
Abstract
In the brain, glutaminase is considered to have a key role in the provision of glutamate, a major excitatory neurotransmitter. Brain slices obtained from wild-type (control) and glutaminase-deficient (GLS1 +/–) mice were incubated without glucose and with 5 or 1 mmol/L [3-13C]glutamine as substrate. At the end of the incubation, substrate removal and product formation were measured by both enzymatic and carbon 13 nuclear magnetic resonance (13C-NMR) techniques. Slices from GLS1 +/– mice consumed less [3-13C]glutamine and accumulated less [3-13C]glutamate. They also produced less 13CO2 but accumulated amounts of 13C-aspartate and 13C-gamma-aminobutyric acid (GABA) that were similar to those found with brain slices from control mice. The newly formed glutamine observed in slices from control mice remained unchanged in slices from GLS1 +/– mice. As expected, flux through glutaminase in slices from GLS1 +/– mice was found diminished. Fluxes through all enzymes of the tricarboxylic acid cycle were also reduced in brain slices from GLS1 +/– mice except through malate dehydrogenase with 5 mmol/L [3-13C]glutamine. The latter diminutions are consistent with the decreases in the production of 13CO2 also observed in the slices from these mice. It is concluded that the genetic approach used in this study confirms the key role of glutaminase for the provision of glutamate.
Introduction
Glutamine is considered to be the preferred precursor of glutamate, the major excitatory neurotransmitter (Fonnum, 1984; Kvamme, 1998; Kvamme et al, 2000). The conversion of glutamine into glutamate is catalyzed by phosphate-activated glutaminase (PAG). It is generally believed that glutamate is mainly formed in neurons from which it is released into the synaptic space. Glutamate binds to postsynaptic receptors to facilitate the transmission of the nerve influx and then, astrocytes take up glutamate, which is known to be neurotoxic, and convert it into glutamine thanks to glutamine synthetase by binding to ammonia. The resulting glutamine is transferred to the neurons closing the so-called glutamate-glutamine cycle (Berl and Clarke, 1983; Van den Berg and Garfinkel, 1971).
Phosphate-activated glutaminase is the most important glutamine-metabolizing enzyme for the synthesis of glutamate (Kvamme et al, 2000). This enzyme is regulated by a variety of activators (Pi, calcium, certain fatty acyl CoA) and inhibitors (proton, glutamate, ammonia, and SH reagents) (Kvamme et al, 2001). Phosphate-activated glutaminase has been shown to be active in neurons and, to a much lesser extent, in astrocytes (Hogstad et al, 1988; Kvamme et al, 2001). It has been shown that PAG is functionally localized in brain mitochondria (Bak et al, 2008; Roberg et al, 1995).
There are two genes encoding PAG. The first one (GLS1) codes for the brain/kidney PAG form; the second one (GLS2) encodes the liver isoform (Aledo et al, 2000; Chung-Bok et al, 1997; Shapiro et al, 1991). It has been shown that mice that are knocked out for GLS1 (GLS1–/–) are not viable and die shortly after birth (Masson et al, 2006). This shows that PAG has a crucial role in brain function. In this study, we took advantage of the availability of glutaminase-deficient mice (GLS1 + /–) that have been shown to survive and grow normally despite a decrease of ∼50% of their cerebral PAG activity (Gaisler-Salomon et al, 2009). In these mice and in the appropriate controls, we studied the cerebral metabolism of glutamine by using our metabolomic approach that previously allowed us to unravel the metabolism of various physiological substrates in renal cells (Chauvin et al, 1994, 1997; Dugelay et al, 1999; Fouque et al, 1996; Vittorelli et al, 2005) and in rat brain slices (El Hage et al, 2011a, b , c ).
Materials and methods
Reagents
Enzymes and coenzymes were supplied by Roche Molecular Biochemicals (Meylan, France).
Animals
All experiments were approved by the Institutional Animal Care and Use Committee of the Lyon 1 University. The wild-type (GLS1 + / +) and glutaminase-deficient (GLS1+/–) mice were kindly provided by Dr J Masson (Masson et al, 2006). At the time of experiments, they weighed 33.8 ± 0.5 (
Preparation of Brain Slices and Incubation
The mice were killed by cervical dislocation. The skin was removed from the skull, which was opened by introducing one blade of scissors into the foramen magnum and by cutting it along its caudal to rostral axis. Then, the brain was quickly dissected. All the steps of preparation were performed in ice-cold oxygenated Krebs—Henseleit buffer (Krebs and Henseleit, 1932). Then, the brain was rapidly cut into 250 μm-thick transverse slices with a McIlwain Tissue Chopper (The Vibratome Company, O'Fallon, MO, USA).
