
Abstract
Transient ischemia causes delayed neurodegeneration in selective brain areas, particularly in the CA1 field of the hippocampus. This is accompanied by neurovascular impairment. It is unknown whether neurodegeneration is the cause or consequence of vascular changes. In an entorhino-hippocampal-organotypic slice culture system with well-preserved blood vessels, we studied the interplay between neurodegeneration and neurovasculature. Short-term oxygen and glucose deprivation (OGD) resulted in upregulation of hypoxic markers and with a delay of 24 to 48 hours in selective nerve cell death in CA1. In parallel, local vessel density decreased as detected by markers of endothelial cells and of the extracellular matrix. Claudin-5, a tight junction protein and marker of the blood–brain barrier was reduced. Preventing neuronal death with tetrodotoxin or 6-cyano-7-nitroquinoxaline-2,3-dione rescued blood vessels, suggesting that vessel loss is not due to OGD per se but a consequence of neuronal death. Induction of excitotoxic neuronal death with AMPA caused widespread neurodegeneration, but vessel reduction was confined to CA1. In dentate gyrus without neuronal loss, vessel density increased. We propose that neuronal stress and death influence maintenance, loss and remodeling of the neurovasculature and that the type of vascular response is in addition determined by local factors within the hippocampus.
Keywords
INTRODUCTION
The cerebral vasculature supplies oxygen and nutrients to the brain as well as eliminates and protects the nervous system from toxins and pathogens. Endothelial cells assembling the inner lining of the vasculature release trophic factors, support the maintenance of the blood–brain barrier (BBB), and are involved in processes of angiogenesis and inflammation.1,2 A decrease in oxygen and glucose by a dysfunction in the cerebral vasculature can lead to neuronal impairment and cell death. Unfortunately, introducing pharmacological neuroprotectants before or after injury to prevent cell death or secondary damage to the central nervous system has so far not met clinical expectations. A better understanding of the proper functioning of the vasculature and its endothelium might provide new ways on how to preserve or restore the vasculature as one of the prerequisites for maintenance of brain activities.
It is a well-known phenomenon that different areas of the brain react differently to ubiquitously present noxious events as hypoxia or exposure to toxins. Reasons for selective vulnerability have been considered to be either due to specific properties of nerve cells (the special pathoclysis concept of Cecile and Oskar Vogt 3 ) or to regional differences in vascularization. 4 This controversial issue has not yet been resolved conclusively. The hippocampus with its different subfields is especially interesting in this context: while the CA1 area exhibits a high susceptibility to hypoxia/ischemia in humans 5 and in animal models,6–8 the CA3 area together with the hilus of the dentate gyrus is much more involved in epilepsy-associated hippocampal sclerosis 9 and in animal models of temporal lobe epilepsy. 10
In case of ischemia, the display of transmitter receptors and ion channels, but also control of oxidative and proteasomal stress11,12 have been proposed as the basis for the differential vulnerability of regions and/or subpopulations of neurons. In addition, local characteristics of glial cells 13 are considered to play a role in the regional selectivity of neurodegeneration. Interestingly, capillary damage and higher susceptibility of BBB leakage in the CA1 region but not the CA3 region in the gerbil and the rat model of cerebral ischemia–reperfusion have been reported.6,14,15 In the case of temporal lobe epilepsy, not vascular loss, but angiogenesis has been described10,16 and the overexpression of vascular endothelial growth factor (VEGF) by neurons and astrocytes has been suggested to be the mediator for BBB damage and vascular remodeling after excitotoxicity. 17 Whether the vasculature is remodeled as a direct effect of neurodegeneration is debatable.
Hippocampal-organotypic slice cultures challenged with oxygen and glucose deprivation (OGD) are in vitro models affording reproducibility and representing closely to in vivo models by mimicking cerebral ischemia that occurs after stroke or cardiac arrest.18,19 Hypoxic conditions in this slice culture system with or without additional glucose deprivation induce a comparable selective neurodegeneration in the CA1 region. 19 We have previously demonstrated the feasibility of the cortical organotypic slice culture as a BBB model system.20,21 In the present studies we have adapted this model to entorhino-hippocampal-organotypic slice cultures (EHOSCs), enabling us to study vascular effects in the CA1 region elicited by OGD and neuronal death.
Up to now, little data are available linking neurodegeneration and neurovascular remodeling. We investigated whether in the hippocampus, remodeling of the cerebral vasculature after ischemia is dependent on the selective neurodegeneration in the CA1 region and whether excitotoxic nerve cell death in other fields of the hippocampal formation has similar effects. A better understanding of not only the mechanisms of neuronal damage but also the diversity in brain regions in their reaction to cerebral injury as well as of the accompanying vascular damage is necessary for proper development of therapeutics.
MATERIALS AND METHODS
Preparation of Entorhino-Hippocampal-Organotypic Slice Cultures
All animal experiments were carried out according to the international guidelines on handling laboratory animals as well as the present Swiss law for animal experiment and were approved by the animal care committee of the Canton of Basel. Cultures were prepared as described previously.20,21 Briefly, C57Bl/6CRL mice were decapitated at postnatal day 4 (P4) and the brains were extracted and placed in ice-cold preparation medium containing minimal essential medium, 1% glutamax (Life Technologies, Zug, CH, Switzerland), pH 7.3. Meninges around the cortical hemispheres were gently removed without disturbing the hippocampus. The hippocampus together with the entorhinal cortex (EC) was dissected and 400 μm transversal hippocampal slices were sectioned using a McIllwain tissue chopper under aseptic conditions. Slices were carefully separated and laid over 0.4 μm Millicell-CM, 30 mm diameter culture inserts (Millipore, Zug, CH, Switzerland) in six-well plates with 1 mL/per well of incubation medium composed of: HEPES-buffered minimal essential medium (minimal essential medium, 50%), Hank's-buffered salt solution (25%), and heat-inactivated horse serum (25%) supplemented with glutamax (Life Technologies), glucose (1 g/L), pH 7.3. Plates were stored in a humidified incubator with 5% CO2 at 37°C. The medium was changed the next day and then every other day up to 1 week.
In Vitro Hypoxia and Oxygen–Glucose Deprivation with Reperfusion
Entorhino-hippocampal-organotypic slice cultures were cultured for 5 days (DIV5) before exposure to hypoxia or OGD. Oxygen and glucose deprivation medium was prepared as detailed by Rytter et al. 19 using Neurobasal medium containing: 2% B27 and 1% glutamax (Life Technologies). Two six-well tissue culture plates were filled with 1 mL/well of OGD medium, perfused with N2 for 1 hour in a hypoxic chamber (Billups-Rothenberg, Del Mar, CA, USA) and then sealed overnight to equilibrate the medium. Anaerobic strips (Sigma-Aldrich, Buchs, CH, Switzerland) were used as hypoxic indicators when the color of the strips changed from pink to white. Before the slices were exposed to OGD, the OGD medium was perfused again with N2 for 30 minutes. Propidium iodide (PI; Sigma-Aldrich, 2 μg/mL) was added to the slices for 30 minutes and only healthy slices were selected for experimentation as indicated by low numbers of PI-positive cells. The schedule of OGD exposure and reperfusion is indicated in Figure 2A. Briefly, slices were either deprived of oxygen only (hypoxia) or oxygen and glucose for 15 minutes, then replaced with oxygenated Neurobasal medium and allowed to recover for 3, 24, or 48 hours. Propidium iodide was added before the insult to establish the baseline of cell death and then added again during the recovery periods.
