
Abstract
Hypoglycemia-induced cerebral neuropathy can occur in patients with diabetes who attempt tight control of blood glucose and may lead to cognitive dysfunction. Accumulating evidence from animal models suggests that hypoglycemia-induced neuronal death is not a simple result of glucose deprivation, but is instead the end result of a multifactorial process. In particular, the excessive activation of poly (ADP-ribose) polymerase-1 (PARP-1) consumes cytosolic nicotinamide adenine dinucleotide (NAD+), resulting in energy failure. In this study, we investigate whether lactate administration in the absence of cytosolic NAD+ affords neuroprotection against hypoglycemia-induced neuronal death. Intraperitoneal injection of sodium
Introduction
Patients with diabetes often face a troubling pair of outcomes in the management of their disease; tight blood glucose control can reduce the risk of diabetic complications, but also increases the risk of dangerous hypoglycemic episodes. Hypoglycemia, a potentially devastating cerebral insult, is usually the result of attempting tight control of blood glucose levels with insulin or other hypoglycemic agents, such as sulphonylurea (Davis and Jones, 1998; Seltzer, 1989). Furthermore, patients suffering frequent hypoglycemic episodes or patients with long-standing type 1 diabetes due to loss of early warning symptoms are at elevated risk of experiencing a severe hypoglycemic episode. Previous studies have shown that the neuronal cell death that occurs after hypoglycemia is not simply a result of lack of glucose supply, but is instead the result of a cell death process that is initiated by the reintroduction of glucose after a sustained period of glucose deprivation (GD) (Auer et al, 1986). We have termed this process ‘glucose reperfusion-induced neuronal death’ after hypoglycemia (Suh et al, 2007). This hypothesis is also applicable in ischemia–reperfusion-induced neuronal death (Suh et al, 2008b). Currently, the only available method for preventing this hypoglycemia-induced neuronal death in the clinical setting is delivery of glucose; a treatment that paradoxically may actually exacerbate the insult. In an attempt to provide clinical recourse, we have suggested several strategies to prevent this neuronal death, including inhibition of poly (ADP-ribose) polymerase-1 (PARP-1) activation, chelation of synaptically released zinc, prevention of superoxide production, and supplementation with pyruvate during the glucose reperfusion period (Suh et al, 2003, 2004, 2005, 2007, 2008a).
It is a commonly held view that glucose is the only energy source usable by neurons. However, several studies have suggested that lactate can also serve as a substrate for neuronal energy metabolism (Schurr et al, 1997b, 1997c; Schurr, 2006). In their astrocyte–neuron lactate shuttle hypothesis, Pellerin and Magistretti (1994) proposed that lactate can be used by neurons (Magistretti et al, 1999; Pellerin et al, 2005). In addition to astrocyte-derived lactate, Boumezbeur et al (2011) and van Hall et al (2009) proposed that plasma-borne lactate may have an important role in supporting oxidative brain metabolism, suggesting that plasma-borne lactate fuels up to 60% of all cerebral metabolism and tricarboxylic acid (TCA) cycle activity even under normal physiological conditions. Consistent with this lactate-fuel hypothesis, several lines of study have also provided evidence that lactate can support energy metabolism during ischemia, which can rescue neuronal death after ischemia or under conditions of oxygen GD (Berthet et al, 2009). Recently, Rinholm et al (2011) reported that lactate can support axonal function in white matter under conditions of energy deprivation and that exogenous
Therefore, a growing body of evidence suggests that lactate is required to sustain neuronal recovery immediately after ischemia or brain trauma; however, the protective effect of lactate administration after hypoglycemia remains untested. Therefore, the aim of this study was to determine whether lactate, when administered as an adjuvant to glucose after hypoglycemia, could reduce neuronal death. Lactate is inexpensive, easily administered, innocuous and already used clinically. Expedient clinical trials may be warranted.
Materials and methods
The surgical and animal care procedures were in accordance with the guidelines of the Institutional Animal Care and Use Committee of the San Francisco Veterans Affairs Medical Center (animal welfare assurance number A3476-01) and of the Hallym University (Hallym 2011-44). This manuscript was written up in accordance with the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines.
Acute/Severe Hypoglycemia Surgery
Hypoglycemia was induced with regular insulin as described by Auer et al (1984) with minor modifications (Suh et al, 2003). In brief, male Sprague-Dawley rats weighing 250 to 350 g were fasted overnight and hypoglycemia was induced by intraperitoneal injection of 10 U/kg of regular insulin (Novolin-R, Novo Nordisk, Clayton, NC, USA). Anesthesia was induced with 3% isoflurane in a 75:25 mixture of nitrous oxide and oxygen (Air Liquid America, Houston, TX, USA). After intubation, rats were ventilated with a small rodent respirator (Harvard Apparatus, South Natick, MA, USA). A femoral artery line was placed for blood sampling and for blood pressure monitoring. Blood pH was measured at 1-hour intervals using an I-STAT machine (I-STAT, Princeton, NJ, USA). Blood pressure and electroencephalogram (EEG) were continuously monitored and recorded (BIOPAC System, Santa Barbara, CA, USA). Core temperature was kept at 36.5°C to 37.5°C with a heating blanket. Blood glucose and lactate were measured every 30 minutes in rat with an YSI 2700 glucose analyzer (YSI, Yellow Spring, OH, USA). Mean arterial blood pressure was maintained between 160 and 200 mm Hg during the entire EEG isoelectric period by adjusting the isoflurane concentration, and bradycardia was prevented with intramuscular injection of atropine (1 mg/kg). Electroencephalogram was monitored using needle electrodes placed in the cortical surface. Burr holes were made in the skull bilaterally over parietal cortex and monopolar electrodes were inserted beneath the dura. A reference needle was placed in neck muscle. The extent of neuronal death after hypoglycemia is tightly correlated with the duration of EEG isoelectricity (iso-EEG) (Auer et al, 1984). To generate a reproducible neuronal death of moderate severity, hypoglycemia was terminated after 30 minutes of iso-EEG (Auer et al, 1984; Suh et al, 2003) by delivery of 25% glucose infusion for 3 hours (1.5 mL/h, intravenously) to maintain blood glucose between 5 and 10 mM. Another group of rats received an intraperitoneal injection of sodium
Histological Evaluations of Neuronal Death
To identify degenerating neurons after hypoglycemia, animals were anesthetized with isoflurane (3%) 7 days after insult and intracardially perfused with 200 mL of 0.9% saline followed by 4% FA (formaldehyde) for 5 minutes. The brains were postfixed for 24 hours in the same fixative solution and immersed in 20% sucrose. Cryostat sections (25 μm) were mounted on superfrosted-coated slides (Fisher Scientific, Pittsburgh, PA, USA). Fluoro-Jade B staining was performed as described by Schmued and Hopkins (2000) (Suh et al, 2003). In brief, the slides were immersed in a basic alcohol solution followed by 0.06% potassium permanganate. The slides were then immersed in 0.0004% Fluoro-Jade B (Histo-Chem, Jefferson, AR, USA) for 20 minutes and washed in distilled water. Sections were photographed with a Leica confocal laser-scanning microscope with blue (450 to 490 nm) excitation light and a barrier filter wavelength of 515 nm. Five coronal sections were collected from each animal by starting 4.0 mm posterior to Bregma, and collecting every fourth section (75 μm intervals) until five sections were in hand. These sections were then coded and given to a second, blinded experimenter who counted the number of degenerating neurons in the hippocampal CA1, subiculum, dentate gyrus, and perirhinal cortex regions of both hemispheres. The total number of degenerating neurons in a region was averaged over the five sections from each brain.
