
Abstract
Phenelzine (PZ) is a scavenger of the lipid peroxidation (LP)-derived reactive aldehyde 4-hydroxynonenal (4-HNE) due to its hydrazine functional group, which can covalently react with 4-HNE. In this study, we first examined the ability of PZ to prevent the respiratory depressant effects of 4-HNE on normal isolated brain cortical mitochondria. Second, in rats subjected to controlled cortical impact traumatic brain injury (CCI-TBI), we evaluated PZ (10 mg/kg subcutaneously at 15 minutes after CCI-TBI) to attenuate 3-hour post-TBI mitochondrial respiratory dysfunction, and in separate animals, to improve cortical tissue sparing at 14 days. While 4-HNE exposure inhibited mitochondrial complex I and II respiration in a concentration-dependent manner, pretreatment with equimolar concentrations of PZ antagonized these effects. Western blot analysis demonstrated a PZ decrease in 4-HNE in mitochondrial proteins. Mitochondria isolated from peri-contusional brain tissue of CCI-TBI rats treated with vehicle at 15 minutes after injury showed a 37% decrease in the respiratory control ratio (RCR) relative to noninjured mitochondria. In PZ-treated rats, RCR suppression was prevented (P < 0.05 versus vehicle). In another cohort, PZ administration increased spared cortical tissue from 86% to 97% (P < 0.03). These results suggest that PZ's neuroprotective effect is due to mitochondrial protection by scavenging of LP-derived 4-HNE.
INTRODUCTION
During the last two decades, researchers have demonstrated that mitochondria play a vital role in traumatic brain injury (TBI).1–4 Brain mitochondrial dysfunction has been firmly associated with excitotoxicity, oxidative damage and metabolic perturbations.5–7 Changes in mitochondrial function, such as suppression of mitochondrial oxidative phosphorylation, accumulation of reactive oxygen species (ROS), loss of mitochondrial membrane potential, and impaired calcium buffering have been shown to play a key role in induction of posttraumatic brain cell death. 8 Recent studies have demonstrated that much of the mitochondrial failure is mediated by ROS-induced lipid peroxidation (LP). 9
4-Hydroxy-2-nonenal (4-HNE) is a major aldehydic LP product of n to 6 polyunsaturated fatty acids that binds to various brain mitochondrial proteins during the first hours after TBI.8–10 Indeed, 4-HNE is a key mediator of oxidative damage-induced mitochondrial failure.1,2 Recently, we confirmed that direct application of 4-HNE to isolated normal brain cortical mitochondria mimics the effects of traumatic brain injury (TBI), which suppresses mitochondrial complex I and II respiratory function in association with 4-HNE accumulation in mitochondrial proteins. 10 In the present study, we first of all confirmed that 4-HNE directly targets brain mitochondria causing altered mitochondrial cellular bioenergetic function in vitro. Second, we examined the effect of the hydrazine-containing compound phenelzine (PZ) on 4-HNE-induced mitochondrial dysfunction using the Seahorse XF 24 analyzer for measuring oxygen consumption rates (OCR). The basis for this experiment was the demonstration by others that hydrazine-containing compounds including PZ can covalently scavenge 4-HNE and other LP-derived aldehydic products11–13 and might be useful for inhibiting the mitochondrial toxic effect of 4-HNE after acute CNS injury. We therefore undertook experiments to determine if the hydrazine function of PZ could also neutralize 4-HNE and thus block its toxicity in isolated rat brain mitochondria. Figure 1 shows the covalent reaction of PZ with 4-HNE that is believed to be the basis for its antioxidant protective effects.
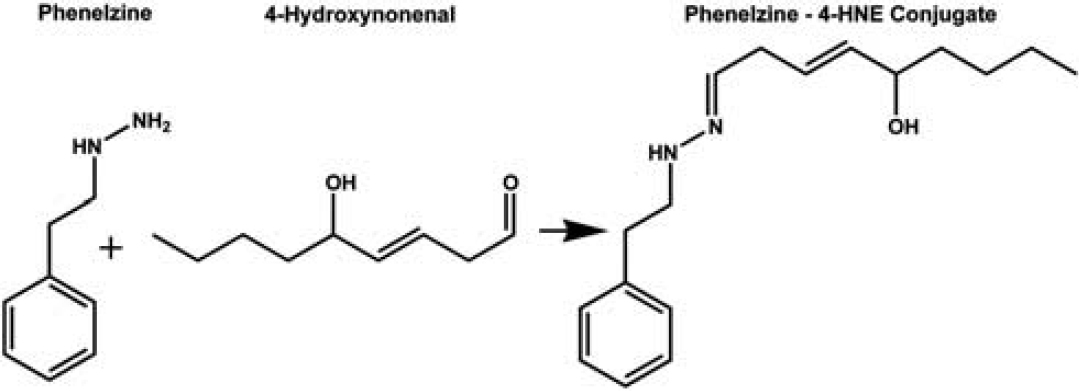
Covalent chemical reaction of 4-HNE with hydrazine moiety of phenelzine (PZ).
Phenelzine is also a nonselective and irreversible inhibitor of the enzyme monoamine oxidase (MAO, both MAO-A and MAO-B), and related to that mechanism of action, PZ will potentiate monoaminergic (dopamine, norepinephrine, serotonin) neurotransmission. Presumably due to that activity, it has long been an FDA-approved drug for the treatment of anxiety, atypical depression, depression that does not respond to other methods of treatment, bulimia, and social anxiety disorder. However, it has been shown to exert a neuroprotective effect in focal and global models of brain ischemia–reperfusion injury due to its ability to scavenge LP-derived aldehydes such as 4-HNE. 13 Accordingly, we quantitatively assessed the protective effects of early postinjury PZ in rats on posttraumatic mitochondrial respiratory function during the first 3 hours after controlled cortical impact (CCI)-TBI as well as its ability to improve cortical tissue sparing at 14 days after injury. Therefore, if it was effective in helping to lessen oxidative damage in the injured brain, its translational use in TBI may be easier to achieve than with a newly discovered compound.