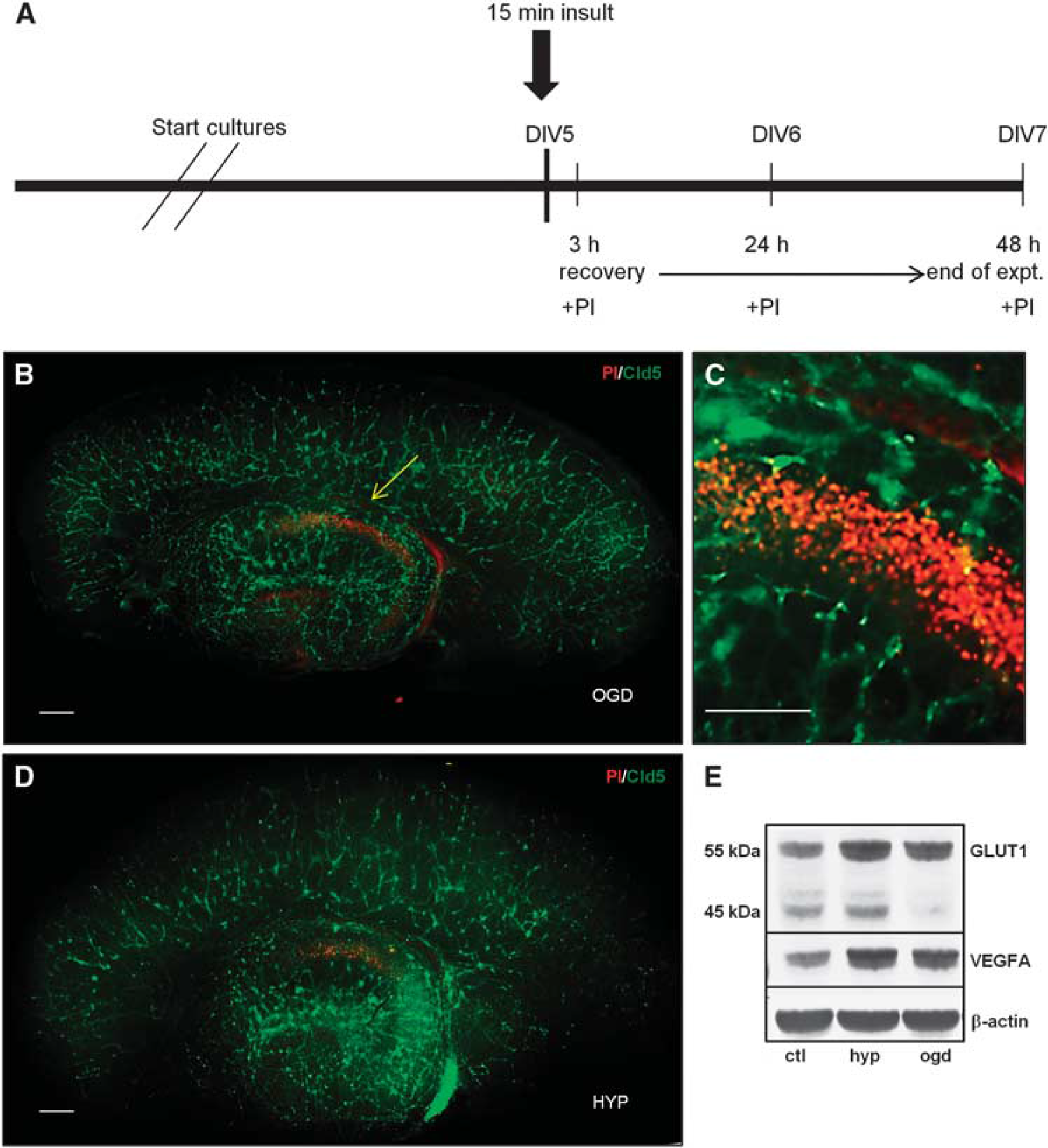
Entorhino-hippocampal-organotypic slice cultures (EHOSCs) submitted to either oxygen and glucose deprivation (OGD) or hypoxia. (
Pharmacological Interventions
At DIV5, EHOSCs with low number of PI-positive cells were selected for exposure with several compounds prepared in Neurobasal medium: 100 μmol/L (RS)-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA; R&D Systems, Oxford, UK), 100 μmol/L 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; R&D Systems), 1 μmol/L tetrodotoxin (TTX; R&D Systems) for 30 minutes in normoxic slices and then kept for 48 hours until fixation and immunohistochemistry. Treatment experiments were carried out with CNQX and TTX during or directly after the insult.
Immunohistochemistry
We used only hippocampal slices of the septal half because they displayed consistent cell death. Slices were fixed in freshly made 4% paraformaldehyde overnight at 4°C. Paraformaldehyde was removed the following day and the slices were washed three to four times with phosphate-buffered saline. Slices were carefully removed from the culture inserts for immunohistochemistry of free-floating sections. Each slice was placed in a well of a 96-well plate containing 100 μL of blocking buffer: 3% normal goat serum in phosphate-buffered saline for blocking and 2% Triton-X100 for permeabilization, respectively. The plates were placed on an orbital shaker and the slices were incubated in the blocking buffer for 2 hours at room temperature. Primary antibodies were prepared in phosphate-buffered saline containing 1% normal goat serum and 0.5% Triton-X100. The primary antibodies used for detecting blood vessels were: polyclonal laminin (Sigma-Aldrich) 1:200, polyclonal rabbit anti-human Von Willebrand Factor (Dako, Glostrup, Denmark) 1:250, polyclonal glucose transporter 1 (Thermo Scientific, Fremont, CA, USA) 1:1,000, and monoclonal Claudin-5 (Life Technologies) 1:50; and for detecting neurons: microtubule-associated protein 2 (MAP2; Abcam, Cambridge, UK) 1:500. DAPI (1500) was occasionally used to verify the total cell population. Incubation with primary antibodies was for 48 hours at 4°C and then washed with 0.5% Triton-X100 for 3 to 4 times. Then, slices were incubated for 3 hours at room temperature with secondary antibody prepared in 1% normal goat serum and 0.5%Triton-X100 of either goat anti-rabbit or goat anti-mouse conjugated to Alexa fluorochromes 350 or 488 (Life Technologies). Afterwards, slices were washed with tris-buffered saline (TBS) three times; mounted on glass slides and cover slipped with Mowiol. The stained slices were viewed by an Olympus AX-70 microscope (Olympus, Hamburg, Germany) installed with a Spot digital camera. The images were recorded and adjusted for brightness by Adobe Photoshop (Adobe Systems, Zurich, Switzerland) or ImageJ (NIH, Bethesda, MD, USA).