Detection of Superoxide
For in vivo studies, dihydroethidium (Molecular Probes, Eugene, OR, USA) was prepared as a 1 mg/mL solution in 1% dimethyl sulfoxide and administered at 1 mg/kg through the intraperitoneal space to rat at the onset of iso-EEG. Animals were euthanized 3 hours after iso-EEG and perfusion fixed with 4% FA. The brains were sectioned on a cryostat and photographed with a confocal fluorescent microscope at excitation 510 to 550 nm and emission >580 nm for detection of ethidium (Et) (Chan et al, 1998). Five 25 μm thickness sections were analyzed from each brain, taken at 75 μm intervals to span the hippocampus. Ethidium signal intensity was expressed as the ratio of the mean fluorescence in neuronal perikaria to fluorescence in the stratum radiatum of hippocampal CA1.
Immunostaining for Evaluation of Microglia Activation
Immunostaining was performed on FA fixed and coronally sectioned brain tissue at a thickness of 40 μm. Three sections were analyzed from each brain, taken at 200 μm intervals to span the hippocampus. After rinsing in 1 mM phosphate-buffered saline, nonspecific protein binding was blocked by a 1-hour incubation in blocking buffer (10% goat serum and 0.1% Triton X-100 in 1 mM phosphate-buffered saline) at room temperature. The sections were then immunostained with a mouse antibody to rat CD11b (Serotec, Raleigh, NC, USA) at a 1:250 dilution. After washing, the sections were incubated with Alexa Fluor 488-conjugated goat anti-mouse IgG secondary antibody (Molecular Probes, Invitrogen, Grand Island, NY, USA) at a dilution of 1:500 for 2 hours at room temperature. Negative controls were performed with secondary antibody alone and showed no staining. Microglial activation was evaluated by a blinded observer. Three sections from each animal were evaluated for scoring. Microglial activation criteria were based on the number of CD11b immunoreactive cells and their morphology (Kauppinen et al, 2008).
Cell Cultures
Glia-free neuron cultures were prepared by plating the embryonic neurons onto a polystyrene surface (Kauppinen and Swanson, 2005). These neurons were cultured in glia-conditioned medium and used at days 10 to 14 in vitro, at which time the cultures contained >95% neurons, as assessed by immunostaining for the neuronal marker microtubule associated protein (MAP)2 and the astrocyte marker glial fibrillary acidic protein (GFAP) (Kauppinen and Swanson, 2005). Glucose concentration in the culture media was 5.5 mM.
Glucose Deprivation in Cell Cultures
The culture medium was exchanged with a bicarbonate-buffered, balanced salt solution containing (in mM) KCl, 3.1; NaCl, 134; CaCl2, 1.2; MgSO4, 1.2; KH2PO4, 0.25; NaHCO3, 15.7; glucose, 0. The pH was adjusted to 7.2, while the solution was equilibrated with 5% CO2 at 37°C. Osmolarity was verified at 290 to 310 mOsm with a Wescor vapor pressure osmometer (Logan, UT, USA). After a 5-minute period to allow complete egress of glucose from the cells, the culture medium was completely exchanged a second time and the cultures were placed in a 37°C, 5% CO2 incubator. Glucose deprivation was terminated at the designated time points by adding 5 mM glucose back to the medium. Control wells received 5 mM glucose replacement (GR) immediately after the initial medium exchange. For cell culture studies, 5 μM dihydroethidium (Budd et al, 1997) was added to the cultures 30 minutes before GD. The cultures were photographed with a fluorescence microscope 1 hour after GR, and the digitized images were analyzed and expressed as percent of neurons with an Et signal at least 50% higher than background. Prior studies have confirmed that the Et signal reflects primarily superoxide under the conditions of hypoglycemia and glucose reperfusion employed here (Suh et al, 2007).
Statistical Analysis
Data were shown as mean values±s.e.m. Statistical significance was assessed by analysis of variance and post hoc testing was accomplished using Scheffe's test. A P value <0.05 was considered to be statistically significant. Microglial activation data were assessed by Kruskal–Wallis nonparametric one-way analysis of variance test followed by Dunn's test for multiple group comparison.
Results
Effects of Lactate Administration on Blood Glucose, Lactate and pH, and Electroencephalogram Before, During, and After Hypoglycemia
Blood glucose, lactate, pH, and EEG were measured for the entire duration of the hypoglycemia experimental period to observe the effects of intraperitoneal administration of lactate on these parameters. Overnight fasting-blood-glucose-level was 3.72±0.15 mM, which dropped to 0.46±0.06 0.5 mM after insulin injection. A silent electroencephalographic trace (iso-EEG) typically appears ∼2.5 hours after insulin injection. Iso-EEG was maintained for 30 minutes and terminated by reintroduction of glucose (femoral vein injection), reinstating blood glucose levels to 7.79±0.35 mM at 30 minutes after glucose reperfusion. Arterial lactate was reduced from 0.94±0.11 mM to 0.31±0.04 mM by insulin injection, returning to 1.50±0.14 mM following glucose reperfusion (Figure 1A) (Table 1). To determine if lactate administration influenced blood glucose or lactate concentrations during the glucose reperfusion period, lactate was intraperitoneally injected 5 minutes before glucose reperfusion (25 minutes after iso-EEG start). Blood lactate level was increased to more than twice its initial concentration in the glucose reperfusion with lactate injection group, compared with the glucose reperfusion with saline group (Figure 1B). Blood glucose concentration also increased in the lactate-injected group during the glucose reperfusion period (1 hour after glucose reperfusion) (Figure 1C). Arterial blood pH was measured before, during, and after iso-EEG. In fasting rats that did not receive insulin injection, blood pH was maintained at 7.43 for the duration of the experiments. In the insulin-injected group, blood pH dropped during the iso-EEG period and further decreased after blood glucose reperfusion. However, blood pH in the lactate-injected rats showed no decrease and even increased during the iso-EEG and the glucose reperfusion period (Figure 1D) (Table 2). To test whether lactate administration could rescue EEG from isoelectricity, lactate was injected 15 minutes before the end of the iso-EEG period. Interestingly, silent EEG (iso-EEG) was not rescued by lactate administration without glucose reinfusion (Figure 1E). Moreover, EEG recovery after glucose reperfusion was delayed by lactate administration (Figure 1F).