MATERIALS AND METHODS
Animals
Young adult male Sprague-Dawley rats (Harlan, Indianapolis, IN, USA) weighing 300 to 350 g were employed for in vivo portions of this study. In vitro experiments described in this study were completed using isolated mitochondria from naive young, adult (8-week-old) male CF-1 mice (Charles River Labs, Portage, MI, USA). Animals received ad libitum access to food and water. All animal protocols followed the guidelines of the National Institutes of Health and complied with the animal study policy and regulations approved by the University of Kentucky Institutional Animal Care and Use Committee, in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Chemicals
Sodium pyruvate, malate, rotenone, carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), magnesium chloride (MgCl2), sucrose, mannitol, EGTA, bovine serum albumin, HEPES potassium salt, Triton, Tris HCl, NaCl, EDTA, glycerol, protease inhibitors (Complete Mini Protease Inhibitor Cocktail tablet, Roche Diagnostics, Indianapolis, IN, USA). potassium phosphate monobasic anhydrous (KH2PO4), were obtained from Sigma-Aldrich (St Louis, MO, USA), and oligomycin was obtained from Enzo Life Sciences (Farmingdale, NY, USA) 4-hydroxy 2-nonenal was purchased from EMD Chemicals (Merck KGaA, Darmstadt, Germany). Phenelzine sulfate salt was obtained from MP Biomedicals (Solon, OH, USA). Working solutions for every experiment were prepared fresh by diluting in respiration buffer for in vitro measurements. All materials and reagents for the XF assays were obtained from Seahorse Biosciences (North Billerica, MA, USA). BCA protein assay kit was purchased from Pierce (Rockford, IL, USA).
Isolation of Ficoll-Purified Mitochondria
Brain mitochondria was isolated as previously described.2,14 Briefly, brain cortical tissue was homogenized using Potter-Elvejhem homogenizers containing ice-cold isolation buffer pH 7.2, which consists of 215 mmol/L mannitol, 75 mmol/L sucrose, 0.1% bovine serum albumin, 20 mmol/L HEPES, and 1 mmol/L EGTA. The crude mitochondrial pellet obtained after differential centrifugation was subjected to nitrogen disruption. The crude mitochondrial pellet was resuspended in isolation buffer and layered on top of a discontinuous 7.5% and 10% Ficoll gradient, and centrifuged at 100,000 × g for 30 minutes at 4°C. The resultant mitochondrial pellet was resuspended in 25 to 50 μL isolation buffer without EGTA to yield a concentration of ~10 mg/mL. The protein concentration was determined using a BCA protein assay kit measuring absorbance at 562 nm with a BioTek Synergy HT plate reader (Winooski, VT, USA). Mitochondria were prepared fresh for every experiment and were used immediately for in vitro respiration assays.
Measurement of In Vitro Mitochondrial Function using the XF24-Extracellular Flux Analyzer
For in vitro mitochondrial respiratory functional assessment, we used the high-throughput XF24 extracellular flux analysis, to quantify the bioenergetic changes that occur in intact and well-coupled Ficoll-isolated brain mitochondria as previously described 14 The Seahorse XF24 Extracellular Flux Analyzer (Seahorse Biosciences) enables multiwell plate based measurement of mitochondrial bioenergetics in isolated mitochondria or cell culture.14–18 The XF24 involves a transient, 7 μL chamber in specialized microplates that allows for the determination of oxygen and proton concentrations in real time.18,19
Based upon our previous studies, 5.0 μg mitochondrial protein was used to study the 4-HNE-induced mitochondrial dysfunction. In other experiments (Miller et al, unpublished data) we exposed isolated brain mitochondria to various concentrations of 4-HNE ranging from 10 to 100 μmol/L. Based on 4-HNE dose curve, a 30 μmol/L 4-HNE dose was selected for the present experiments. After basal oxygen consumption measurements using XF24 analyzer, the 4-HNE was injected through port A to give a final concentration of 30 μmol/L whereas control wells received no 4-HNE. Complex I (ADP rate using pyruvate + malate substrate) and complex II (succinate substrate)-driven OCR (pmoles O2/min) were assessed after 10 minutes of exposure to 4-HNE. Baseline mitochondrial oxygen consumption and respiratory control rates (RCR) were measured before 4-HNE treatment and verified again at the end of every experiment. The OCR measurements were performed as previously described. 14 Briefly, the area under the curve was generated by an algorithm executed by the Seahorse device and these values were compared between experimental and control groups. Using the Excel software package (Microsoft), point-by-point rates are generated using the AKOS algorithm written by AKOS Gerencser in collaboration with Seahorse Bioscience.