Western Blot
Four to five slices of the septal half of the hippocampal region were sonicated and lysed in 1 × RIPA buffer: 50 mmol/L Tris–HCl (pH 7.4), 1% NP-40, 025% deoxycholate, 150 mmol/L NaCl, 1 mmol/L EGTA. In total, 150 μg of protein was boiled for 5 minutes in Laemmli buffer and then loaded in an sodium dodecyl sulfate polyacrylamide gel electrophoresis. The gel was transferred onto a nitrocellulose membrane. The membrane was incubated at room temperature on an orbital shaker for 1 hour in blocking solution containing TBST (TBS and 0.1% Tween20), and 5% bovine serum (Sigma-Aldrich). After blocking, the membrane was probed with either polyclonal GLUT1 (Thermo Scientific) 1:5,000, polyclonal VEGF 164 (VEGFA; Abcam) 1:1,000, or monoclonal β-actin (Millipore) 1:8,000 overnight at 4°C and washed the next day for 5 to 10 minutes with TBST before incubating in secondary goat anti-rabbit or goat anti-mouse conjugated to AP (alkaline phosphatase; Jackson ImmunoResearch Europe, Suffolk, UK) for 1 hour. Then, the secondary antibody was washed several times with TBST for 30 minutes and the signal was detected by AP Conjugate Substrate Kit system (Bio-Rad, Cressier, FR, Switzerland).
Vessel Density and Statistics
The vessel density was quantified based on vessel crossings as described by Bendfeldt et al. 20 Recorded images of × 10 magnification were superimposed with three 6 × 6 grids, where each grid is equal to 100 × 100 μm per square and a total field of 0.25 mm2 over the CA1, CA3, DG, and EC region (Figure 1C). The average of the crossings from the three grids for each region was calculated. Countings were based on a minimum of nine animals (three independent experiments, which each included data from three animals, each providing three slices). Statistical analysis was performed by analysis of variance with Bonferroni correction as post hoc test (P < 0.05 was defined as significant).
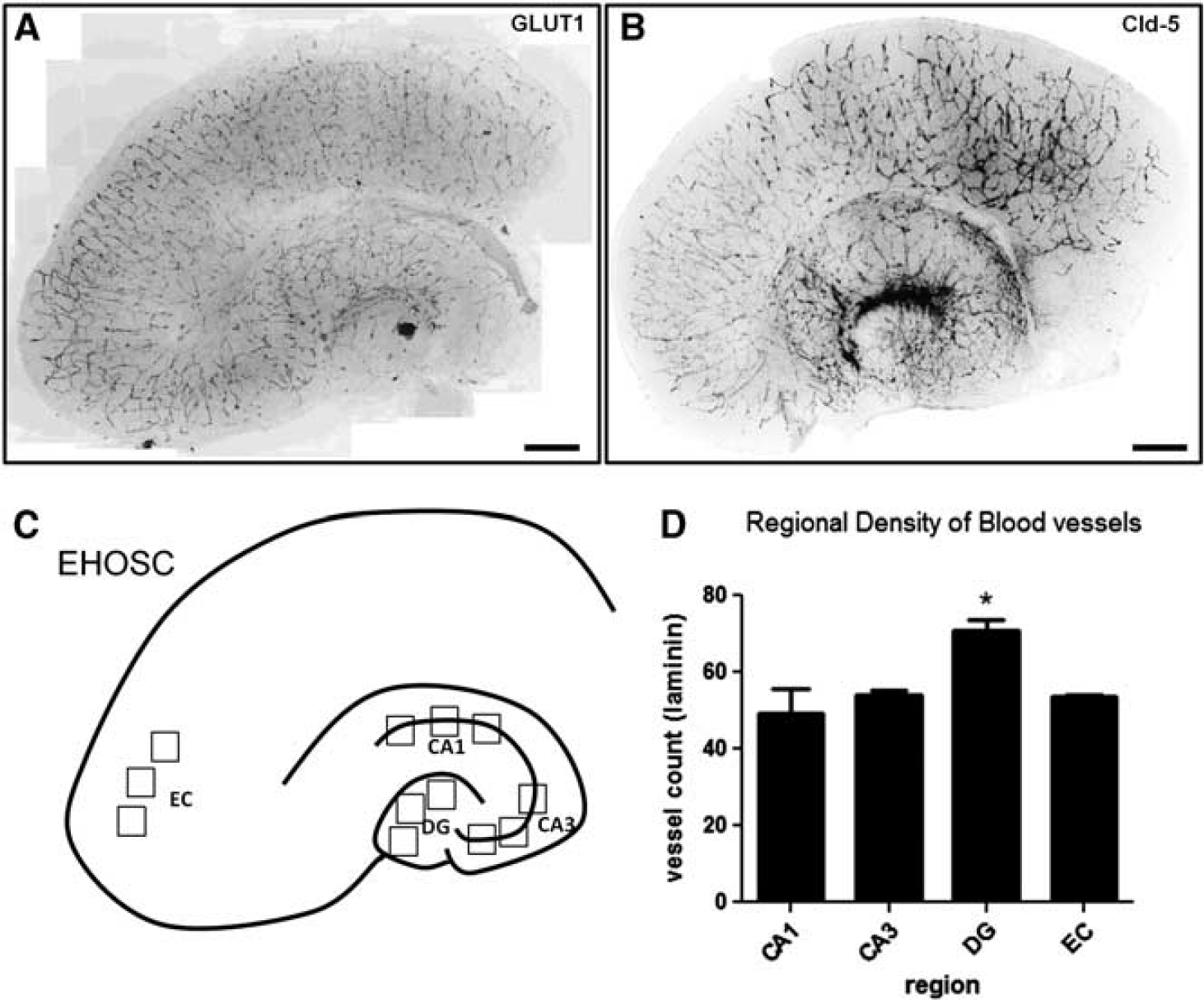
Preservation of blood vessels in entorhino-hippocampal-organotypic slice cultures (EHOSCs). (
RESULTS
Blood Vessels and Tight Junction Markers Are Preserved Under Normal Culture Conditions in Entorhino-Hippocampal-Organotypic Slice Cultures
Entorhino-hippocampal-organotypic slice cultures prepared of postnatal day 4 (P4) C57Bl/6CRL mice showed preservation of local blood vessels, which exhibited a stable expression of glucose transporter 1 (GLUT1) and claudin-5 (Cld-5) for 1 week (DIV7) in culture (Figures 1A and 1B). Vessel counts were performed in the single subfields of the EHOSCs (Figure 1C). Vessel densities as examined by laminin staining were similar for the CA1, CA3, and EC region but significantly higher in DG (Figure 1D).
Entorhino-Hippocampal-Organotypic Slice Culture as a Model for Studying the Blood–Brain Barrier in Short-Term Oxygen and Glucose Deprivation with 48 hours of Recovery
Since the presence of tight junction proteins and blood vessel densities remained stable for > 1 week in culture, we conducted all experiments within this time period (Figure 2A). Organotypic slices were cultured for 4 to 5 days and cell death was assayed in living slices by PI signal. Only slices with low constitutive PI staining were selected for experimentation. Oxygen and glucose deprivation caused cell death along the pyramidal layer of the CA1 sector, where strong PI staining was observed at 48 hours of recovery (Figures 2B and 2C). In parallel, staining for markers of the cerebral vasculature as Cld-5 (Figures 2B and 2C), von Willebrand factor (not shown) and laminin (see Figure 3) was massively reduced in the CA1 sector only. For all other subfields of the EHOSCs, the integrity of blood vessels appeared intact and resistant to short-term OGD. Hypoxia had the same effect on EHOSC but to a lesser extent: nerve cell death in CA1 was less pronounced and vessel reduction less severe (Figure 2D).