All slices obtained from each complete brain were incubated with shaking (80 cycles/min) at 37°C in 100 mL Erlenmeyer flasks containing an atmosphere of O2/CO2 (19:1) and 8 mL of Krebs—Henseleit buffer. Brain slices were incubated for 60 minutes in the absence of glucose and in the presence of glutamine at a 5 or 1 mmol/L concentration. The 5 mmol/L glutamine concentration was chosen to facilitate the observation of small carbon 13 nuclear magnetic resonance (13C-NMR) because 13C-NMR spectroscopy is a relatively insensitive technique. The 1 mmol/L glutamine concentration better mimicked the
Zero-time samples were obtained by adding perchloric acid to the incubation medium before the freshly prepared slices.
After removal of the denaturated protein by centrifugation, the supernatant was neutralized with a mixture of 20% (w/v) KOH and 1% (v/v) H3PO4 (8 mol/L) for metabolite determination and NMR spectrometry. The pellet of brain slices was dried for 48 hours at 60°C and then for 1 week at 37°C for determination of their dry weight.
Analytical Methods
The concentrations of metabolites were measured by enzymatic methods whereas the 13C enrichment of the individual metabolite carbons was measured by 13C-NMR spectrometry.
Glutamine, glutamate, alanine, aspartate, and gamma-aminobutyric acid (GABA) were determined according to the methods of Passonneau and Lowry (1993).
13C-Nuclear Magnetic Resonance Techniques
The NMR measurements were performed and the data were recorded as indicated previously (Chauvin et al, 1994, 1997) at 125.17 MHz on a Bruker AM-500 WB spectrometer (Bruker Biospin, Wissembourg, France) using a 5-mm broadband probe thermostatically maintained at 8 ± 0.5°C. In brief, magnet homogeneity was adjusted using the deuterium lock signal. To obtain absolute quantitative results, special care was taken in data acquisition. Metabolites were dissolved in D2O and acquisition parameters were as follows: spectral width, 25,000 Hz; tilt angle, 90°; data size, 32 K; repetition time, 50 seconds; number of scans, 420.
We used a standard (Waltz-16) pulse sequence for inverse-gated proton decoupling (Shaka et al, 1983). We did not use nuclear Overhauser enhancement during proton decoupling to avoid the use of corresponding correction factors. A 1-Hz line broadening was applied. [2-13C]glycine was used as internal standard. Chemical shifts were expressed as p.p.m. (parts per million) relative to tetramethylsilane. Assignments were made by comparing the chemical shifts obtained with those given in the literature (Howarth and Lilley, 1978). We used a Lorentzian deconvolution (line shape fitting) for spectra analysis included in Bruker Topspin 2.1 (Bruker Biospin, Wissembourg, France).
Calculations and Statistical Analysis
Net substrate utilization and product formation were calculated as the difference between the total flask contents (tissue plus medium) at the start (zero-time flasks) and after the period of incubation. The net metabolic rates, reported as means ± s.e.m., are expressed in μmoles of metabolite removed or produced per gram of brain tissue dry weight per hour.
We used a mathematical model derived from a model previously validated and published for glutamine (Chauvin et al, 1997; El Hage et al, 2011b). The model, which is based on the incorporation of 13C into various metabolites and combined with the distribution of the enzymes in the neuronal and glial cells, allows the calculation of rates of the labeled substrate uptake, and fluxes through glutaminase, glutamine synthetase, glutamate decarboxylase, succinate dehydrogenase, malate dehydrogenase, malic enzyme, aspartate aminotransferase, pyruvate kinase, alanine aminotransferase, pyruvate dehydrogenase, pyruvate carboxylase, and citrate synthase.
The 13CO2 production was calculated as the difference between the removal of the labeled substrate and the sum of the labeled nonvolatile products calculated from the 13C NMR spectra. The results were analyzed by the Student's
Results
Table 1 shows the results obtained when glutamine metabolism was studied by enzymatic methods. The removal of glutamine in brain slices from glutaminase-deficient mice was statistically different from that in brain slices from control animals only when glutamine was used at a near-physiological (1 mmol/L) concentration. The removal of glutamine was not accompanied by the accumulation of glutamate, but rather by the removal of part of the glutamate present at the start of the incubation period; this removal was higher in brain slices from glutaminase-deficient mice than in brain slices from control mice only with 5 mmol/L glutamine as substrate. The small amount of GABA present at the start of incubation was also partly removed at similar rates in brain slices from both glutaminase-deficient and control mice (Table 1). A small amount of alanine and a large amount of aspartate were found to accumulate in brain slices from both glutaminase-deficient and control mice; with 1 mmol/L glutamine as substrate, less alanine accumulated in brain slices from glutaminase-deficient than from control animals.