Arterial blood glucose and lactate change before, during, and after hypoglycemia (mM)
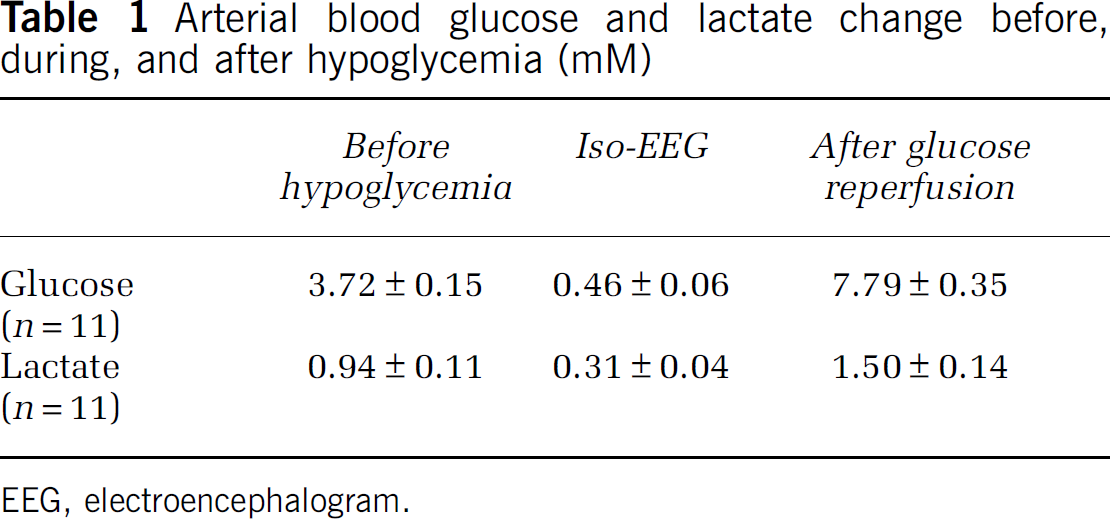
EEG, electroencephalogram.
Arterial blood pH change before, during, and after hypoglycemia
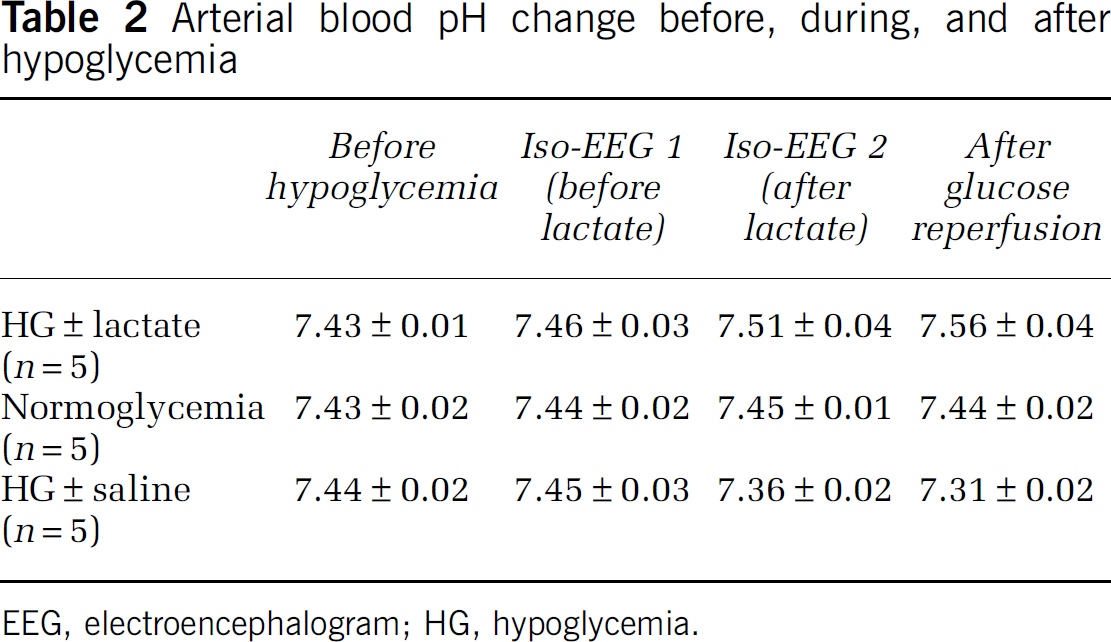
EEG, electroencephalogram; HG, hypoglycemia.