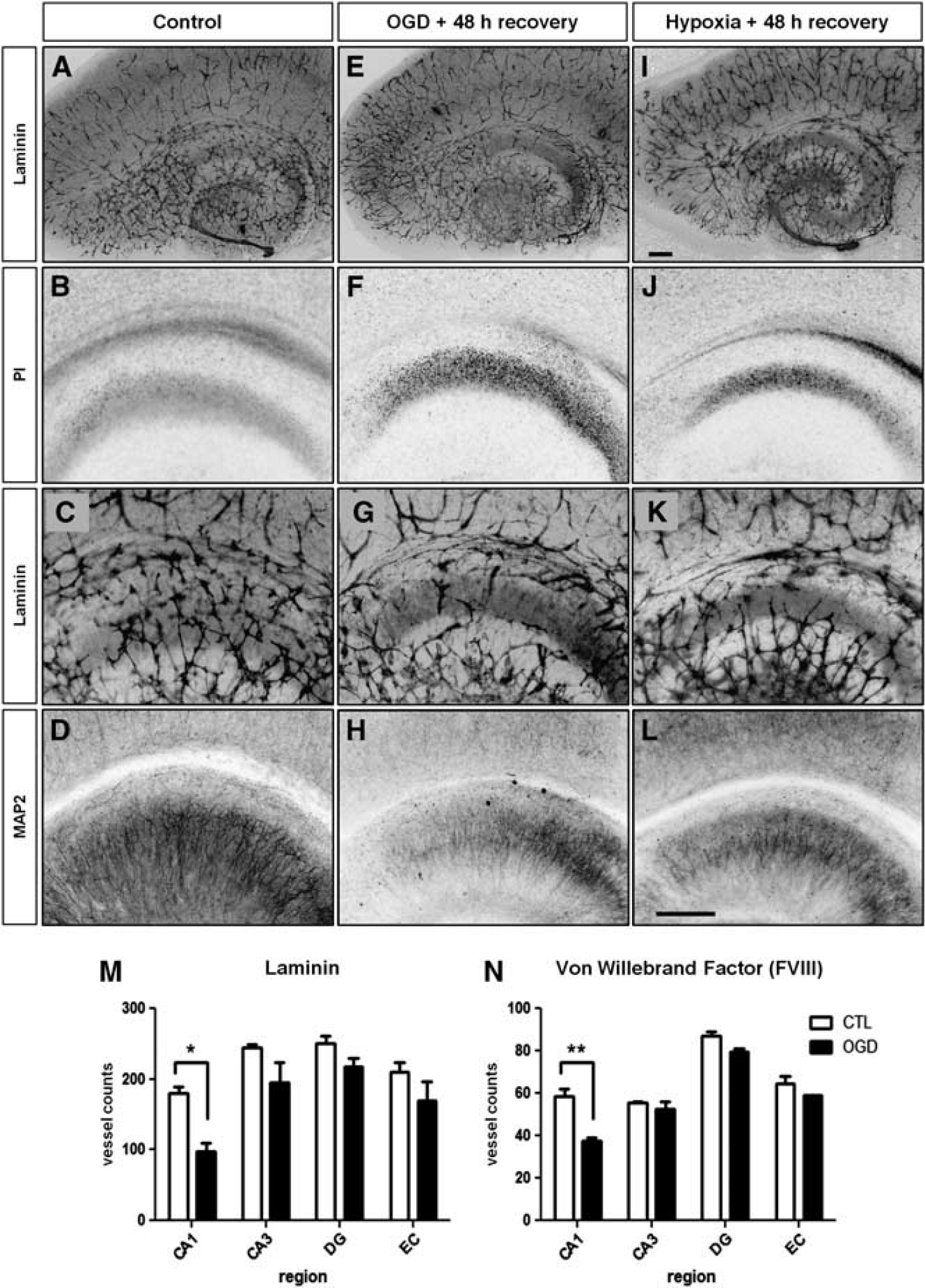
Oxygen and glucose deprivation (OGD) induces cell death, along with neuronal loss and vessel reduction in the CA1 region of the hippocampus. (
The protein expression in the EHOSCs of vascular endothelial growth factor-A (VEGFA) and the 55-kDa GLUT1 isoform were upregulated after short-term hypoxia or OGD followed by 48 hours recovery (Figure 2E). Both proteins are well known to be hypoxia regulated. Interestingly, the 45-kDa GLUT1 isoform that is expressed in glial cells was found to be downregulated specifically after OGD but not after hypoxia (Figure 2E).
Selective Vessel Loss in the CA1 Region Upon Short-Term Oxygen and Glucose Deprivation Followed by 48 hours Recovery
Despite the induction of cell death in the CA1 region by OGD or hypoxia in our model, overall the integrity of blood vessels in EHOSCs was preserved: the vasculature appeared well branched as observed by the abundance of Cld-5 or laminin immunolabeling (Figures 2B, 2D, 3A, 3E, and 3I). Solely in the CA1 region, blood vessels were reduced compared with the control (Figures 3C, Figures 3G, and 3K). Cell death seen with PI staining was pronounced in CA1 of the OGD or hypoxia-treated samples compared with the control (Figures 3B, Figures 3F, and 3J) and in parallel pyramidal neurons exhibited a disintegration of cytoskeleton as visualized with less MAP2 immunostaining (Figures 3D, Figures 3H, and 3L). These effects in the CA1 field were milder after hypoxia than OGD (Figures 3J, Figures 3K, and 3L). Other regions of the EHOSC (CA3, DG, EC) were not affected, neither PI increase nor MAP2 decrease was found there after short-term OGD or hypoxia followed by 48 hours recovery. The quantitation of vessel density for CA1, CA3, DG, and EC indicated a general decrease in the vessel number for all hippocampal regions, but the reduction of vessels became statistically significant only in the CA1 region where ~50% of the vessels were lost 48 hours after short-term OGD (Figures 3M and 3N). This holds true for the endothelial marker, von Willebrand factor (FVIII), as well as the extracellular matrix marker of blood vessels, laminin.
Since short-term OGD did affect vessels specifically in the CA1 region, we performed a time course analysis to see whether the change in vessel architecture began before, in parallel, or was preceded by neuronal death. Immunohistochemistry of Cld-5 revealed that at 3 hours of recovery, vascular architecture in CA1 did not differ from controls showing the presence of highly branched vessels and no PI-stained cells (Figures 4A–4C). As the recovery period lengthened to 24 hours, cell death became evident in the OGD-treated slices as PI-positive cells were appearing and, in parallel, the network of vessels started to become disintegrated (Figures 4E and 4F). At 48 hours of recovery, the CA1 region was virtually cleared of vessel coverage (Figures 4H and 4I). The temporal increase of PI signal from 24 to 48 hours suggests an inverse relationship occurring between the development of cell death and the decrease of vessels. No changes were evident in the control slices (Figures 4A, Figures 4D, and 4G). These findings indicate that the vessel changes neither occur before or after, but rather developed in parallel with a close temporal correlation to the development of neuronal death.