Metabolism of 5 and 1 mmol/L [3-13C]glutamine in brain slices of wild-type (WT) and glutaminase-deficient (GLS1+/–) mice (Enzymatic data)
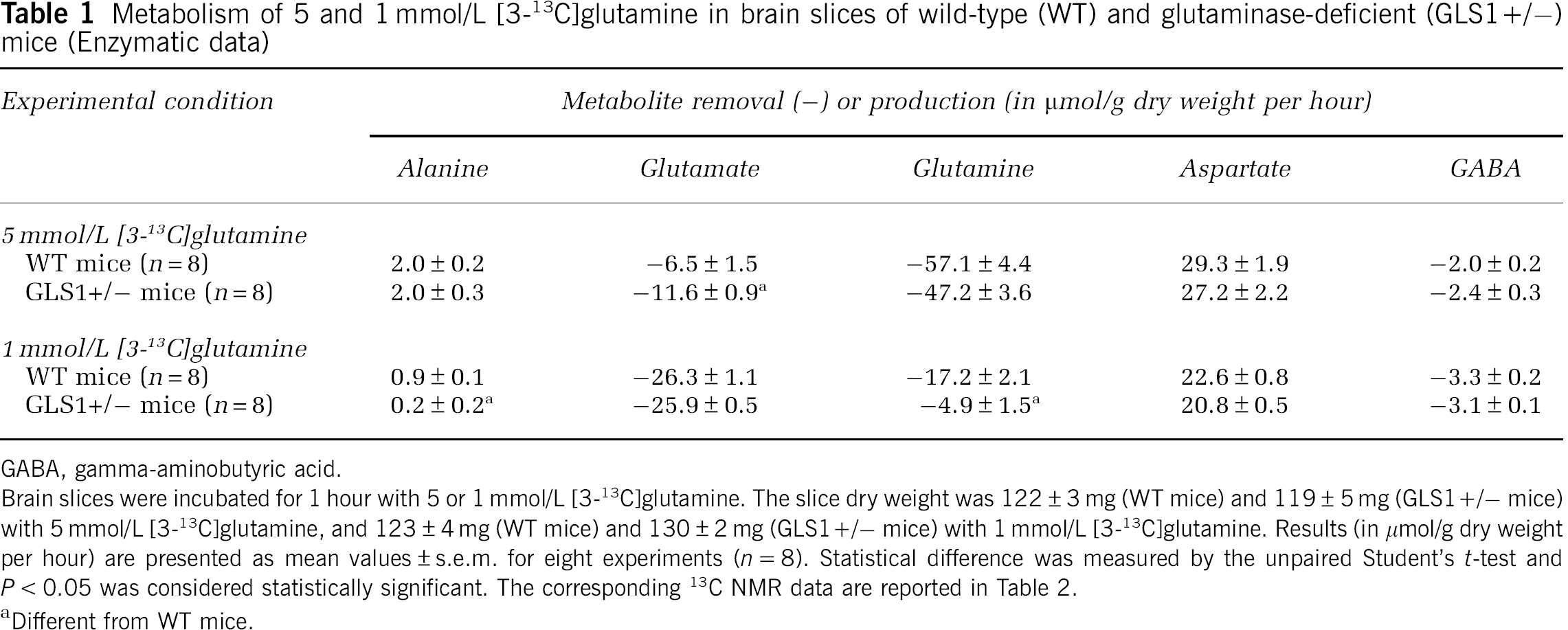
GABA, gamma-aminobutyric acid. Brain slices were incubated for 1 hour with 5 or 1 mmol/L [3-13C]glutamine. The slice dry weight was 122 ± 3 mg (WT mice) and 119 ± 5 mg (GLS1+/– mice) with 5 mmol/L [3-13C]glutamine, and 123 ± 4 mg (WT mice) and 130 ± 2 mg (GLS1+/– mice) with 1 mmol/L [3-13C]glutamine. Results (in μmol/g dry weight per hour) are presented as mean values ± s.e.m. for eight experiments (
Different from WT mice.
Figure 1 shows representative 13C-NMR spectra obtained after incubation of brain slices from a control (Figure 1A) and a glutaminase-deficient (Figure 1B) mice when 5 mmol/L [3-13C]glutamine was the substrate. It can be seen that all significant peaks could be assigned and corresponded to carbons of the products determined by enzymatic methods. From these spectra and those obtained with 1 mmol/L [3-13C]glutamine as substrate (not shown), it was possible to calculate the amounts of labeled products after correction for the 1.1% 13C natural abundance.