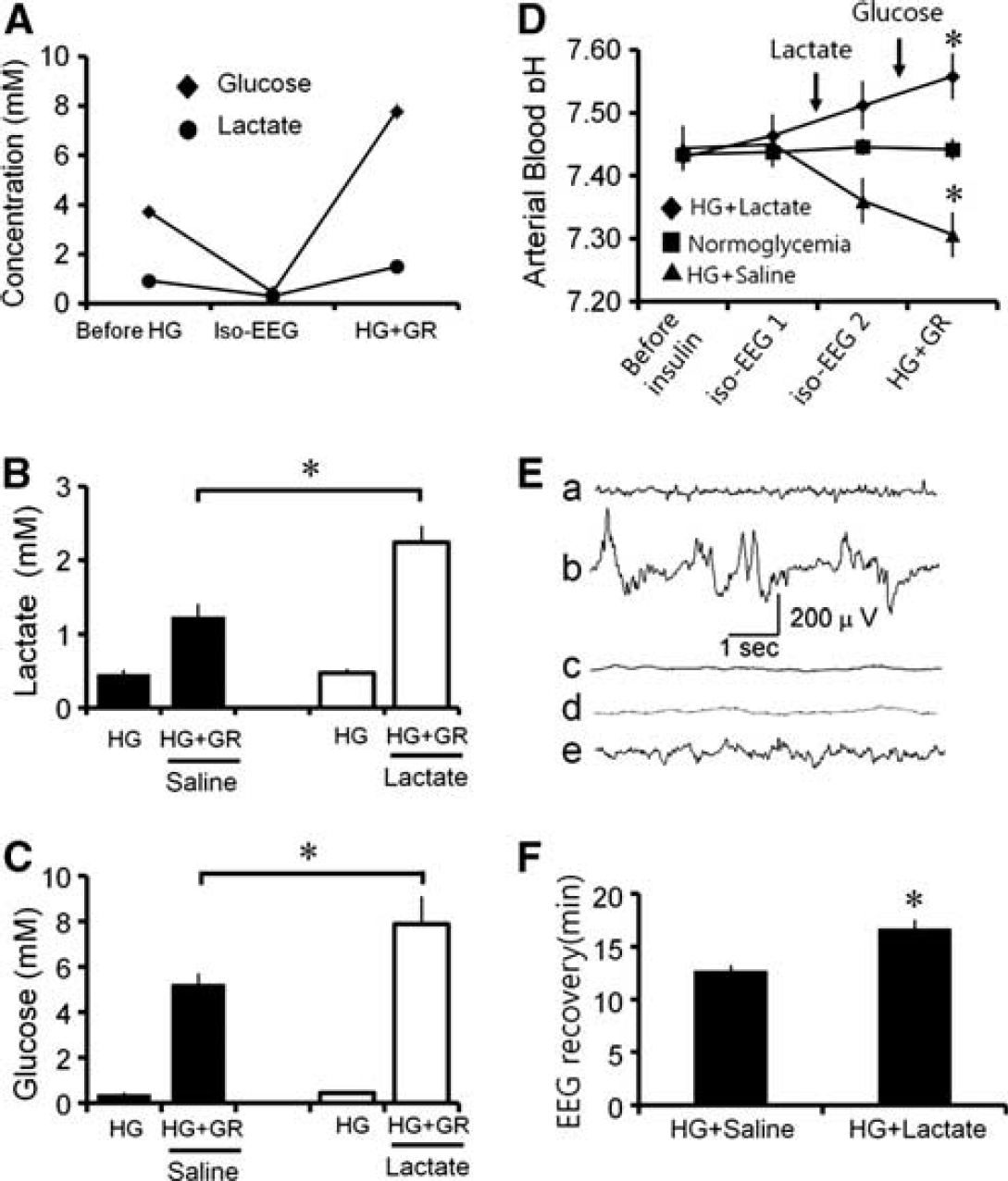
Effects of lactate administration on blood glucose, lactate and pH, and electroencephalogram (EEG) before, during, and after hypoglycemia. Blood glucose, lactate, pH, and EEG were measured for the duration of the hypoglycemia experimental period to determine the effects of intraperitoneal administration of lactate. (
Lactate Reduces Neuronal Death After Hypoglycemia
Hypoglycemia induces a reproducible pattern of neuronal death when evaluated at 7 days after surgery. Fluoro-Jade B staining showed widespread neuronal death in the hippocampal CA1, subiculum (Sub), and dentate gyrus area (Figure 2). Fluoro-Jade B staining provides a selective, unambiguous marker of neuronal degeneration. Compared with rats receiving glucose alone, rats that received glucose plus lactate (500 mg/kg, intraperitoneally) at the end of the iso-EEG period showed a marked reduction in neuronal death (Figure 2A). As quantified in Figure 2B, rats receiving glucose plus lactate had around 90% fewer degenerating neurons than the rats given only glucose plus vehicle (saline).
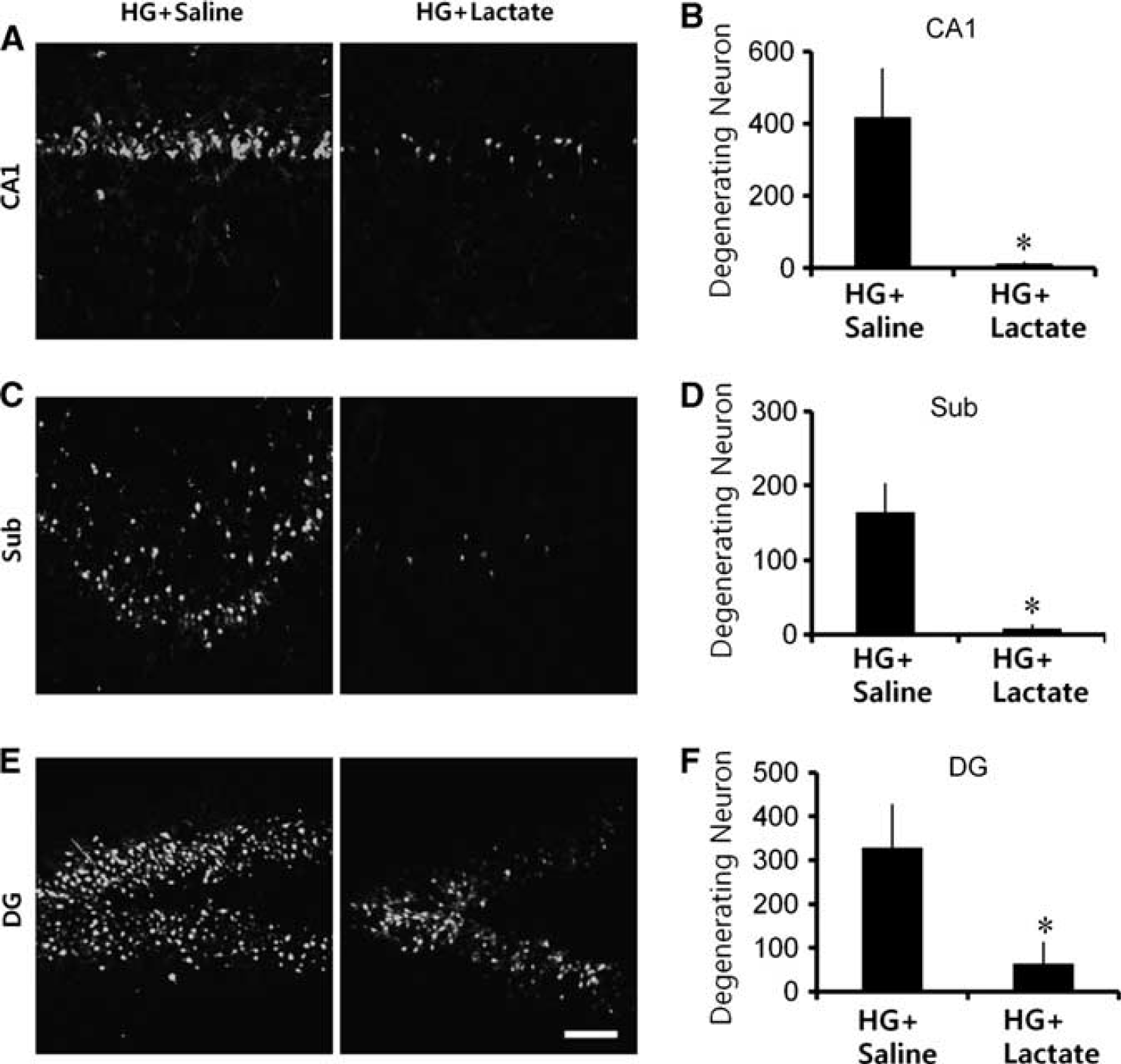
Hypoglycemia (HG)-induced neuron death is prevented by lactate administration. (
Lactate Prevents Hypoglycemia-Induced Superoxide Production
To image superoxide production after hypoglycemia, we evaluated the production of reactive oxygen species with dihydroethidium (Murakami et al, 1998). Dihydroethidium is oxidized by superoxide and superoxide reaction products to form fluorescent Et species (Zhao et al, 2003), which are then trapped within cells by DNA binding. Rats subject to 30 minutes of hypoglycemia followed by a subsequent 30-minute interval of glucose reperfusion showed a several-fold increase in neuronal Et fluorescence (Figure 3A). To determine the effects of intraperitoneal administration of lactate on hypoglycemia-induced superoxide production, we examined Et fluorescent intensity in the hippocampal CA1, subiculum, and dentate gyrus. Lactate injection resulted in a much smaller increase in Et fluorescence in hippocampal neurons after glucose reperfusion, relative to the saline-injected group (Figures 3A and 3B).