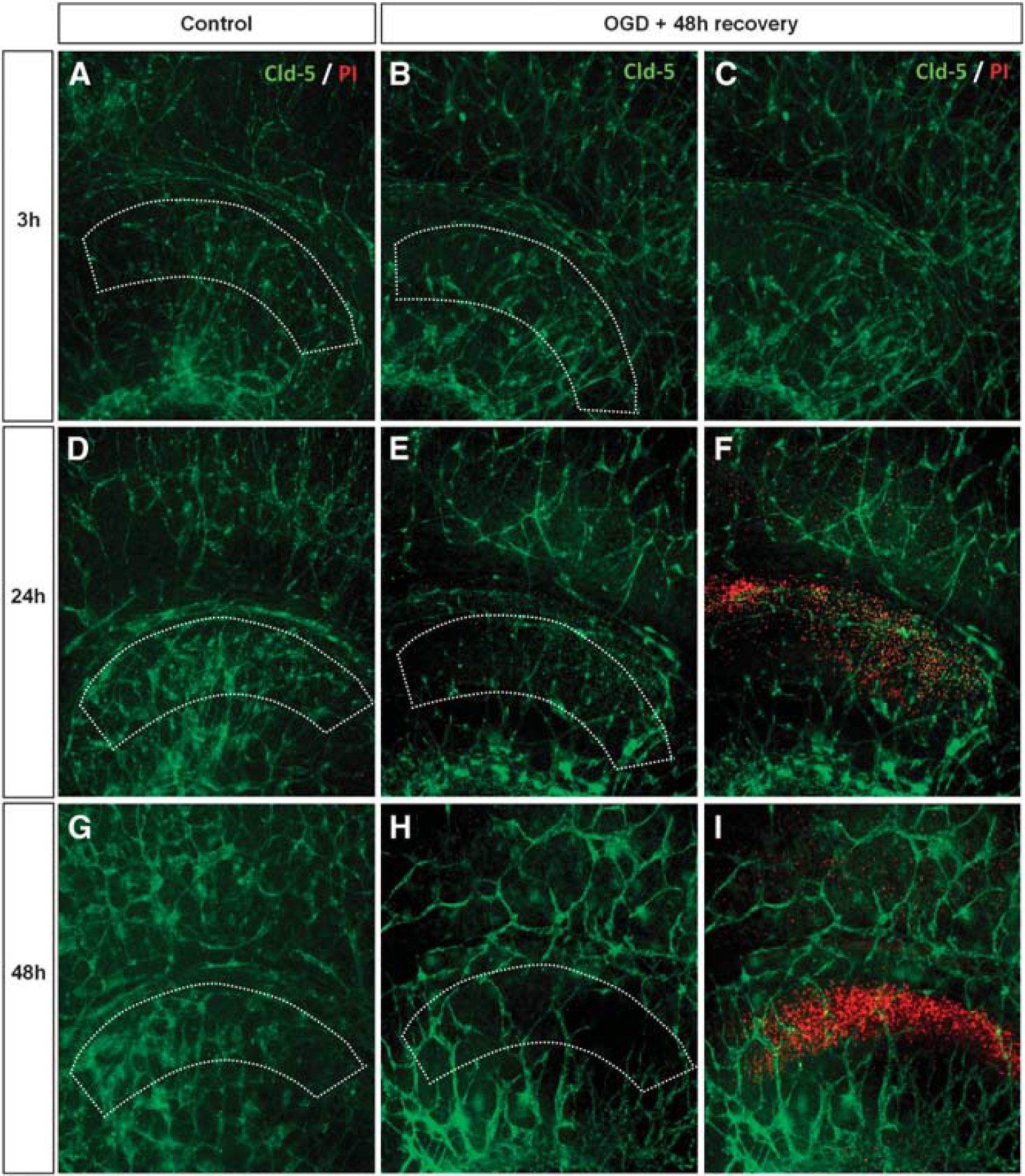
Time course of cell death and change in the vessel architecture at 3, 24, and 48 hours after oxygen and glucose deprivation (OGD). (
Protection of Neurons and Vasculature by Tetrodotoxin and CNQX After Short-Term Oxygen and Glucose Deprivation with 48 hours of Recovery
Next, we questioned whether the loss of vasculature after OGD is due to a direct effect of OGD on the vessels or due to the OGD-induced neurodegeneration. Furthermore, is blockade of neuronal activity sufficient to induce vessel loss, or must neurodegeneration take place to cause a disintegration of the vasculature? We tested this by using the blocker of fast sodium spikes TTX or the AMPA receptor antagonist CNQX. In control slices with TTX only, with neuronal activity silenced, vessel architecture was similar to controls (Figures 5A, Figures 5B, 5E and 5F). With 1 μmol/L TTX, CA1 neurons were strongly protected from cell death induced by OGD and the vessel architecture did not change compared with the OGD only (Figures 5C, Figures 5D, 5G and 5H). In control slices with CNQX only, the vessel architecture was also not affected (Figures 5I and 5J). Very similarly, 100 μmol/L CNQX also protected neurons from OGD (Figures 5C, Figures 5D, and 5K, and 5L). Quantification of vessel density clearly established that both neuroprotecting compounds, TTX and CNQX preserved the blood vessel morphology and density (Figure 5M). Furthermore, vessel density in CA1 did not change between controls and slices treated with TTX, where neuronal activity was blocked for 48 hours (Figure 5N). These data clearly show that vascular loss after OGD is due to neurodegeneration.
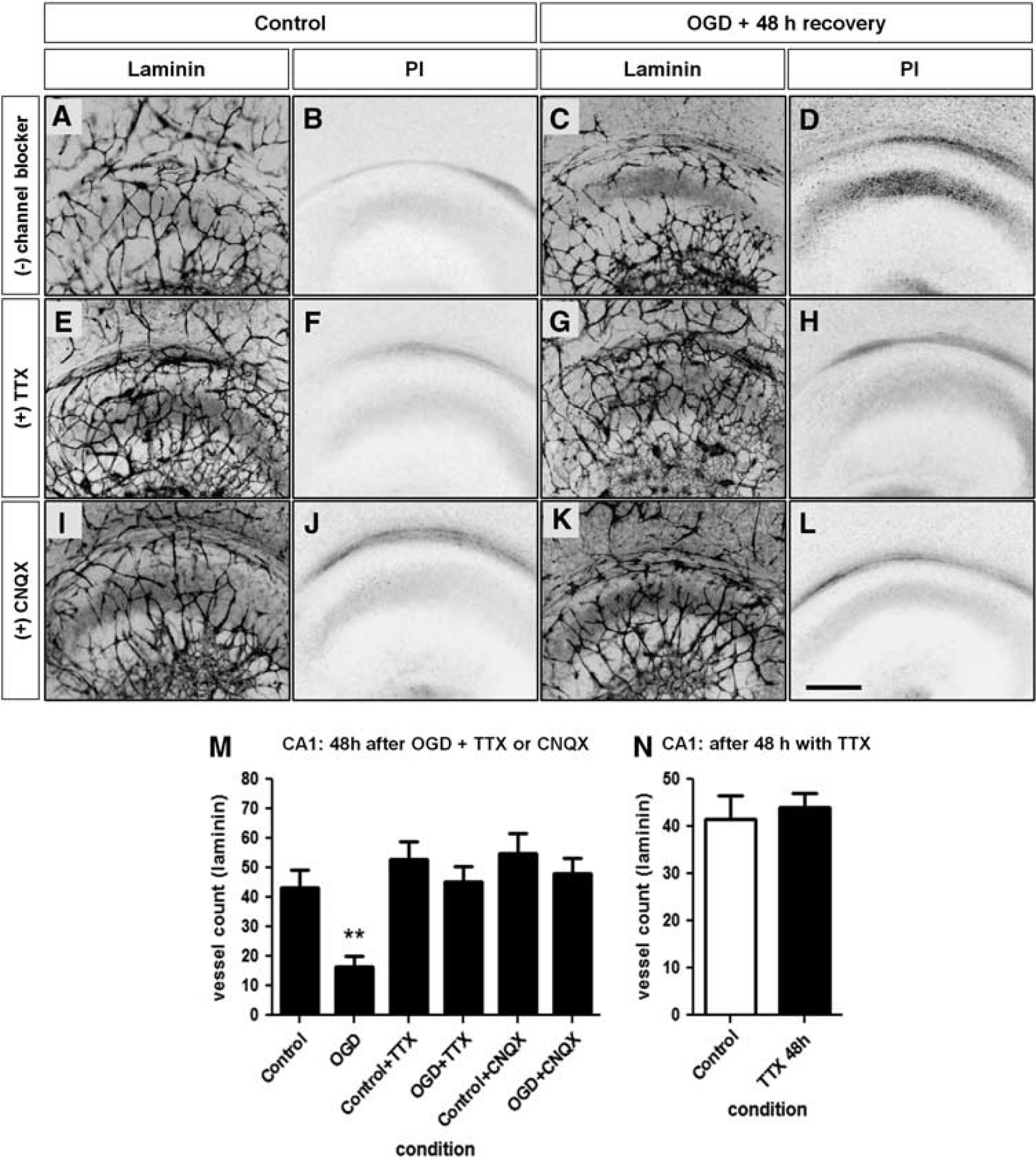