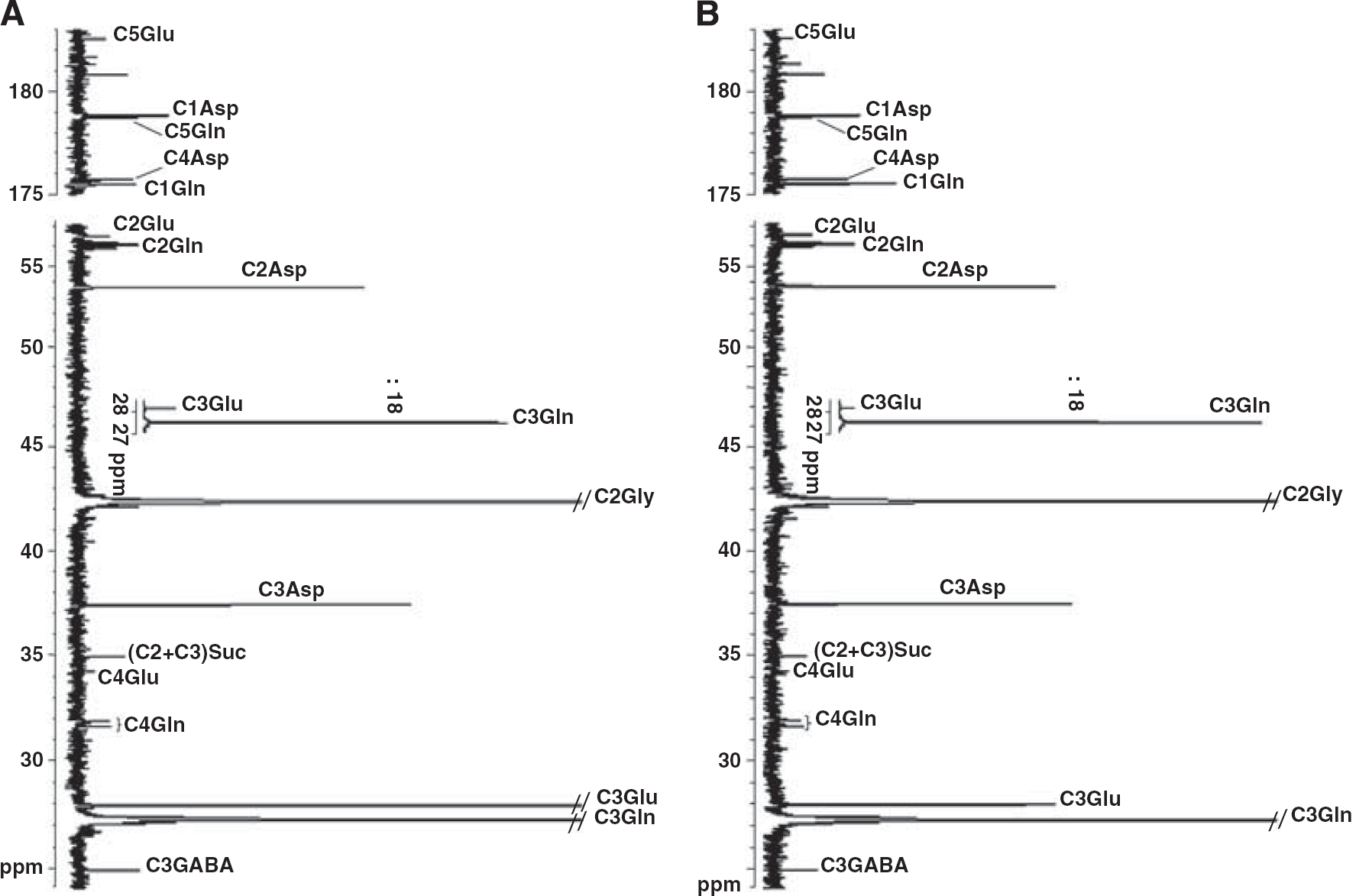
13C-NMR spectra of neutralized HCIO4 extracts obtained from rat brain slices incubated for 1 hour with 5 mmol/L [3-13C]glutamine from a control (
Table 2 reports the results obtained when the metabolism of glutamine was studied by 13C NMR spectrometry. With both 1 and 5 mmol/L [3-13C]glutamine as substrate, the removal of [3-13C]glutamine measured was much greater than that measured enzymatically (see Table 1) in brain slices from both control and glutaminase-deficient mice. This indicates that the removal of [3-13C]glutamine was partially masked by the concomitant appearance of newly formed unlabeled glutamine. This newly formed glutamine could be calculated as the difference between the removal of [3-13C]glutamine and the removal of glutamine measured enzymatically; the corresponding values are 38.8 ± 6.5 and 28.1 ± 4.0 (not significant) μmol/g dry weight per hour in brain slices from control and glutaminase-deficient mice, respectively, with 5 mmol/L [3-13C]glutamine; with 1 mmol/L [3-13C]glutamine, they were 13.2 ± 2.7 and 17.9 ± 2.0 (not significant) μmol/g dry weight per hour. Table 2 also shows that the removal of [3-13C]glutamine was significantly lower in brain slices from glutaminase-deficient mice than in those from control mice with both 1 and 5 mmol/L [3-13C]glutamine as substrate.