Western Blotting Detection of 4-HNE-Induced Oxidative Damage
The protein concentration for each sample was determined using a BCA protein assay kit (Pierce). Western blots were performed using 15 μg aliquots of protein, resolved on 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels. Proteins were transferred to polyvinylidene fluoride Immobilon-P membranes (Millipore, Durham, UK) by electro-blotting. Membranes were blocked at room temperature for 1 hour with 5% dry skim milk in tris-buffered-saline containing 1% Tween-20 (TBS-T, Sigma-Aldrich, Dorset, UK). The membranes were then incubated with the primary antibody overnight at 4°C. Anti-4-hydroxynonenal (HNE-J2 monoclonal antibody; 1:100, JalCA Japan Institute for the Control of Aging, Nikken SEIL, Shizuoka, Japan), in 5% milk in TBS-T was added, and incubated at 4°C overnight. Membranes were washed in TBS-T, incubated for 2 hours with an infrared labeled secondary antibody goat anti-mouse IRDye 800 (Li-Cor, Lincoln, NE, USA) was added to bind to the primary antibody. The bound complex was detected using the Odyssey Infrared Imaging System (Li-Cor). The images were analyzed using the Odyssey Application Software, version 1.2 (Li-Cor) to obtain the integrated intensities.
Controlled Cortical Impact Traumatic Brain Injury
The young adult male Sprague-Dawley rats (300 to 350 g) were subjected to a unilateral cortical contusion (2.2 mm) using an electronically controlled pneumatic impact device (TBI 0310; Precision Systems & Instrumentation, Fairfax Station, VA, USA) as previously described.1,3,19,20 All animals were anesthetized with isoflurane (2%) and placed in a stereotaxic frame (Kopf Instruments, Tujunga, CA, USA) before CCI injury. Before performing CCI, the skin was retracted and a 6-mm unilateral craniotomy was performed that was centered between the bregma and the lambda. The skull cap was removed without disruption of the underlying dura. The exposed brain was injured using a 5-mm beveled tip that compressed the cortex at 3.5 m/sec to a depth of 2.2 mm. Following injury, surgical foam was laid upon the dura, the skull cap was replaced, and a thin coat of dental acrylic was spread over the craniotomy site and allowed to dry before the wound was stapled closed. A rectal probe and heating blanket were employed to maintain body temperature at 37.0°C until the rats recovered their righting reflex.
Phenelzine Administration
The PZ concentrations chosen for the in vitro experiments in isolated brain mitochondria were based on previously examined concentrations in cell culture systems.21,22 For the in vivo TBI experiments, PZ sulfate was dissolved in 0.9% saline and administered as a single subcutaneous dose of 10 mg/kg in a concentration of 2.5 mg/0.5 mL at 15 minutes after CCI-TBI. The PZ dose used for the TBI experiments was based upon prior use of a similar dose by other investigators in an ischemia-reperfusion brain injury model. 13
Measurement of Ex Vivo Mitochondrial Respiratory Function After Traumatic Brain Injury Using a Clark-Type Electrode
For studies of mitochondrial respiratory function in isolated mitochondria from the ipsilateral injured cortex from rats subjected to CCI-TBI, we used a Clark-type oxygen electrode in a sealed, thermostatically controlled, and continuously stirred chamber (Oxytherm System, Hansatech Instruments, Norfolk, UK), maintained at 37°C as described previously.1,2 The respiratory control ratio (RCR) was calculated by dividing state III oxygen consumption (defined as the rate of respiration in the presence of ADP, second bolus addition) by the state IV oxygen consumption (rate obtained in the presence of oligomycin).
Tissue Processing and Measurements of Cortical Tissue Sparing
After a survival of 14 days, the animals were anesthetized by an overdose of pentobarbital (150 mg/kg body weight intraperitoneally) and transcardially perfused with physiological saline followed by 10% buffered formalin (w/v) at pH 7.4. The brains were removed, postfixed for 24 hours, and subsequently placed in formalin-sucrose (15%) for an additional 24 hours. Coronal sections 50 μm thick were cut with a freezing microtome throughout the rostral caudal extent of the damaged hemisphere, extending from the septal area (intra aural level 10.7) to the most posterior extent of the hippocampus (intra aural level to 0.3). Sections were stained with cresyl violet and subjected to image analysis (ImageJ, NIH, Bethesda, MD, USA). The extent of tissue sparing following TBI was assessed blindly with respect to being from vehicle or PZ-treated rats. The analysis was carried out according to the employed an unbiased Cavalieri stereological protocol as previously described.23,24 Briefly, 12 sections were selected through the injured brain area that were separated by a known distance (d). The distance d is known because it is the mean measured section thickness multiplied by the number of sections between the sampled sections. On each section, the total cortical area, defined as the dorsal aspect of lamina I to the dorsal aspect of the corpus callosum was determined for each hemisphere. Each area was multiplied by d to calculate a subvolume, the subvolumes were then summed to yield the total volume. The volume of ipsilateral hemisphere was then compared with the volume of tissue measured in the contralateral hemisphere and the results expressed as percentage of tissue spared (ipsilateral/contralateral × 100). These methods eliminated the need to adjust values due to possible differential shrinkage resulting from fixation and tissue processing.
Statistical Analysis
Statistical analysis was performed with Prism version 5.0 (Graph Pad, San Diego, CA, USA). Statistical analysis for the in vitro and ex vivo mitochondrial respiration studies involved an initial one-way analysis of variance (ANOVA) followed by Student–Newman–Keuls (SNK) post hoc analysis. For the oxidative damage (4-HNE bound proteins) studies, the two-tailed unpaired Student's t-test was employed. The analysis of spared tissue volume was also performed to compare vehicle- versus PZ-treated animals using a repeated-measures ANOVA across all 12 brain sections and by a two-tailed Mann–Whitney test to compare the overall volume of spared cortical tissue expressed as a percent of the contralateral uninjured cerebral cortex. In all cases, a P < 0.05 was required for statistical significance. All data are expressed as mean ± s.d. except in Figure 5D in which the data are shown in a dot plot with median indicated.