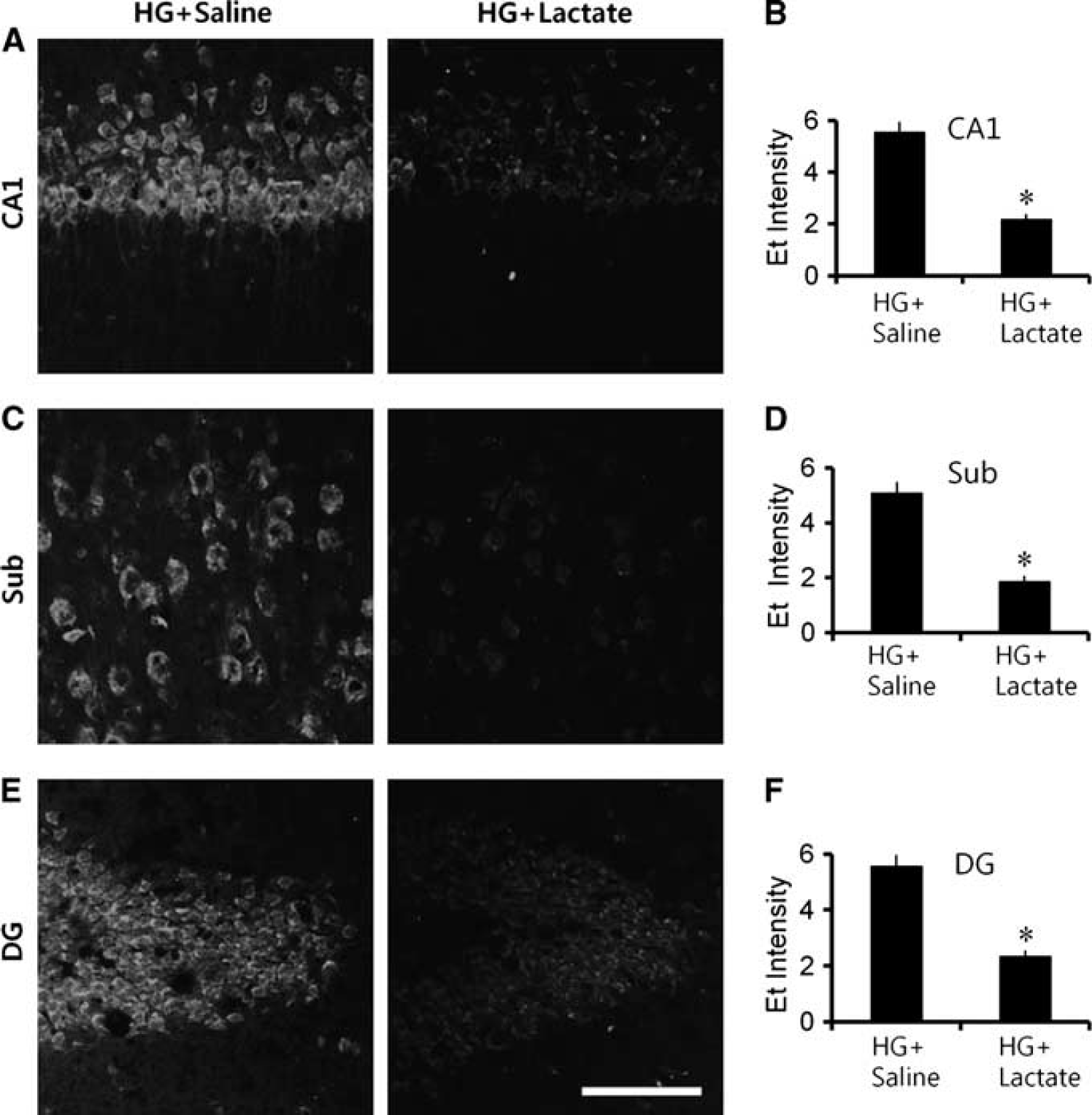
Hypoglycemia (HG)-induced reactive oxygen species (ROS) production is prevented by lactate administration. (
Lactate Prevents Hypoglycemia-Induced Microglia Activation
After hypoglycemia, neuronal deprivation of glucose and its resulting sequelae may not only impose neuronal damage via neuron-specific effects, but may also promote neuronal injury indirectly via microglial activation. Hallmarks of microglial activation include a gradual change in morphology from a highly ramified to an amoeboid shape, proliferation, migration to the site of injury, increased expression of surface molecules, increased secretion of cytokines, chemokines, free radicals and proteases, and phagocytotic activity (Kreutzberg, 1996). We investigated the degree of microglial activation after hypoglycemia with or without lactate administration. Compared with saline-treated rats, microglial activation was significantly reduced by lactate administration in the hippocampus (Figures 4A–4C).