Metabolism of 5 and 1 mmol/L [3-13C]glutamine in brain slices of wild-type (WT) and glutaminase-deficient (GLS1+/–) mice (nuclear magnetic resonance (NMR) data)
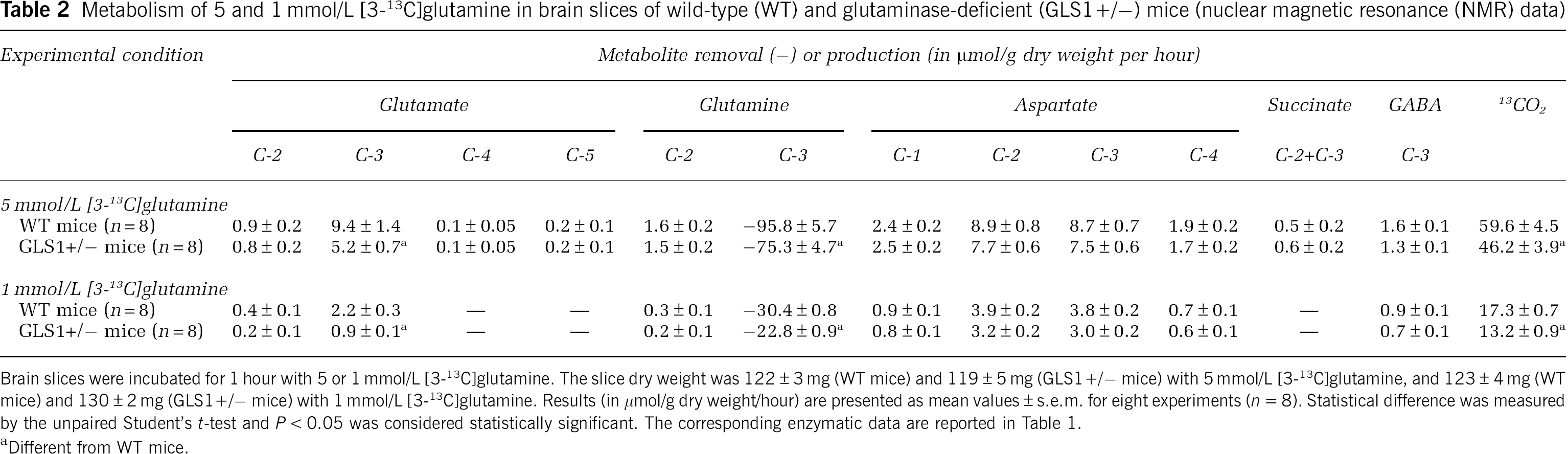
Brain slices were incubated for 1 hour with 5 or 1 mmol/L [3-13C]glutamine. The slice dry weight was 122 ± 3 mg (WT mice) and 119 ± 5 mg (GLS1+/– mice) with 5 mmol/L [3-13C]glutamine, and 123 ± 4 mg (WT mice) and 130 ± 2 mg (GLS1+/– mice) with 1 mmol/L [3-13C]glutamine. Results (in μmol/g dry weight/hour) are presented as mean values ± s.e.m. for eight experiments (
Different from WT mice.
The lower removal of [3-13C]glutamine in brain slices from glutaminase-deficient mice was associated with a lower accumulation of [3-13C]glutamate, the carbon product of the glutaminase reaction. Interestingly, glutamate and glutamine were found to be labeled on carbons other than their carbon 3, which means that resynthesis of both glutamate and glutamine occurred during the metabolism of the removed [3-13C]glutamine. As expected, because their labeling was already low with 5 mmol/L [3-13C]glutamine, the labeling of the C4 and the C5 of glutamate and that of the C2 + C3 of succinate could not be detected with 1 mmol/L [3-13C]glutamine as substrate.