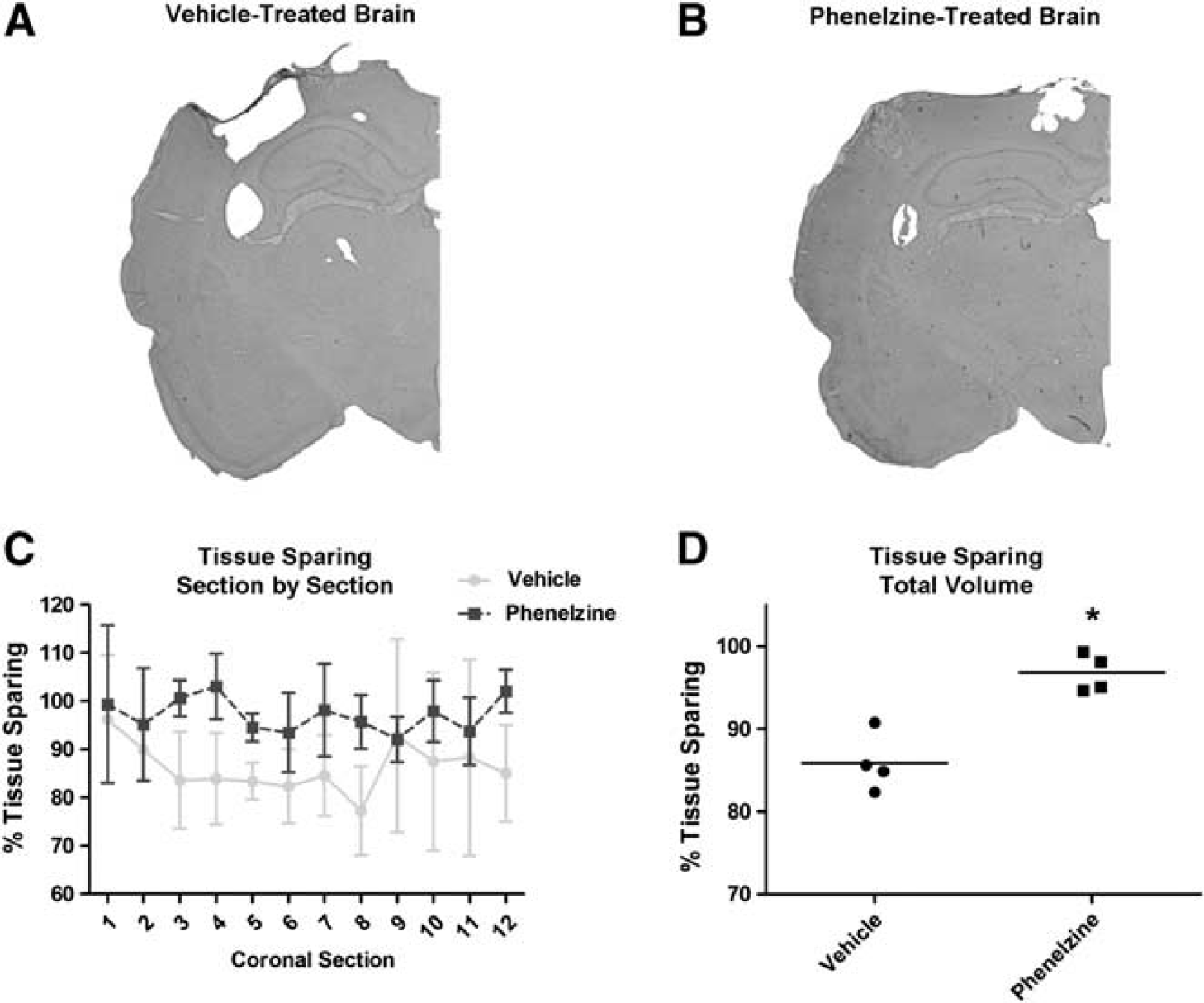
Effect of phenelzine to increase 14-day posttraumatic cortical tissue sparing. (
RESULTS
Phenelzine Protection Against 4-HNE-Impaired Brain Mitochondrial Bioenergetic Dysfunction in vitro
We previously reported the potency of the LP by-product, 4-HNE to produce brain mitochondrial bioenergetic dysfunction in isolated mitochondria in vitro using a Clark-type electrode which required 70 μg of mitochondrial protein. 10 We recently replicated the detrimental concentration-related effects of 4-HNE on isolated mitochondria using the more sensitive and high-resolution extracellular flux method developed by Seahorse Biosciences in which we used 5.0 μg of mitochondrial protein. Those studies confirmed that 4-HNE produced a progressive dose-related reduction in pyruvate/malate/ADP-driven complex I and succinate-driven complex II function third (Miller et al, in press). From those experiments, we employed a 30 μmol/L concentration of 4-HNE against which to test the ability of PZ to prevent 4-HNE mitochondrial impairment. As shown in Figure 2A, 30 μmol/L 4-HNE was observed to produce a 37% reduction in complex I respiration whereas, pretreatment of the mitochondria 5 minutes before 4-HNE exposure with a 30-μM concentration of PZ largely prevented this detrimental effect (ANOVA: P < 0.01, F = 20.6, df = 2,9). Post hoc analysis (SNK) showed that 30 μmol/L 4-HNE attenuated complex I function by 37% (P < 0.05 versus non-4-HNE treated mitochondria), PZ pretreatment reduced this to 17% (P < 0.05 versus 4-HNE alone) although the PZ-treated group was still significantly less than the untreated. Figure 2B also shows significant difference across the three groups complex II (ANOVA: P < 0.01, F = 9.1, df = 2,9). Complex II was inhibited by 24% by 4-HNE (P < 0.05 versus non-HNE treated; t-test), which was completely prevented by PZ pretreatment (P < 0.05 versus 4-HNE treated; t-test), to the point that there was no significant difference between the 4-HNE group treated with PZ and the non-HNE treated mitochondria.
