
Abstract
The present antithrombotic drugs used to treat or prevent ischemic stroke have significant limitations: either they show only moderate efficacy (platelet inhibitors), or they significantly increase the risk for hemorrhages (thrombolytics, anticoagulants). Although most strokes are caused by thrombotic or embolic vessel occlusions, the pathophysiological role of platelets and coagulation is largely unclear. The introduction of novel transgenic mouse models and specific coagulation inhibitors facilitated a detailed analysis of molecular pathways mediating thrombus formation in models of acute ischemic stroke. Prevention of early platelet adhesion to the damaged vessel wall by blocking platelet surface receptors glycoprotein Ib alpha (GPIb
Introduction
Recombinant tissue plasminogen activator (tPA) is the only approved medication in acute ischemic stroke. However, <20% of all stroke patients receive tPA (Weimar et al, 2006), a situation mainly owed to the narrow treatment time window of 4.5 hours. Another significant limitation of tPA is bleeding-related adverse events. Moreover, as time increases between symptom onset and treatment, the efficacy of tPA decreases (Lees et al, 2010). In the case of stroke prevention, the situation is not different. Platelet inhibitors (PIs) and anticoagulants are commonly used in (early) secondary prophylaxis, but often with limited efficacy (CAST, 1997; IST, 1997).
It is known for many years that during ischemic stroke platelets become locally activated and adhere to the cerebral endothelium, thereby promoting thrombus formation and subsequent thrombus growth (del Zoppo, 1998; Okada et al, 1994). Consequently, in some of the early experimental studies, the prototype of PIs, acetylsalicylic acid (ASA), was used in an attempt to salvage brain tissue by counteracting detrimental microvascular thrombosis. In rats, ASA was effective only after temporary, but not permanent, occlusion of the MCA (Berger et al, 2007). In mice, multiple applications of very high doses (6 × 40 mg/kg, which corresponds to roughly 19 g for humans) were needed to significantly reduce infarct volumes (Berger et al, 2004). Even then, no amelioration of functional deficits could be observed, whereas lower doses of ASA remained inefficient. The limited success of ASA in experimental stroke is in agreement with modest results in clinical trials for early (within the first 48 hours) secondary prophylaxis after ischemic stroke (CAST, 1997; IST, 1997). For example, one has to treat 100 stroke patients with ASA over 1 year to prevent one further stroke. Other platelet antagonists were also studied in experimental stroke. In baboons, Ticlopidin, an adenosine diphosphate receptor antagonist, which functions similarly to Clopidogrel by inhibiting the P2Y12 receptor on platelets, reduced the formation of microthrombi in the basal ganglia (del Zoppo, 1998). However, to achieve this effect, combination of Ticlopidin together with heparin was necessary, which complicates the interpretation of the results, that is, the differentiation of the pathways involved (platelet activation or thrombin activity). An important issue with the use of PIs in stroke is the risk of increased bleeding, reducing the benefit-hazard ratio (Hankey and Eikelboom, 2010).
The classic approach to inhibit coagulation-driven thrombus formation in models of acute ischemic stroke was to apply unfractionated or low-molecular-weight heparins (Libersan et al, 1998; Mary et al, 2001; Pratt et al, 1998; Yanaka et al, 1996a, 1996b). Although these enhancers of antithrombin III activity to some extent reduced infarct volumes and neurological deficits, the main drawback of unfractionated heparins was an increased risk of bleeding. For instance, severe intracranial hemorrhages occurred in up to 25% of animals treated with heparin (Mary et al, 2001; Yanaka et al, 1996b). While some studies found a positive benefit-hazard ratio for low-molecular heparins such as enoxaparin (Mary et al, 2001), various studies in the clinical setting showed that the net benefit of heparins in general was outweighed by an increased risk of severe bleedings (Sandercock et al, 2008). Consequently, full-intensity parenteral anticoagulation with heparins either unfractionated or low-molecular weight is no longer recommended in the acute phase of cerebral ischemia (guidelines of the European Stroke Organisation (ESO): http://www.eso-stroke.org/pdf/ESO%20Guidelines_update_Jan_2009.pdf).