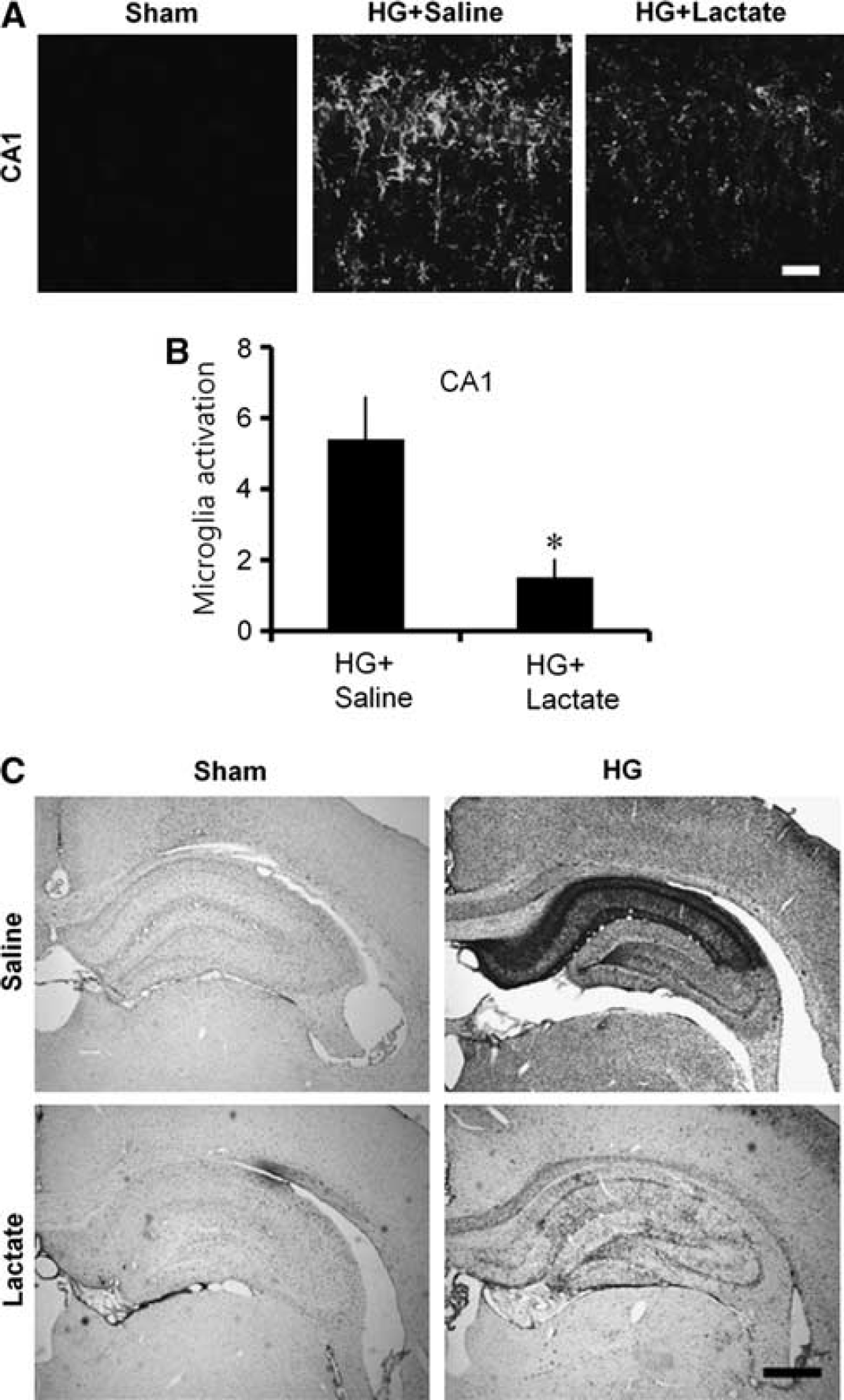
Hypoglycemia (HG)-induced microglia activation is prevented by lactate administration. (
Lactate Prevents Glucose Deprivation-Induced Superoxide Production and Subsequent Neuronal Death in Cultured Neurons
To identify the effects of lactate on superoxide production, we subjected cortical neuron cultures to 2 hours of GD followed by 1 hour of GR. As observed during in vivo hypoglycemia, GD alone for 3 hours caused only a negligible increase in the Et signal, but GD (2 hours) + GR (1 hour) produced a rapid increase in neuronal Et fluorescence (Figures 5A and 5B). Control cultures (5 mM glucose was maintained throughout the experiment) exhibited a negligible Et signal (Cont). However, neurons treated with lactate (5 mM) generated much less superoxide after GR than cultures treated with GD (2 hours) + GR (1 hour) (Figures 5A and 5B) and showed reduced neuronal death when evaluated 24 hours after GD/GR (Figures 5C and 5D). We found that lactate treatment alone showed no neuroprotection, which suggests that only combining lactate administration with glucose serves as a neuroprotective strategy, as seen in an additional study (Cater et al, 2003).