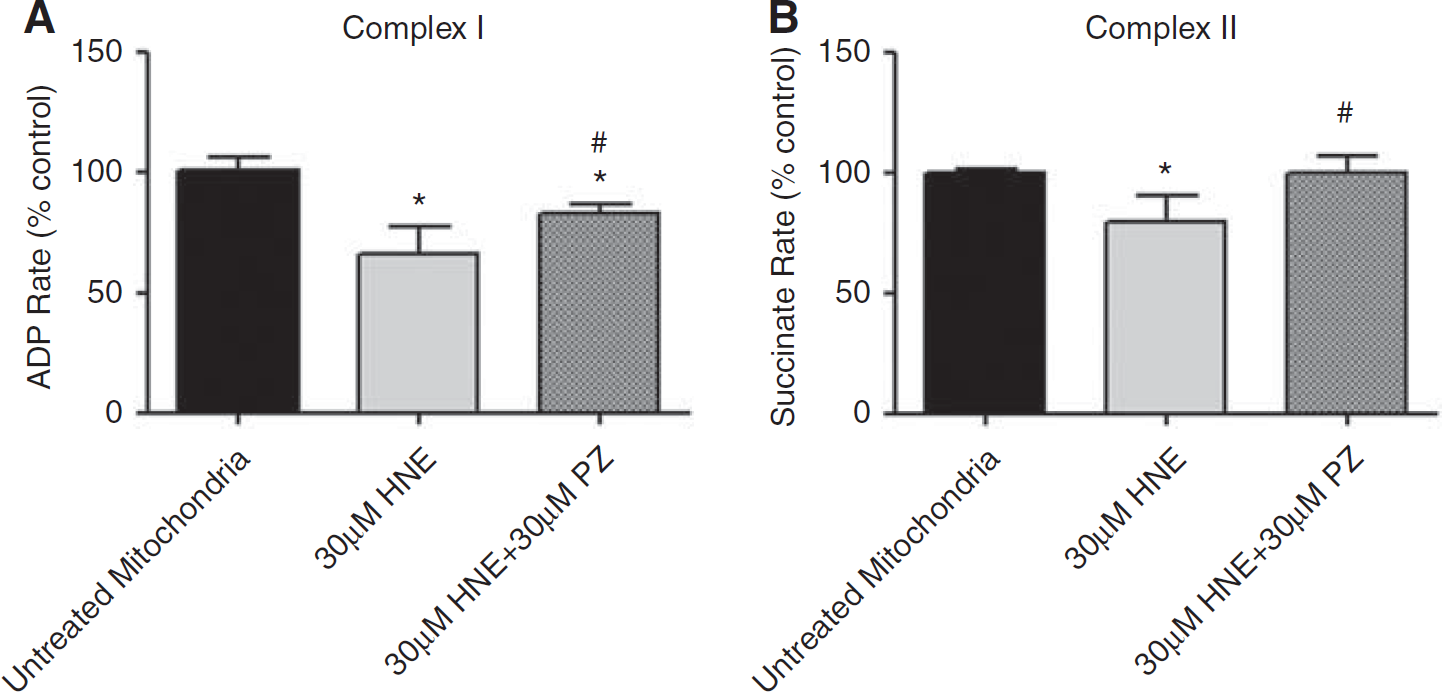
Protection by phenelzine (PZ) against 4-HNE-induced mitochondrial dysfunction in vitro in terms of complex I (A) and complex II (B) activities. Each analysis involved an initial one-way analysis of variance (ANOVA) followed by Student–Newman–Keuls (SNK) multiple comparison testing. For all experiments, significance was set at P < 0.05. Values represent mean ± s.d. n = 4/group.
Phenelzine Attenuation of 4-HNE-Induced Oxidative Damage in Vitro
In a parallel in vitro experiment, Ficoll-purified brain mitochondria were isolated and treated with 30 μmol/L 4-HNE in vitro for 10 minutes. As shown in Figure 3A, a large concentration-related increase able increase in protein-4-HNE adducts was observed when brain mitochondria were preincubated with 3, 10, or 30 μmol/L 4-HNE in vitro for 10 minutes at 37°C as examined by western blot analysis. This protein-4-HNE adduct increase was mostly prevented protected by pretreatment with an equimolar concentration of PZ. Quantitative analysis of the westerns, shown in Figure 3B, revealed a statistically significant near complete protection of 30 μmol/L 4-HNE-induced mitochondrial oxidative damage when mitochondria were pretreated with 30 μmol/L PZ for 5 minutes (P < 0.01 versus 4-HNE alone; t-test).