While new PIs (thromoboxane receptor antagonists, phosphodiesterase inhibitors) and oral anticoagulants (direct thrombin inhibitors, factor Xa inhibitors) have recently been tested in humans or have even found their way into the clinic, this efficacy versus safety (bleeding risk) dilemma remains a burning issue (Bousser et al, 2011; Shinohara et al, 2010; Connolly et al, 2009). Hence, there is still a large unmet medical need for more effective and safer antithrombotics in stroke therapy. Platelets and coagulation factors increasingly gain attention as prime targets in stroke therapy. Yet, much remains unknown about the molecular pathways that initiate and perpetuate cerebral thrombus formation (Stoll et al, 2008; Kraft et al, 2011; Pham et al, 2012). This review summarizes novel pathophysiological insights into the role of platelets and coagulation in mouse models of stroke. Interestingly, while elucidating critical pathways of hemostasis and thrombosis in the brain these basic studies have led to the intriguing concept of ‘bleeding-free antithrombosis’ in stroke, which now awaits validation in future clinical trials.
Translational hurdles in the implementation of novel anithrombotics: it's nice, but it's (still) mice
In the search for new targets for improved antithrombotic treatment in ischemic stroke, it is important to recognize several translational caveats (reviewed in Braeuninger and Kleinschnitz (2009)). Indeed, efficient translation of novel experimental therapies into effective treatments for stoke patients has so far not been successful. One important consideration is that rodents are different from humans, with significant physiological, neuro-anatomical, and metabolic differences (Ishihara et al, 1998; Rowley et al, 2011). Although rodents classically are the first line of animals for experimental stroke studies, their use may bias results towards rodent physiology, hindering successful translation to humans. Also, when using genetically modified animals, for example, knockout or transgenic mice that have no counterpart in normal humans, one must take into account compensatory upregulation of alternative mechanisms that could alter cerebral vascular supply or tissue responses (Barone et al, 1993). Moreover, cerebral vasculature, hemostasis, and thrombosis may have specific characteristics that differ from established systemic concepts. For example, a particular phenomenon in ischemic stroke is infarct progression despite sustained early reperfusion of previously occluded blood vessels, a process known as ‘reperfusion injury’ (Nieswandt et al, 2011). On reperfusion of previously ischemic tissue, adherent platelets and leukocytes can obstruct microvessels and hinder efficient reperfusion, referred to as ‘no reflow’ (del Zoppo and Mabuchi, 2003). Collectively, it is clear that one has to be cautious when extrapolating promising findings in mouse models of stroke to human therapies. Preclinical follow-up studies in larger animal models, preferably gyrencephalic non-human primates, are therefore recommended as translational proof of concept before starting clinical trials (Cook et al, 2012).
The coagulation cascade as novel target in stroke therapy
The coagulation cascade consists of several serially connected serine proteases. Activation of the cascade leads to thrombin generation and ultimately to fibrin formation, which stabilizes growing platelet thrombi. Whereas it is well established that the extrinsic coagulation pathway is triggered by tissue factor at sites of vascular damage, the physiological trigger for the intrinsic coagulation cascade remained elusive until recently. Indeed, recent elegant work showed that negatively charged ribonucleic acid or polyphosphates are potent activators of factor XII (FXII, Hageman factor)
Blocking specific components of the coagulation cascade, such as FIXa and FXa, resulted in hopeful findings. In a rat model of thromboembolic stroke, inhibition of FXa was shown to reduce infarct size and improve neurological outcome without increasing risk of bleeding (Wang et al, 2003). Similar findings were obtained when blocking FIX (Toomey et al, 2002). Another study in mice also showed that low doses of FIX inhibitor were protective in ischemic stroke without inducing intracranial hemorrhage, while higher doses resulted in more cerebral bleeding, similar to heparin and tPA (Choudhri et al, 1999). Larger studies are needed to further validate these central components of the coagulation cascade as safe and efficient targets in stroke therapy.