Aspartate labeling explained ∼20% and 30% of the fate of the carbon 3 of the [3-13C]glutamine metabolized with 5 and 1 mmol/L substrate concentration, respectively. It was not modified in brain slices from glutaminase-deficient mice when compared with that in brain slices from control mice. Such labeling occurred mainly and in equal amounts on its inner carbons (the C2 and the C3) and, to a much lesser extent, on its outer carbons (the C1 and the C4). Gamma-aminobutyric acid was found to be slightly labeled on its carbon 3 in brain slices from both control and glutaminase-deficient mice whereas alanine, whose formation requires malic enzyme or phosphoenolpyruvate carboxylase activity, was not found to be labeled.
The production of 13CO2 (calculated as the difference between the [3-13C]glutamine removed and the sum of the 13C resonances found in the spectra), which was lower in brain slices from glutaminase-deficient mice than in brain slices from control mice, was the major fate of the C3 of the [3-13C]glutamine metabolized. Indeed, with 5 mmol/L [3-13C]glutamine, 13CO2 represented 62.2% and 61.3% of the C3 of the [3-13C]glutamine metabolized in brain slices from control and glutaminase-deficient mice, respectively; with 1 mmol/L [3-13C]glutamine, the corresponding values were 56.9% and 58.3%, respectively.
Table 3 shows the fluxes through enzymes of glutamine metabolism whose scheme is presented in Figure 2. Only fluxes through glutaminase, succinate dehydrogenase, and citrate synthase (and through other enzymes of the tricarboxylic acid cycle, i.e., aconitase, isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, and malate dehydrogenase only with 1 mmol/L [3-13C]glutamine) were statistically diminished in brain slices from glutaminase-deficient mice when compared with brain slices from control mice with both 5 and 1 mmol/L [3-13C]glutamine as substrate.
Fluxes through pathways of 5 and 1 mmol/L [3-13C]glutamine metabolism in brain slices of wild-type (WT) and glutaminase-deficient (GLS1+/–) mice
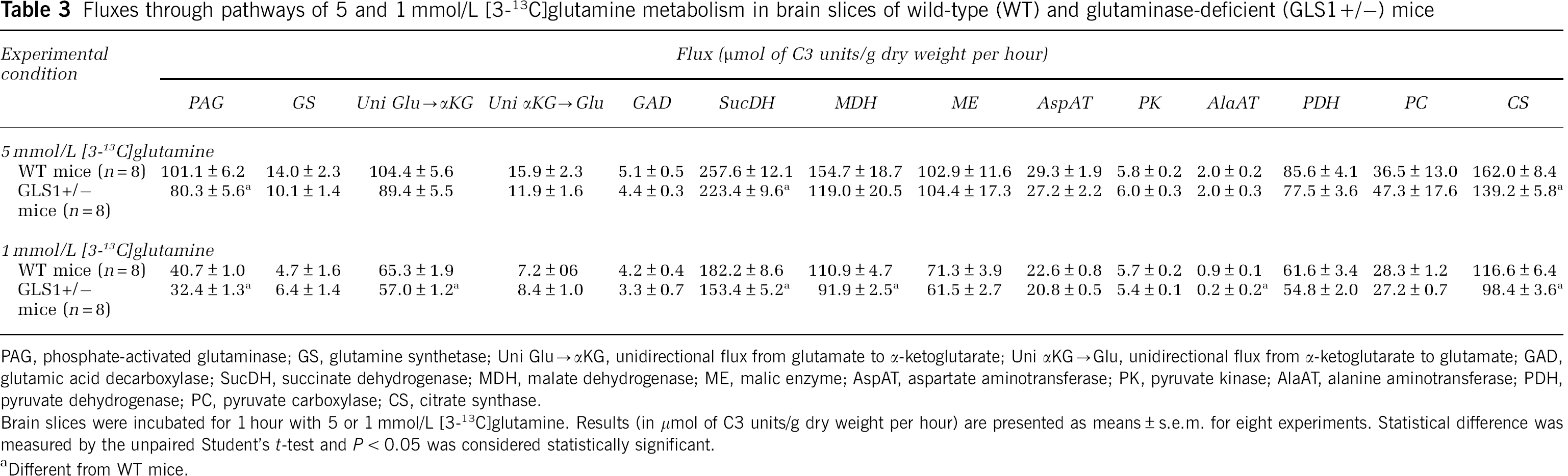
PAG, phosphate-activated glutaminase; GS, glutamine synthetase; Uni Glu→αKG, unidirectional flux from glutamate to α-ketoglutarate; Uni αKG→Glu, unidirectional flux from α-ketoglutarate to glutamate; GAD, glutamic acid decarboxylase; SucDH, succinate dehydrogenase; MDH, malate dehydrogenase; ME, malic enzyme; AspAT, aspartate aminotransferase; PK, pyruvate kinase; AlaAT, alanine aminotransferase; PDH, pyruvate dehydrogenase; PC, pyruvate carboxylase; CS, citrate synthase.