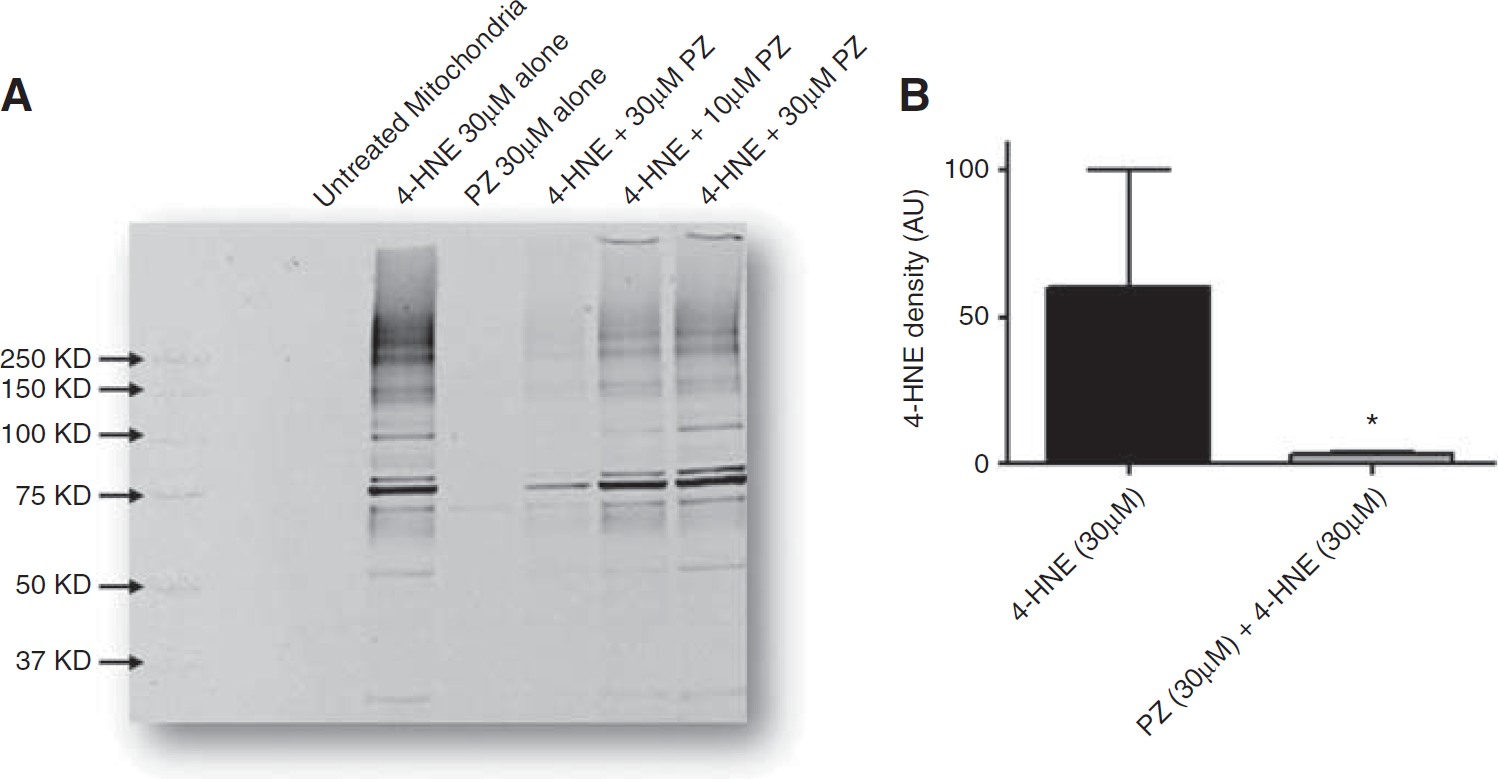
(
Phenelzine Inhibition of Post-Traumatic Brain Injury Cortical Mitochondrial Dysfunction and Oxidative Damage
In a another set of experiments, we explored the ability of PZ to preserve posttraumatic mitochondrial bioenergetics (RCR and respiration rates) by administering 10 mg/kg of PZ subcutaneously or vehicle 15 minutes after injury and measuring mitochondrial respiration ex vivo in cortical Ficoll-purified samples from the CCI-TBI epicenter and immediately surrounding peri-contusional area using a Clarke-type oxygen electrode at 3 hours after injury. Figure 4 indicates the respiratory rates compared between the sham vehicle-treated, injured vehicle-treated, and injured PZ-treated groups. For the RCR (Figure 4A), there was a significant difference across the noninjured, injured vehicle and injured PZ groups (ANOVA: P < 0.0001, F = 25.7, df = 2,23). Post hoc (SNK) analysis showed that injured vehicle-treated mitochondria displayed a significantly decreased RCR compared with noninjured mitochondria (P < 0.05), which was completely prevented by PZ treatment (P < 0.05 versus vehicle treated). The RCR is calculated as the ratio of complex I ADP-driven state III OCR to the state IV oligomycin-inhibited OCR. Examination of the effects of CCI-TBI on state III with and without PZ treatment (Figure 4B) failed to reveal a significant difference across the groups (ANOVA: P = 0.07, F = 3.0, df = 2,23). Although this precluded a post hoc analysis, a mean post-TBI decrease in state III of 40% was observed which certainly play a role in the decrease in the RCR (Figure 4A) considering that there was no apparent effect of injury to increase on state IV respiration (Figure 4C). In summary, an effect of posttraumatic PZ treatment was observed in terms of a significant preservation of complex I RCR.