Neuronal cell death and vessel loss are rescued by applying either 1 μmol/L tetrodotoxin (TTX) or 100 μmol/L 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) during oxygen and glucose deprivation (OGD). (
CA1 Vessel Loss by Short-Term Exposure to AMPA
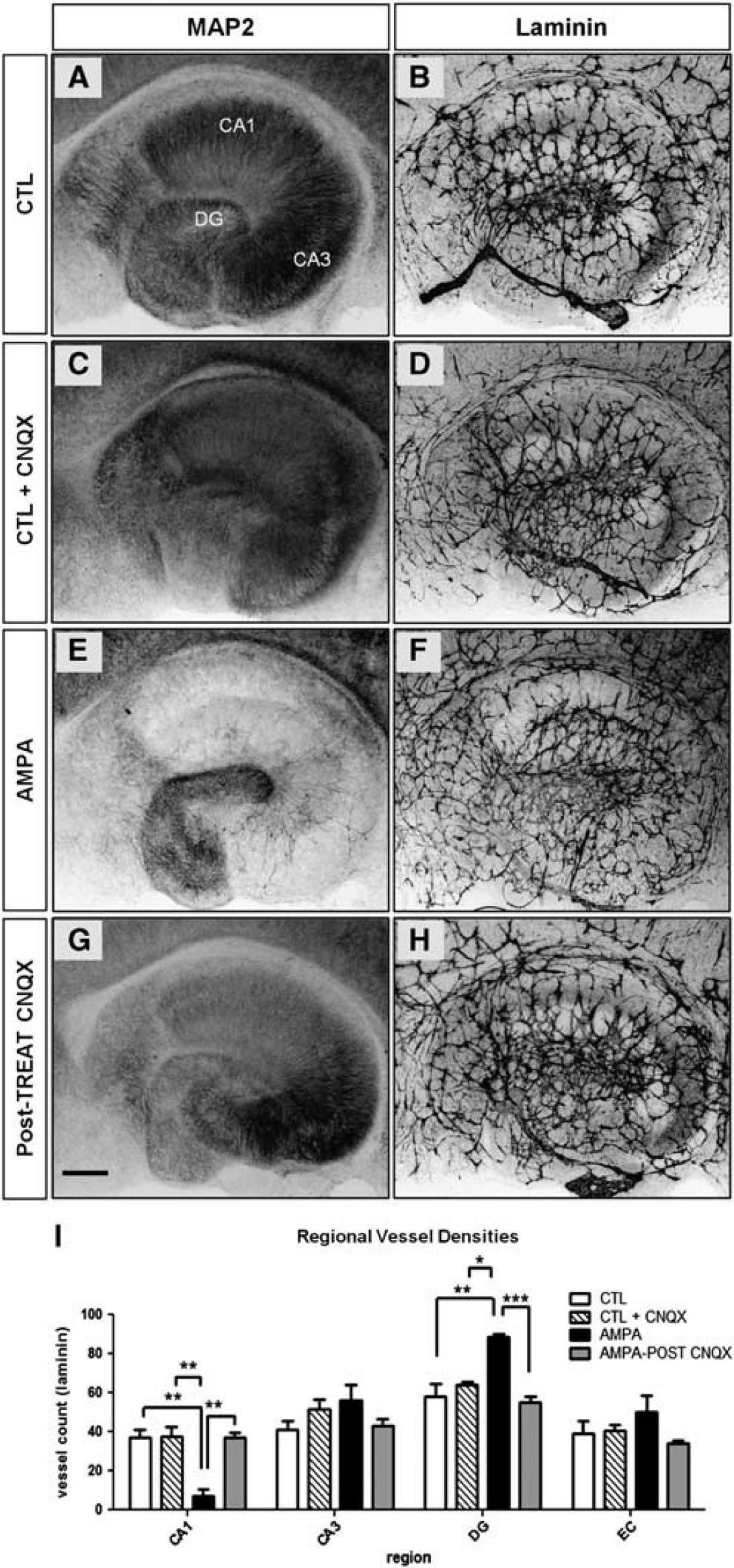
Since TTX and CNQX could rescue neurons and blood vessels from OGD-induced cell death and disintegration, we wondered whether AMPA receptor-triggered excitotoxicity in the absence of OGD might also induce blood vessel disintegration. Microtubule-associated protein 2 and laminin staining show a well-preserved appearance of neuronal cytoskeletal and vascular architecture, respectively, in a control slice culture (Figures 6A and 6B). A dose response for AMPA indicated strong cell death in the CA1 region to occur after a 30-minute exposure to 100 μmol/L (Figure 6G). In contrast to hypoxia or OGD, there was no delay in nerve cell death; it was already present after 3 hours (data not shown) to a massive degree. A 30-minute exposure to AMPA followed by a 48-hour recovery period had eliminated the majority of MAP2-positive neurons in the cornu ammonis (Figures 6A, Figures 6D, 7A and 7E). At the same time, there was also a clear reduction of vessel density in CA1, indicating that the AMPA-treatment induced neurodegeneration was sufficient to provoke vessel loss (Figures 6B, 6C, 6E, 6F, 7B, and 7F). Vessel counts confirmed that excitotoxic nerve cell death reduced the vessel density in the CA1 region (Figure 6H). Blocking the AMPA-induced excitotoxicity by 100 μmol/L of CNQX partially rescued neurons and preserved the vessels almost to control levels, even when applied as posttreatment (Figures 7C, Figures 7D, 7G and 7H, quantification in Figure 7I). Taken together, these experiments prove that vessel loss in the hippocampal CA1 field is due to excitotoxic neurodegeneration.