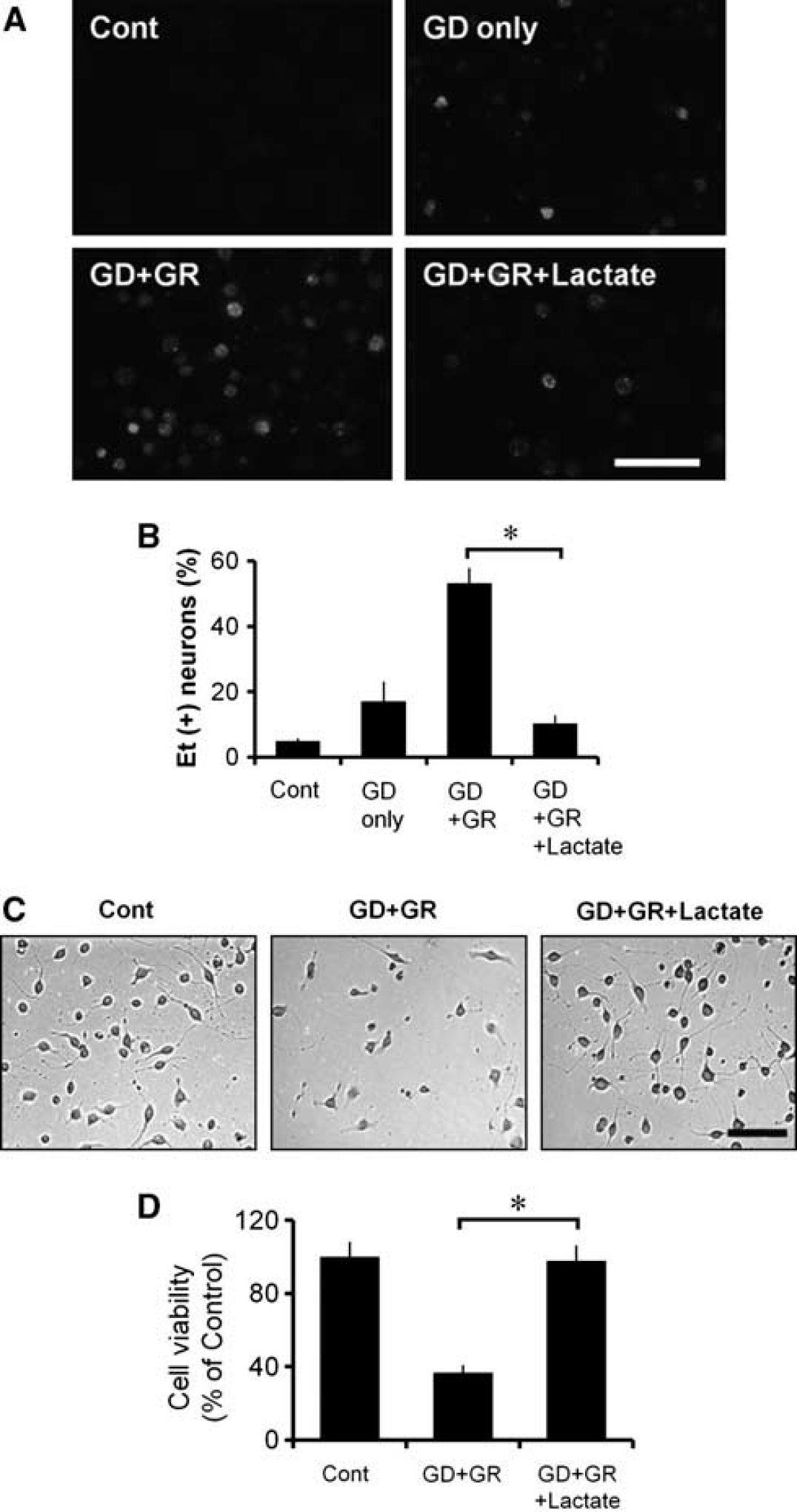
Neuronal superoxide production and neuronal death after glucose deprivation/glucose replacement (GD/GR) is prevented by lactate. (
Long-Term Neuroprotective Effects of Lactate After Hypoglycemia
Assessment of the brains 8 weeks after the hypoglycemic insult revealed atrophy and loss of neurons in the CA1 region of the hippocampus (Figure 6). These changes were substantially attenuated in rats treated with lactate after hypoglycemia, suggesting that neuronal death is prevented rather than simply delayed by this intervention.
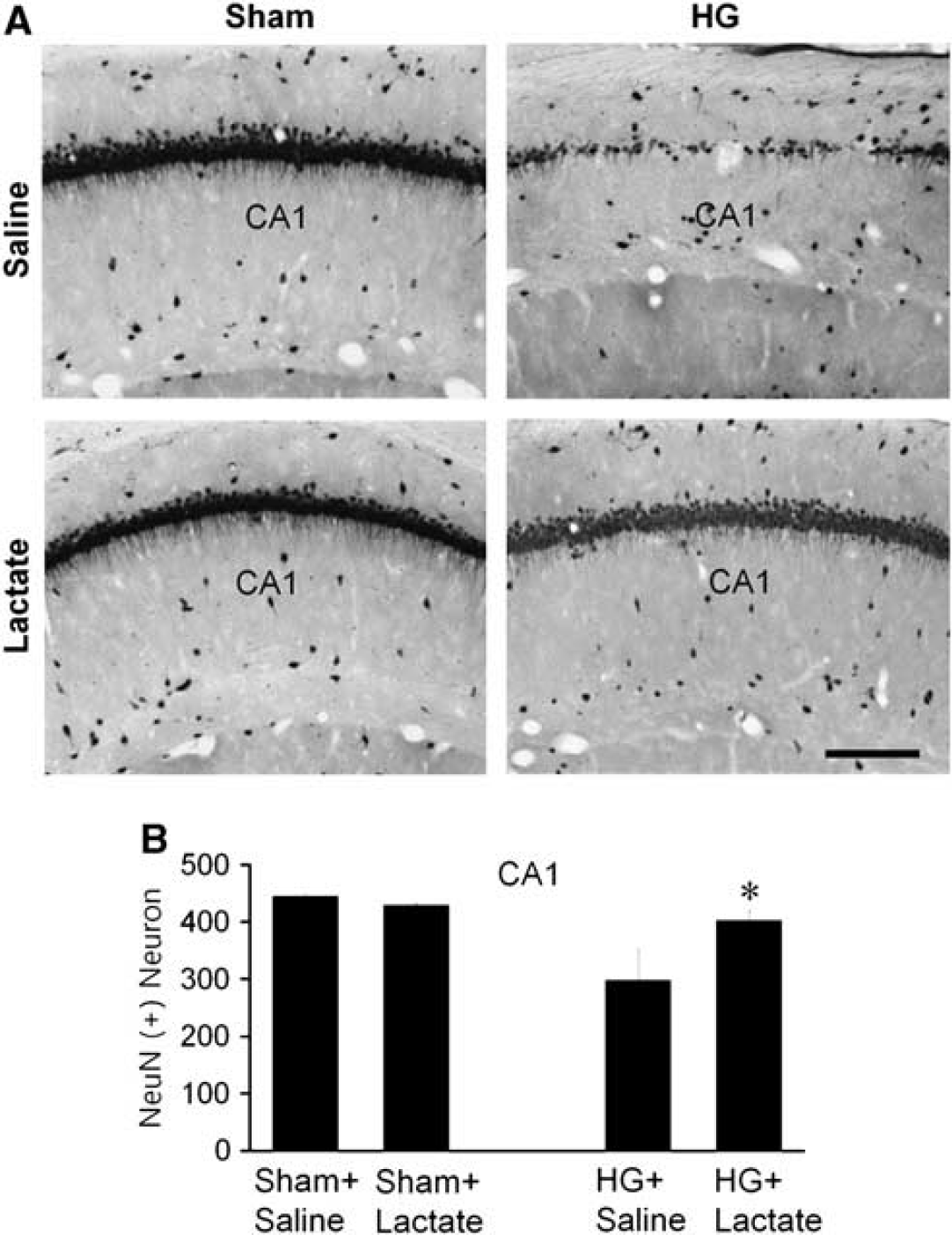
Long-term neuroprotective effect of lactate after hypoglycemia (HG). (
Discussion
The development of clinically useful techniques to reduce the neuronal death that results from severe hypoglycemia is greatly needed. The present study demonstrates that intraperitoneal administration of lactate significantly reduced hypoglycemia-induced neuronal death in the CA1, dentate granule cell, and subiculum of the hippocampus that would occur without intervention. Furthermore, this improvement in neuronal survival was also detected 8 weeks after the hypoglycemic insult, indicating that neuronal death is fully prevented, not just delayed, by lactate administration.