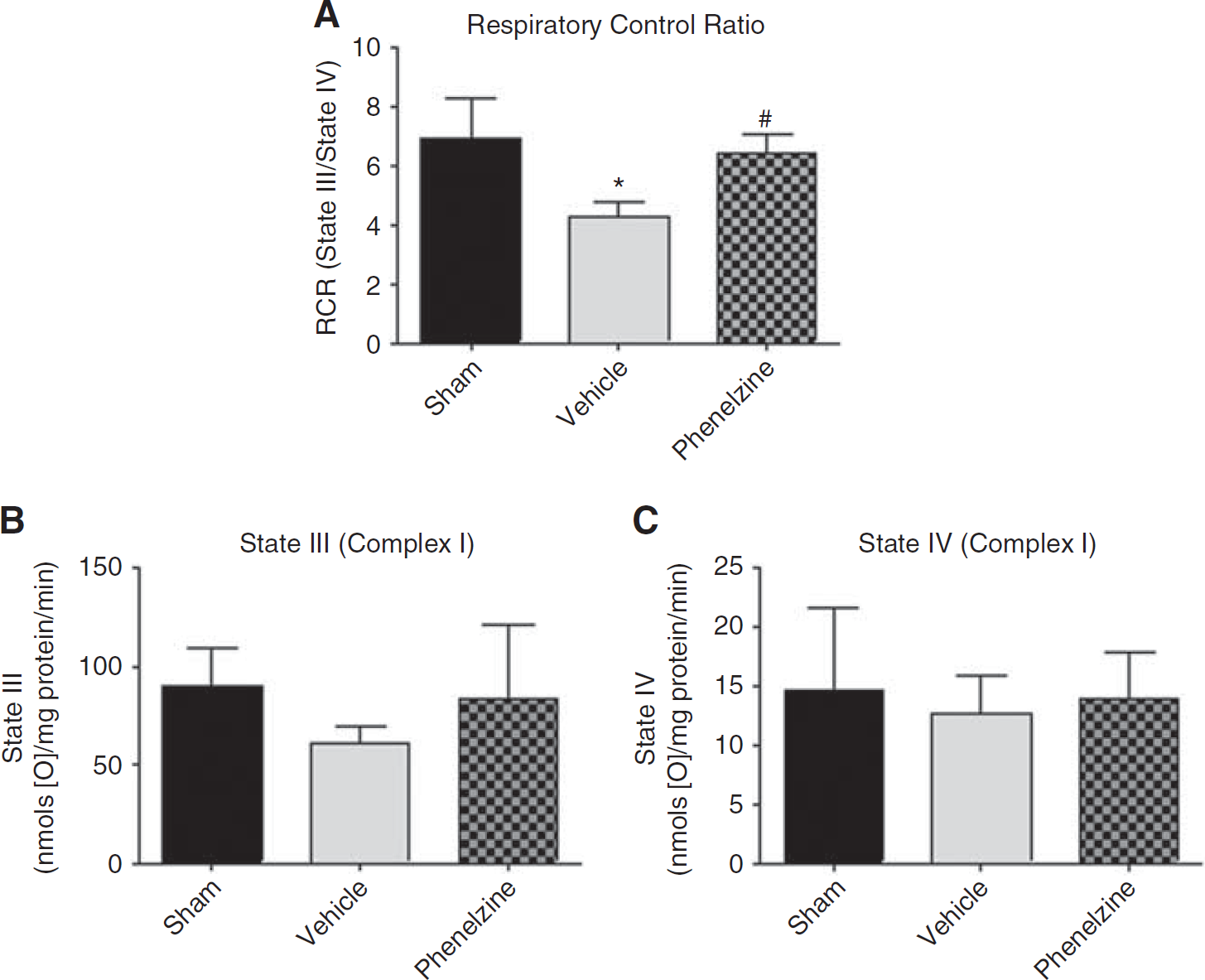
Effects of phenelzine (PZ) on cortical mitochondrial bioenergetics at 3 hours following severe controlled cortical impact traumatic brain injury: (
Phenelzine Improvement in Post-Traumatic Brain Injury Cortical Tissue Sparing
In a second set of CCI-TBI experiments, we examined whether a single subcutaneous dose of PZ could improve cortical tissue sparing at 14 days after injury. As shown in Figure 5, PZ dosing at 15 minutes after CCI-TBI significantly increased the volume of spared cortical tissue from 86% in vehicle-treated rats to 97% in PZ-treated animals (P < 0.03 versus vehicle, Mann–Whitney test). Conversely, there was a 14% loss in cortical tissue compared with the contralateral uninjured cortex in vehicle-treated animals that was reduced to only 3% in PZ-treated rats. Additionally, repeated-measures ANOVA also showed a significant PZ treatment main effect across the 12 brain sections included in the analysis (P < 0.01, F = 21.6, df = 1,6).