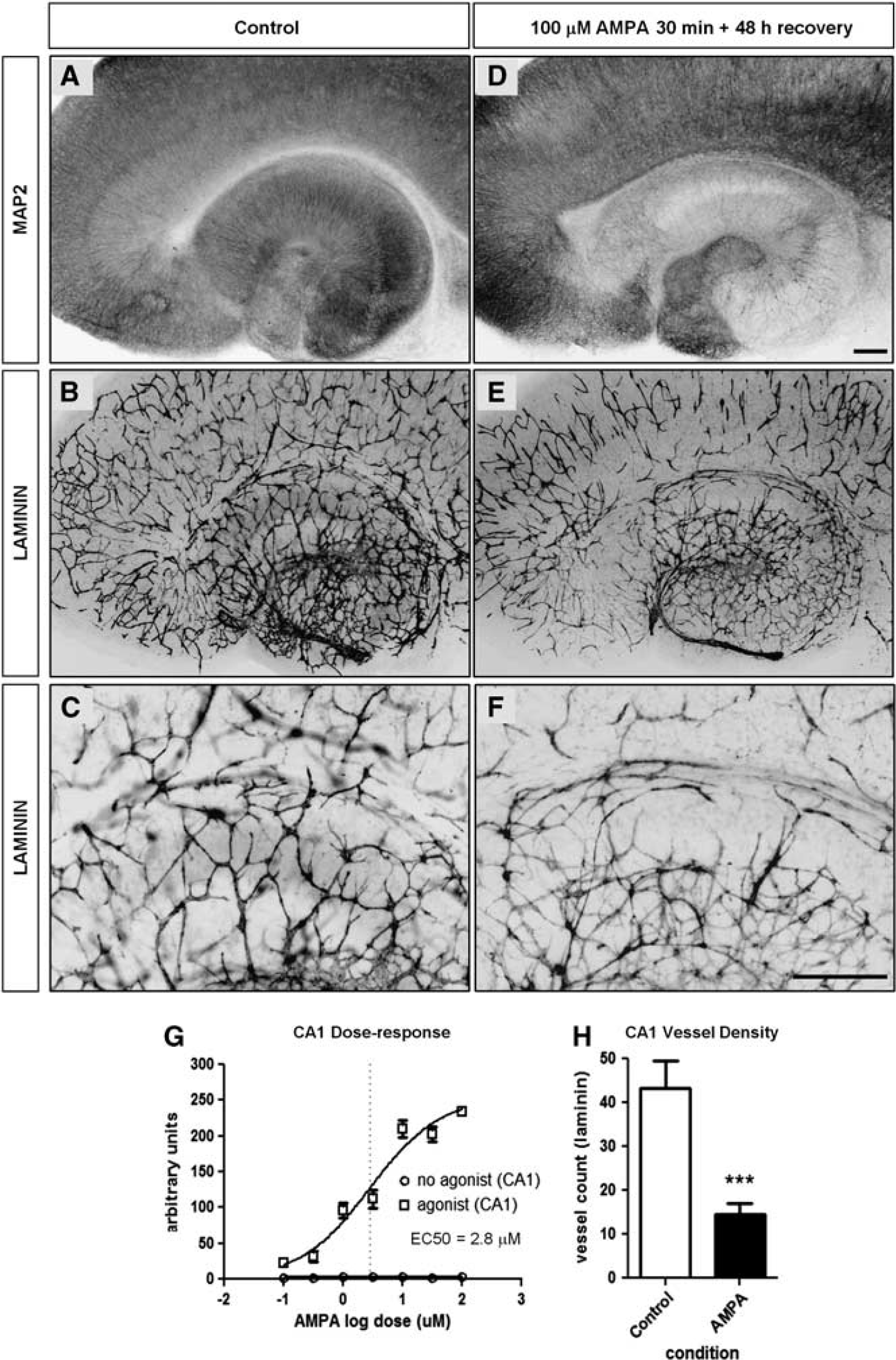
Entorhino-hippocampal-organotypic slice cultures (EHOSCs) treated with amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) for 30 minutes and allowed to recover for 48 hours. (
6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX) applied after exposure to amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) maintains the vascular architecture and blocks neuronal death in the hippocampus proper. (
Regional Variations in Blood Vessel Plasticity After AMPA-Induced Excitotoxicity
Neurodegeneration induced by AMPA is not confined to the CA1 region. Microtubule-associated protein 2 staining revealed loss of neurons in CA3 and survival of neurons in the dentate gyrus (Figures 7A and 7E). In contrast to CA1, blood vessels were not lost or were even enriched with more numerous branches in CA3 (Figures 7B and 7F, quantification in Figure 7I). In the DG, there was a consistent increase in blood vessel density, which could be blocked by posttreatment with CNQX (Figures 7F and 7H, quantification in Figure 7I). Thus, nerve cell death is not always accompanied by vessel loss but rather can induce a variety of changes in neurovasculature. The actual type of modification is probably determined by regional features together with the mode of induction of neurodegeneration.
DISCUSSION
We have adapted an in vitro neocortical BBB model20,21 to the hippocampal formation to investigate the effects of ischemia and recovery on neurovascular integrity in the most vulnerable brain region, the hippocampal CA1. Entorhino-hippocampalorganotypic slice cultures behave similar to cortical organotypic slice cultures with respect to the preservation of blood vessels in the control situation. Short-term hypoxia or OGD induced a selective cell death in the CA1 region, delayed by 24 to 48 hours. This is accompanied by a significant reduction in vessel density. The blocker of fast sodium spikes TTX or the AMPA receptor antagonist, CNQX, prevented both, neuronal death and vascular loss in the CA1 region. Induction of excitotoxicity by AMPA caused neuronal death in CA1 and CA3 regions. In contrast to a diminished vasculature in neuron-depleted CA1, in CA3 nerve cell loss did not reduce vessel density. Nerve cells in the EC and DG were resistant to excitotoxic cell death and in DG the vessel density even increased. These contrasting effects could be prevented by CNQX and TTX. We conclude that neuronal stress and neuronal death control maintenance, loss and remodeling of the neurovasculature and that the vascular response is in addition determined by region-specific factors.
Studying Hypoxia/Ischemia Using Entorhino-Hippocampal-Organotypic Slice Cultures
Hippocampal-organotypic slice cultures have been used to study neural development, neural function, neurodegeneration, as well as neuroprotection.18,19,22 Ischemia induced in in vitro models produce consistent cell death in specific regions within the hippocampus.18,19 In our study, we used the OGD medium proposed by Rytter et al., 19 which caused a delayed and selective cell death to CA1 without damaging CA3 after 15 minutes of OGD exposure. Septal and temporal regions have been known to function distinctively from each other. 23 Notably, the vulnerability to cell death appears to be more associated with the septal half, a phenomenon that we also observed (preliminary data). To avoid a possible interference from the position of the slice along the septotemporal axis, we used only hippocampal slices of the septal half because they displayed consistent cell death in addition to the reduced vessel density.
In models of hypoxia or ischemia, an increase in induction of VEGF and GLUT1 has been reported. 24 In fact, we were able to observe VEGFA and GLUT1 upregulation after hypoxia or OGD. Intriguingly, we found that unlike the increase in expression of the 55-kDa GLUT1 isoform that resides in endothelial cells, the expression of the 45-kDa GLUT1 isoform that resides in glial cells 25 was decreased after OGD. The functional relevance of the GLUT1 isoforms is largely unknown, but the glycosylated form facilitates glucose transport. 26 Nevertheless, the observed decrease in expression of the 45-kDa GLUT1 isoform suggests an impairment of glucose transport capacity in glial cells, adding to the potency of the OGD medium proposed by Rytter et al.: 19 it was reported to induce stronger cell death after glucose was reintroduced in the recovery period. Furthermore, the increase of the 55-kDa GLUT1 isoform indicates that despite the absence of blood flow, endothelial cells respond to OGD, rendering their functionality in EHOSCs. All in all, the results of this study suggest that our in vitro model of short-term OGD using EHOSCs is valid and representative of hypoxia or ischemia occurring in vivo.