The mature brain is one of the few organs that depend on glucose as its primary metabolic substrate. Once in brain, glucose may be metabolized directly by neurons and glia. Glucose may be metabolized to lactate by glia and the lactate subsequently shuttled to neurons for oxidative metabolism (Dienel and Cruz, 2004; Pellerin and Magistretti, 1994; Schurr et al, 1997b; Wender et al, 2000). Lactate may act as a neuronal oxidative energy substrate (Schurr, 2006). While pyruvate, lactate and other ketone bodies are known to be comparable to glucose in their capacity to support neuron metabolism in vitro, the blood–brain barrier in the mature brain preferentially transports glucose over other energy substrates. Blood–brain barrier transport of lactate and other substrates occurs via both facilitated transport and diffusion. Therefore, increases in plasma concentration of these compounds should substantially increase their transport into the brain (Boumezbeur et al, 2011; van Hall et al, 2009). The dose of lactate used in the present studies is estimated to generate a lactate plasma concentration of ∼2 to 3 mM, which is about twofold to threefold higher than under physiological conditions, likely increasing brain lactate concentrations accordingly. Lactate appears to have a crucial role in improving postischemic outcomes. Schurr et al (1997a, 2001a, 2001b) have published several articles reporting the neuroprotective effect of lactate following hypoxia or following ischemia, which involves GD. Blockade of lactate transport into neurons prevents its utilization and, consequently, exacerbates delayed ischemic neuronal damage (Schurr et al, 2001b). Although previous studies demonstrated the beneficial effects of lactate on ischemia-induced neuron death, no direct study was performed to evaluate neuroprotective effects of lactate after hypoglycemia, where oxygen levels are maintained at ambient levels. Our present study shows that lactate also prevents neuronal death after severe hypoglycemia under normoxic conditions. Rather than acting as an oxidative energy substrate, several studies have suggested that lactate may also have antioxidant activities. When lactate is converted to pyruvate by mitochondrial lactate dehydrogenase (LDH) (when ATP levels are very low and thus when glucose cannot be metabolized), there is a simultaneous conversion of nicotinamide adenine dinucleotide (NAD) to reduced form of NAD (NADH), which is known to have antioxidant activity (Heinemann et al, 2002; Kirsch and De Groot, 2001).
Our studies indicate that a cascade of events is precipitated by the lack of energy substrate during hypoglycemia, which then leads to neuronal demise through a series of sequential pathological reactions. Key steps in this cell death pathway include glutamate release, zinc translocation, and PARP-1 activation. A major effect of this cascade is the consumption of cytosolic NAD+ (Engelsen et al, 1986; Suh et al, 2004; Wieloch, 1985), the depletion of which prevents metabolism of glucose. In settings where glucose is the chief metabolic substrate, this leads to mitochondrial dysfunction and cell death (Alano et al, 2004; Ying et al, 2003; Zong et al, 2004). Interestingly, repletion of NAD+ after PARP-1 activation has been shown to restore glycolytic capacity and prevent cell death (Ying et al, 2003, 2005) in primary neuronal cultures. Similarly, providing nonglucose substrates such as pyruvate and α-ketoglutarate that can be metabolized without the need for cytosolic NAD+ also preserves cell viability, even when delivered hours after PARP-1 activation (Alano et al, 2004; Ying et al, 2002). However, to be used oxidatively, lactate must be first converted to pyruvate by lactate dehydrogenase, which also requires NAD+ and thus competes with glycolysis for NAD+. Therefore, given this fact, how can lactate administration have a neuroprotective effect in this situation? We suggest the following explanation: it has been previously suggested that PARP-1 consumes the cytosolic NAD+ pool, but does not consume the mitochondrial NAD+ pool until the point that mitochondrial permeability transition occurs (Alano et al, 2010). Thus, during the first few hours or so after extensive PARP-1 activation, the mitochondria remain capable of respiration but the cytosol cannot metabolize glucose. Thus, when given nonglucose substrates such as lactate, the mitochondria can make ATP (Alano et al, 2007, 2010; Ying et al, 2005). Once enough ATP is generated, glucose can again become the main energy substrate. In agreement with these cell culture studies, the present findings suggest that lactate promotes neuronal survival after hypoglycemia by circumventing a sustained impairment in glycolysis induced by PARP-1 activation. This further supports the idea that hypoglycemia causes a sustained impairment in glucose metabolism that persists after glucose reperfusion and that combining lactate administration along with glucose to terminate hypoglycemia serves as a neuroprotective strategy.
Increasing blood lactate after ischemia or hypoglycemia has traditionally been believed to be toxic through promotion of acidosis. Acid-sensing ion channels are activated after ischemia (Xiong et al, 2004) and their inhibition is neuroprotective (Pignataro et al, 2007). However, in the present study, sodium
Using the same hypoglycemic brain injury model, very similar levels of neuroprotection in vulnerable brain regions, as well as prevention of cognitive deficits, were previously achieved by administration of PARP inhibitors at the termination of hypoglycemia (Suh et al, 2003, 2005). Since pathological overactivation of PARP is known to induce cell death indirectly by sequestering intracellular NAD+ needed to maintain glycolysis at sufficient levels, it is tempting to explain the protection afforded by lactate in terms of this relationship. While further study is needed to directly establish how lactate achieves comparable neuroprotection to PARP inhibition, one parsimonious explanation is that a concomitant increase in brain lactate offsets the decrease in NAD+ due to overactivation of PARP by acting as an alternative energy substrate that can effectively bypass glycolysis and be fed directly to the citric acid cycle to maintain cellular ATP levels (Ying et al, 2005). Lactate is inexpensive, readily available, and unlike directly inhibiting PARP activation, does not have the theoretical limitation of potentially impairing DNA repair mechanisms (Nagayama et al, 2000). Results of this study suggest that lactate may be an effective intervention for patients with severe hypoglycemia.
Footnotes
Acknowledgements
The authors thank Aaron M Hamby, University of California, Berkeley, for help with preparing the manuscript.
SJW, BGJ, and HKS researched the data and reviewed/edited the manuscript. BHY, MWL, BYC, and JHK researched the data. MS performed data analysis and reviewed/edited the manuscript. SWS contributed to discussion, reviewed/edited the manuscript, wrote the manuscript, and took full responsibility for the manuscript and its originality.
Disclosure/conflict of interest
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.