DISCUSSION
Lipid peroxidation is initiated by the attack of highly reactive free radical species (e.g., •OH, •NO2, •CO3) that are generated from the breakdown of nonradical ROS such as hydrogen peroxide or reactive nitrogen species such as PN. The subsequent LP self-propagating chain reactions involving the formation of lipid peroxyl (LOO•) and alkoxyl (LO•) radicals lead to the disruption of the integrity of cellular and intracellular membrane polyunsaturated fatty acids (e.g., arachidonic acid), which increases cellular ionic permeabilities and disrupts the function of phospholipid membranes. This in turn compromises the function of phospholipid-dependent membrane-localized proteins such as the active transport ionic pumps (Ca++, Na+, and K+). Although LP can occur in all tissues, neural tissue is exquisitely sensitive to LP due to its having a high content of polyunsaturated fatty acids that are susceptible to free radical attack as well as a high content of iron, which plays an important role in the initiation and propagation of LP reactions. 9
In addition to membrane phospholipid destruction, LP chain reactions ultimately lead to fragmentation of the peroxidized polyunsaturated fatty acid and generation of aldehydic end-products. Multiple aldehydic (i.e., carbonyl-containing) end-products of LP are generated during oxidative damage depending upon the particular polyunsaturated fatty acid that is involved. These aldehydic products include malondialdehyde (MDA), 2-propanal (acrolein) and 4-HNE. While each of these can bind to cellular proteins, protein-bound MDA is mainly a nontoxic tombstone, the levels of which measured in diseased or injured tissues have some direct relationship to the amount of LP that has occurred. In contrast, while the levels of acrolein and 4-HNE (which has been the focus of this study) are also indices of the amount of LP that has occurred, they are highly toxic to cells in their own right. 25 It has recently been demonstrated that brain and spinal cord mitochondria are highly susceptible to 4-HNE's toxic actions. 10 While the molecular mechanisms of 4-HNE mitochondrial toxicity still remain unclear, it has been observed demonstrated that the generation of additional ROS and additional LP initiation is involved in the mechanism(s) of 4-HNE-induced neurotoxicity.26,27 Whatever the intricacies of 4-HNE neurotoxicity are, the loss of mitochondrial integrity is the pivotal event leading to neuronal cell death in the ischemic penumbra in stroke models 13 and in models of acute neurological injury including TBI1,28 and spinal cord injury.29,30
Our in vitro data demonstrate protective effects of PZ against 4-HNE-induced bioenergetic function and oxidative damage in isolated mitochondria that tracks with a decrease in 4-HNE modification of mitochondrial proteins. Moreover, the idea that these two effects are mechanistically related comes partially from other studies reporting protective effects of PZ in primary neurons and astrocytes against formaldehyde-induced toxicity, 21 and some of these protective properties of PZ have been confirmed to be due to its ability to sequester aldehydes. 31 In our in vitro experiments, 4-HNE produced a concentration-dependent decrease in complex I (ADP rate) and complex II (succinate rate) oxygen consumption that was directly related to an increase in the amount of 4-HNE bound to mitochondrial proteins. In contrast, both effects were attenuated by PZ.
It should be noted that the 30 μmol/L concentration of 4-HNE that we had to apply to the isolated mitochondria to see respiratory impairment we believe is higher than what would be needed to cause mitochondrial respiratory impairment in vivo. This is for two reasons. First, 4-HNE, as well as the other LP-derived aldehydes (carbonyl compounds) rapidly react with a wide variety of proteins of a broad range of molecular weights. While we can determine the relative amount of protein-bound 4-HNE by quantitative western blot in vehicle versus PZ-treated rats, it is impossible to convert that to a molar concentration of 4-HNE in brain tissue after TBI, or any other insult. Second, in our in vitro mitochondrial studies, the applied 4-HNE attacks the outer mitochondrial membrane first while some portion diffuses across and comes into contact with the inner mitochondrial membrane where it binds to various proteins that are involved in oxidative phosphorylation. However, in the mitochondria in the injured brain, the 4-HNE is generated within the injury-stressed mitochondrion and is therefore able to impair respiratory function at a much lower concentration compared with the amount we have to add extra-mitochondrially in our in vitro studies. Thus, while we cannot determine with any precision the molar concentration that causes mitochondrial impairment in vivo we expect that it is considerably less than the amount we needed to apply to the isolated mitochondria to produce impairment.
Although we have previously shown that the toxic aldehyde acrolein is even more potent than 4-HNE in impairment of brain mitochondrial function in vitro, 10 we did not investigate whether PZ can inhibit it's effects. However, PZ has been shown by others to attenuate acrolein toxicity in cultured retinal ganglion cells by sequestration of acrolein. 13
This effect of PZ caused us to investigate whether PZ could similarly protect against early mitochondrial failure during the first 3 hours after TBI. As shown, a single subcutaneous dose of PZ at 15 minutes after TBI significantly reduced the degree of post-TBI respiratory compromise, and this effect appears to be related to a decrease in 4-HNE modified mitochondrial proteins in the contusion and immediate peri-contusion area (the latter data not shown). More importantly, we have shown that the same PZ dose is able to significantly improve cortical tissue sparing at 14 days after TBI. Surprisingly, these in vivo neuroprotective effects are obtained with only a single subcutaneous dose of PZ administered at 15 minutes after TBI.
The present experiment, which has shown that a single fixed dose of PZ can preserve cortical mitochondrial function and improve cortical tissue sparing after TBI, is consistent with the work of others who have demonstrated neuroprotective effects of PZ in a focal cerebral ischemia model purportedly due to scavenging of LP-derived aldehydes. 13 However, this finding requires much additional investigation. Indeed, ongoing studies are exploring a more complete pharmacological analysis including a definition of the full dose-response, the post-TBI therapeutic efficacy window and the optimal repeated dosing regimen. Most importantly, we need to determine in a larger sample size if the cortical neuroprotective effects of PZ are consistent and are associated with an improvement in sensorimotor recovery, and whether the protection occurs in the injured hippocampus which would be expected to produce an improvement in cognitive recovery assessments. We have recently completed this type of detailed analysis with the radical scavenger tempol, 32 the lipid peroxyl radical scavenger U-83836E20,33 and the mitochondrial protectants cyclosporine A and NIM-811.34,35 Thus, based upon logical need to inhibit posttraumatic oxidative damage by multiple complimentary mechanisms, we plan to investigate inhibition of LP oxidative damage in the injured brain at multiple points by combining the aldehyde scavenging compound PZ with a second compound that acts upstream to either limit the initiation and/or propagation of post-TBI LP chain reactions to possibly achieve an additive or perhaps synergistic mitochondrial and neuronal protective effect.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.