In recent years, also another factor, FXIIa, has gained increasing interest as an attractive candidate to prevent thrombus formation in the ischemic brain. Factor XII has long been thought to be physiologically irrelevant for thrombus formation as hereditary FXII deficiency is not linked with hemorrhagic diathesis, not even after trauma (Mackman, 2004). However, this paradigm had to be fundamentally revised in the last few years. The generation and characterization of FXII-deficient mice revealed that these animals built fewer and less stable thrombi after artificial vessel wall injury, but not at the cost of an increased rate of hemorrhages (Renné et al, 2005). FXII-deficient mice have normal tail bleeding times, despite their increased activated partial thromboplastin time. Lack of FXIIa has also interesting consequences in experimental stroke:
Findings obtained from transgenic mouse models are interesting from a mechanistic perspective, but do not represent the clinical situation. If one wants to treat a patient, a pharmacological inhibitor is needed. A very selective inhibitor of FXIIa is recombinant protein rHA-Infestin-4 (CSL Behring GmbH, Marburg, Germany), a Kazal-type serine protease inhibitor originally isolated from the gut of the sanguivorous kissing bug
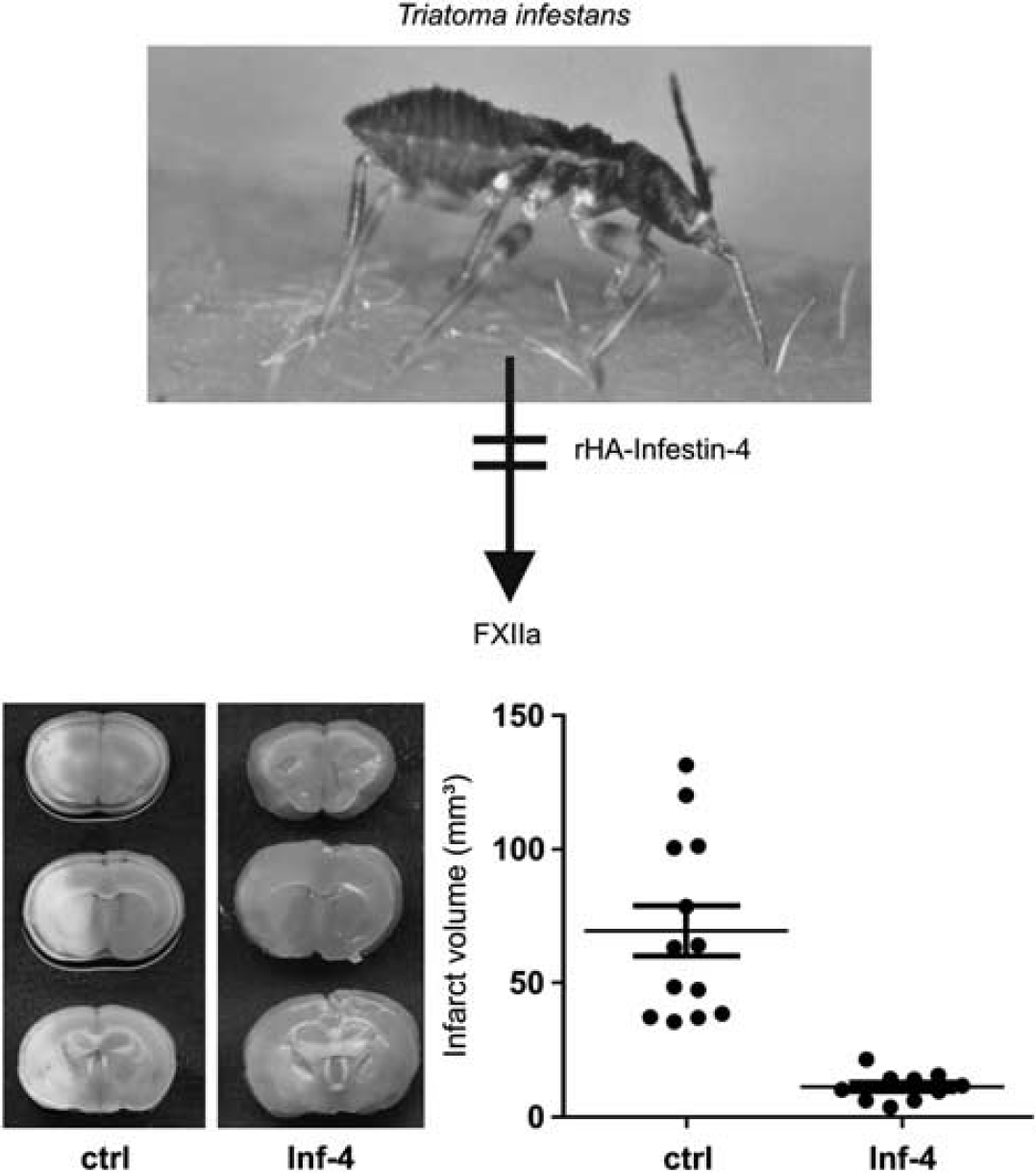
rHA-Infestin-4 protects from ischemic stroke in mice. rHA-Infestin-4 is a Kazal-type serine protease inhibitor originally isolated from the gut of the sanguivorous kissing bug
Despite promising preclinical data, the relevance of FXII in the pathophysiology of human stroke cannot be judged so far. Interestingly, the index patient John Hageman died from pulmonary embolism (albeit after a preceding accident) and since then FXII deficiency was for a long time assumed to act prothrombotic (Gailani and Renné, 2007). Overall, evidence from epidemiological or clinical studies is conflicting and difficult to interpret: FXIIa levels above the 90th percentile were associated with a 2.1-fold increased probability of stroke in younger women taking oral contraceptives (Siegerink et al, 2010), and 21 patients with severe FXII deficiency developed no arterio-embolic events within a 15-year period (Girolami et al, 2005). In contrast, a case–control study showed that low FXIIa levels in middle-aged men were associated with an increased risk for coronary artery disease and stroke (Govers-Riemslag et al, 2007). Finally, deficiency of