Brain slices were incubated for 1 hour with 5 or 1 mmol/L [3-13C]glutamine. Results (in μmol of C3 units/g dry weight per hour) are presented as means ± s.e.m. for eight experiments. Statistical difference was measured by the unpaired Student's
Different from WT mice.
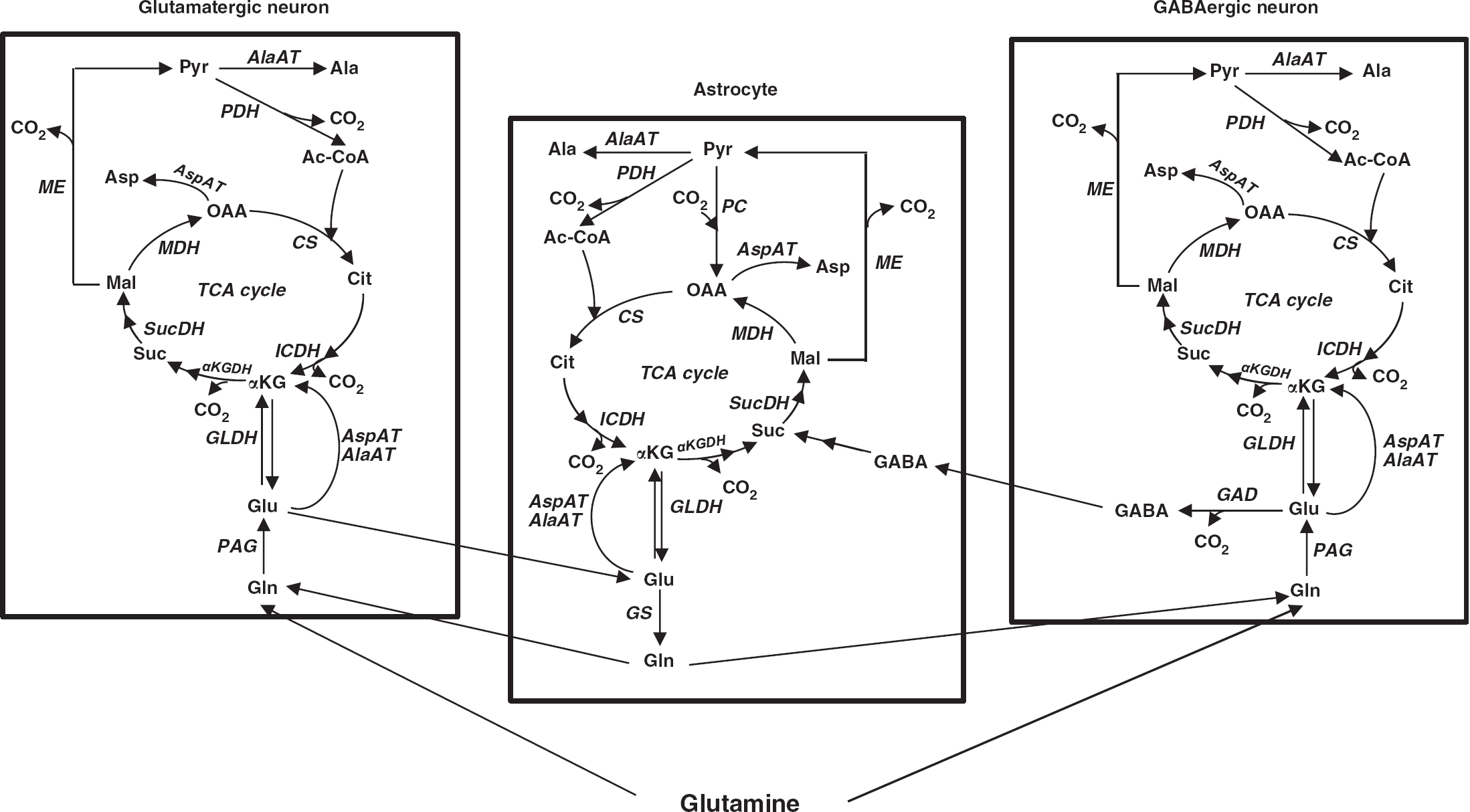
Scheme of glutamine metabolism in the brain. PAG, phosphate-activated glutaminase; GS, glutamine synthetase; GLDH, glutamate dehydrogenase; AlaAT, alanine aminotransferase; AspAT, aspartate aminotransferase; GAD, glutamic acid decarboxylase; GABA-T, gamma aminobutyric acid aminotransferase; αKGDH, α-ketoglutarate dehydrogenase; SucDH, succinate dehydrogenase; MDH, malate dehydrogenase; ME, malic enzyme; PDH, pyruvate dehydrogenase; PC, pyruvate carboxylase; CS, citrate synthase; ICDH, isocitrate dehydrogenase; TCA cycle, tricarboxylic acid cycle; Gln, glutamine; Glu, glutamate; αKG, α-ketoglutarate; Suc, succinate; Mal, malate; OAA, oxaloacetate; Cit, citrate; Asp, aspartate; Pyr, pyruvate; Ala, alanine; Ac-CoA, acetyl coenzyme A; GABA, gamma-aminobutyric acid.