Selective Cell Death and Vessel Loss in CA1
Neurodegeneration and BBB damage often cooccur after cerebral ischemia, but it is not clear whether the events are interconnected or separate entities. Neuronal death has been shown to be associated with BBB leakage in the context of transient ischemia,6,14,15 suggesting neuronal damage as the primary event of injury. However, this idea was challenged by the finding that intracerebral injection of kainic acid could induce BBB damage without neuronal death in knockout mice resistant to excitotoxic injury, supporting that BBB damage and neuronal death are separate events. 27 Thus, depending on the context of CNS injury, the question of whether neuronal death and BBB leakage are separate or linked events is debatable, which encouraged us to perform the present study.
In brain slices, techniques for measuring the permeability of the BBB do not exist. Indirect measures are the detection of tight junction proteins. In addition, investigating the expression of components of the cellular matrix of the neurovascular unit is reasonable, since functioning of the BBB also depends on the survival of the endothelium, basal lamina, astrocytes, and pericytes.2,28,29 In the present study, we have provided evidence of a link between neuronal death and impairment of BBB: along with the glial impairment in glucose transport and the upregulation of GLUT1 in the endothelium, the main tight junction protein of the BBB, Cld-5, is markedly reduced in CA1 24 hours after OGD, that is, at the onset of neurodegeneration, with a further reduction at 48 hours. Yet, not only tight junction proteins are reduced with neurodegeneration, the whole neurovascular unit is impaired as evidenced by the loss of the extracellular matrix protein laminin. Previous studies on rodents submitted to cerebrovascular ischemia have also seen capillary damage in the CA1 region. 14 Earlier studies in vessel density in control animals have indicated fewer vessels in CA1 than CA314,30,31 and more recently a higher vascular density in the molecular layer of the dentate gyrus than in the rest of the hippocampus was demonstrated. 32 Differences in vessel density have also been observed across the septotemporal axis, 33 suggesting that the organization of the vasculature in the hippocampus is dependent on regional factors. The regional variation in vessel density has been considered to be caused by differences in glucose utilization. 30 Also in so-called neurovascular niches, such as in the DG, angiogenesis is more frequent as it relies on the same trophic factors that are involved in neurogenesis. 32
In organotypic cultures, without a need for blood flow as oxygen and glucose are equally distributed to all areas, regional differences in vessel density were nevertheless still apparent. The cellular organization in the single brain region might determine vessel density. Since pyramidal cells in CA1 and CA3 are different in terms of calcium load, mitochondrial function, glutamate receptor expression, and proteosomal degradation, it might be possible that factors produced by or associated within CA1 influence vessel density. Because in our in vitro model of global ischemia, vessel loss was specifically observed in CA1, we propose that the loss of vessels arises as a consequence of CA1 pyramidal cell death.
Blocking Neuronal Death and Vascular Loss in CA1
Neuronal death after hypoxia or ischemia is mainly contributed to glutamate excitotoxicity. 34 The glutamate release has been linked to sodium and calcium influx as a consequence of ionic imbalance and neuronal depolarization. Blocking the activation of either sodium channels by TTX or ionic glutamate receptors with NMDA or AMPA antagonists before OGD improves neuronal resistance. 35 In our OGD model, TTX as well as the AMPA receptor antagonist CNQX prevented neuronal death proving that it is due to glutamate excitotoxicity. This neuroprotective effect of TTX or CNQX was associated with a preservation of blood vessels. Moreover, in control experiments with TTX only, the vessel density was similar to controls. Since TTX can inhibit neuronal activity without affecting cell viability, the stability in vessel expression in the presence of TTX implied that it is not the absence of neuronal activity but that indeed neuronal death is responsible for vessel loss.
We used an AMPA antagonist, CNQX, rather than a NMDA antagonist in our study, as it has been shown to be more effective in preventing delayed neuronal death. 36 Moreover, we wanted to use an antagonist that was specifically affecting neurons. Evidence for the presence and function of glutamate receptors in cerebral endothelial cells is controversial. Cerebral endothelial cells derived from rat or human brain have been contended as having no expression or function of glutamate receptors, 37 but subsequent studies showed glutamate receptors in cerebral blood vessels and even suggested NMDA receptors to be involved in BBB regulation. 38 On the other hand, AMPA receptors are only expressed at very low levels in cerebral endothelial cells and the application of DNQX, an AMPA receptor antagonist, but not MK-801, an NMDA receptor antagonist, failed to counteract BBB permeability induced by glutamate excitotoxicity, suggesting that AMPA receptors may have little importance in BBB regulation. 38 In light of these published findings, we presumed that CNQX had little direct effect on vessels, if at all.
Glutamate Excitotoxicity Leads to a Selective Modulation of Vessel Architecture in the Hippocampal Formation
Since AMPA receptors seemed to be involved in OGD-induced neurodegeneration, we challenged this finding by directly exposing EHOSCs to a high dose of (RS)-AMPA agonist for a short duration followed by 48 hours of recovery to mimic the glutamate excitotoxicity induced by OGD. A widespread neuronal death together with vessel loss in the CA1 region was observed. Both neurodegeneration and vessel loss could be reversed when the agonist was blocked by CNQX providing additional evidence that excitotoxic neuronal death in the CA1 region is accompanied by a concomitant loss of blood vessels. However, the other subfields of the hippocampus did not react in the same way. In the CA3 region, excitotoxic neuronal death did not induce major changes of blood vessel density or architecture. In the DG with neuronal survival, there was an increase of blood vessel density and in the EC neurons survived and there were no evident changes in the blood vessels. Interestingly, in four different areas we thus have observed four different types of neuronal and vascular responses to excitotoxic challenge highlighting the importance of local factors for determining the final cellular responses.
In in vivo and in vitro models of epilepsy, an increase of vessel density has been demonstrated.10,16,17,32 It has been linked to seizure-induced mossy fiber sprouting and neurogenesis, which is prominent in the neurovascular niche of the DG. 39 An upregulation of VEGF expression by neurons and astrocytes as well as its receptor, VEGFR2, in endothelial cells was present in in vitro kainate seizure-induced vessel remodeling. 17 In our model, we also did observe VEGF expression to be upregulated. Further work is necessary to determine whether the selective AMPA-induced excitation mimics on the one hand in DG seizures resulting in angiogenesis, and on the other hand in CA1 ischemia-induced glutamate toxicity resulting in massive neurodegeneration and accompanying vessel loss.
Possible Mechanisms of Vascular Damage Induced by Neuronal Death
A primary mechanism underlying ischemia-induced neuronal death of pyramidal cells is excitotoxicity, which we observed 48 hours after injury by OGD and by excessive stimulation of AMPA-type glutamate receptors. However, the neuronal death was accompanied by microvessel loss confined to the CA1 region. Ischemia-induced neuronal death has been characterized by a mixture of apoptosis and necrosis. Neuronal death includes a large number of changes such as the loss of ATP, production of free radicals, loss of mitochondrial membrane potential, release of cytochrome c, and production of reactive oxygen species. 40 The mechanisms by which death of pyramidal neurons mediate vessel loss, are not known. The present results lay the groundwork for further study of the biological crosstalk between pyramidal neurons and the vasculature, in particular, the role for AMPARs in ischemia-induced neuronal death. Understanding the regional differences of the hippocampus and especially the interaction between pyramidal cells and their surrounding vasculature might be an important aspect for developing better treatments for neurovascular disorders in particular cerebral ischemia after stroke or short-term cardiac arrest.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.