to be resolved. One is that this study did not clarify to what extent PARL and HtrA2 signaling are crucial in ischemic neuronal injury. However, we conducted a study of PARL inhibition, and this inhibition study alone cannot clarify the exact role of PARL in ischemic neuronal injury. Overexpression studies of both proteins should be done to clarify their exact roles. The other important issue is that the specificity of PARL and HtrA2 signaling in ischemic neuronal death is not obvious. In this study, we evaluated the roles of this signaling in the mouse striatum. In an earlier study, we found that ischemic neuronal injury is consistently induced after prolonged transient global ischemia. 24 In addition to ischemic vulnerability, the striatum is reported to be one of the selected areas where severe neuronal loss is observed in PARL knockout and HtrA2 mutant or knockout mice.11,17,18 Accordingly, the roles of PARL and HtrA2 signaling in the other lesions are obscure. Further studies using other cerebral ischemia models that produce cortical neuronal injury and death, such as a middle cerebral artery occlusion model, are needed to resolve this issue.
Based on our results, we propose the roles of PARL and HtrA2 after cerebral ischemia to be as shown in Figure 7. Processed HtrA2, maintaining mitochondrial integrity under physiologic conditions, decreases after ischemia caused by the downregulation of PARL, which results in mitochondrial dysfunction and, finally, ischemic neuronal injury. In addition, residual processed HtrA2 is released into the cytosol, which induces apoptosis via interacting XIAP.
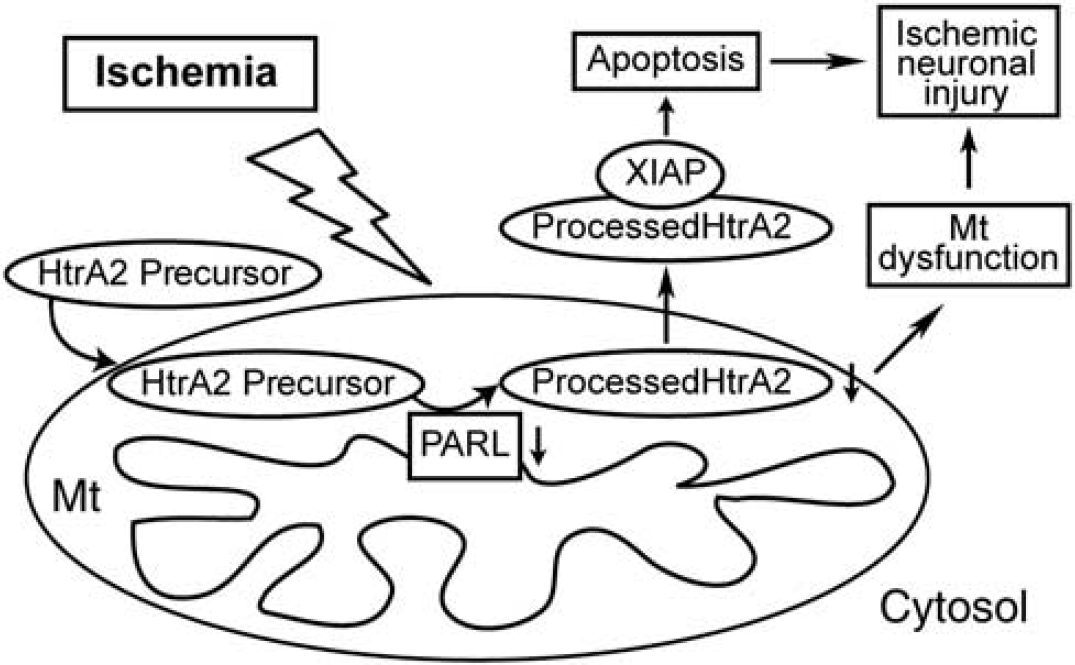
Summary of the roles of high temperature requirement factor A2 (HtrA2) and presenilin-associated rhomboid-like (PARL) protein in ischemic neuronal injury. Processed HtrA2, maintaining mitochondrial integrity under physiologic conditions, decreased after ischemia caused by the downregulation of PARL, which results in mitochondria (Mt) dysfunction and, finally, ischemic neuronal injury. In addition, the residual processed HtrA2 is released into the cytosol, which induces apoptosis via interacting X chromosome-linked inhibitor-of-apoptosis protein (XIAP).
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
ACKNOWLEDGEMENTS
The authors thank Liza Reola and Bernard Calagui for technical assistance, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for assistance with the figures.