Discussion
Characteristics of Glutamine Metabolism in Brain Slices from Glutaminase-Deficient Mice
It is noteworthy that, taking a dry weight/protein ratio equal to 1.3 (Antal et al, 1984), in both control and glutaminase-deficient mice, flux through glutaminase represented only 0.7% and 1.1% (with 5 mmol/L [3-13C]glutamine) and 0.3% and 0.4% (with 1 mmol/L [3-13C]glutamine) of the corresponding maximal glutaminase activity measured in brain homogenates of these mice (Gaisler-Salomon et al, 2009). Thus, despite this, our study shows that a reduction of the maximal activity of glutaminase in the brain is accompanied by a reduction of flux through glutaminase.
It is also interesting to note that, like rat brain slices metabolizing glutamine (El Hage et al, 2011b), mouse brain slices concomitantly degraded and synthesized glutamine (see Tables 1 and 2 and the corresponding comments in Results section).
It is interesting to mention that, despite a diminution of the formation of glutamate from glutamine in brain slices from glutaminase-deficient mice, there were no changes in the accumulation of 13C-labeled aspartate and GABA. This suggests that the preservation of the production of these two important neurotransmitters represents a priority for the brain. It is of interest to mention here that cultured neurons from GLS1–/– mice released GABA like neurons from normal mice (Masson et al, 2006). Note that the C1 and C4 of aspartate became labeled after the formation of the C1 and C4 of oxaloacetate resulting from a complete tricarboxylic acid cycle turn involving the formation of [1-13C]succinyl-coenzyme A synthesized from [2-13C]α-ketoglutarate.
Limitations of the Results Obtained
It should be underlined that the results presented are those obtained after 60 minutes of incubation and that the combination of these results with the model developed and used in the present study provides only mean rates over the incubation period used. In this respect, it is possible and likely that certain product accumulations and labelings reached steady states before others. Further studies would allow to examine this question but it should be underlined that time course experiments are difficult to perform because of the small amount of cerebral tissue available from each mouse and because of the limited number of genetically modified mice available. Another limitation is that, with 1 mmol/L [3-13C]glutamine as substrate, the 13C resonances obtained and measured are much smaller and therefore much less reliable than those obtained with 5 mmol/L [3-13C]glutamine as substrate.
It should also be emphasized that, although we cannot assign fluxes through all enzymes to a specific compartment, we believe it is of interest to know these fluxes in the brain considered as a single compartment, which contains functional astrocytes, glutamatergic and GABAergic neurons.
It is also important to underline that our slices were incubated with [3-13C]glutamine as substrate but in the absence of glucose. Although we did not incubate mouse brain slices with [3-13C]glutamine plus glucose, we did so with rat brain slices (unpublished results). In the latter experiments, we observed that the addition of 5 mmol/L glucose to the incubation medium containing 5 mmol/L [3-13C]glutamine did not significantly modify glutamine utilization nor fluxes through glutaminase and glutamine synthetase. As expected, glucose addition greatly diminished aspartate accumulation because the oxaloacetate available was diverted from aspartate to citrate synthesis as a result of an increased formation of acetyl-coA in the presence of glucose. It is therefore very likely that the high rate of aspartate production observed in the present study was due to the absence of glucose in the incubation medium.
Functional Consequences of a Decreased Glutaminase Activity
This study shows that,
It is important to mention here that a relationship has recently been shown between the genetic variation in GLS1 and the brain glutamine-to-glutamate ratio measured
Studies by other authors have shown that a complete deficit of glutaminase leads to mice which die shortly after birth (Masson et al, 2006) whereas GLS1 + /– mice do not display any change in their growth, development, and behavior but have a hippocampal hypoactivity and are resistant to the effects of pro-psychotic challenges (Gaisler-Salomon et al, 2009).
Finally, one cannot rule out the possibility that major metabolic changes observed in the present study in GLS1 + /– mice are amplified in GLS1–/– mice and contribute to their early death.
Footnotes
The authors declare no conflict of interest.