FXI, the prime substrate of FXIIa, protected from stroke but not myocardial infarction in a Jewish population (Salomon et al, 2008). Available evidence points towards a dose-dependent, maybe even a U-shaped correlation between FXIIa levels and the likelihood of thrombosis. However, prospective epidemiological studies that are adequately powered are difficult to realize, as people with FXII deficiency are ‘phenotypically healthy’ except for a prolonged activated partial thromboplastin time.
The quest for novel inhibitors of platelet function
The increasing availability of transgenic mouse strains and specific blocking agents paved the way for identification of novel molecules that control platelet function and activation (Stegner and Nieswandt, 2010; Stoll et al, 2008). Use of these tools in experimental stroke settings has much improved our understanding of detrimental thrombus formation in the ischemic brain. Classically, arterial thrombus formation is considered a multistep process, involving platelet rolling over vascular lesions, followed by firm adhesion, platelet activation, and eventually stable platelet aggregate formation (Figure 2). Under conditions of high shear stress (>800/s, found in normal arteriolar circulation or stenosed arteries), platelets need to be slowed down to allow firm arrest at sites of vascular injury. This is accomplished by the reversible interaction of the platelet-specific glycoprotein (GP) Ib-V–IX receptor complex with immobilized von Willebrand factor (VWF) present in the subendothelium, or immobilized from plasma onto exposed collagen, or present on endothelium as VWF strings after release by activated endothelial cells (De Meyer et al, 2012a), resulting in the so-called ‘tethering’ or ‘rolling’ of platelets in the direction of the blood flow. This permits platelets to establish more stable interactions that lead to definitive arrest, such as binding to collagen with their collagen receptors. Among the two main collagen receptors on platelets, GPVI and GPIa/IIa, the former is considered the most functionally relevant, eliciting strong activation signals on engagement (Nieswandt and Watson, 2003; Broos et al, 2011). Platelet activation finally results in a conformational change of the GPIIb/IIIa receptor. Activated GPIIb/IIIa then promotes platelet aggregation by binding fibrinogen (and to a lesser extent VWF), which will act as a substrate for recruitment of additional platelets.
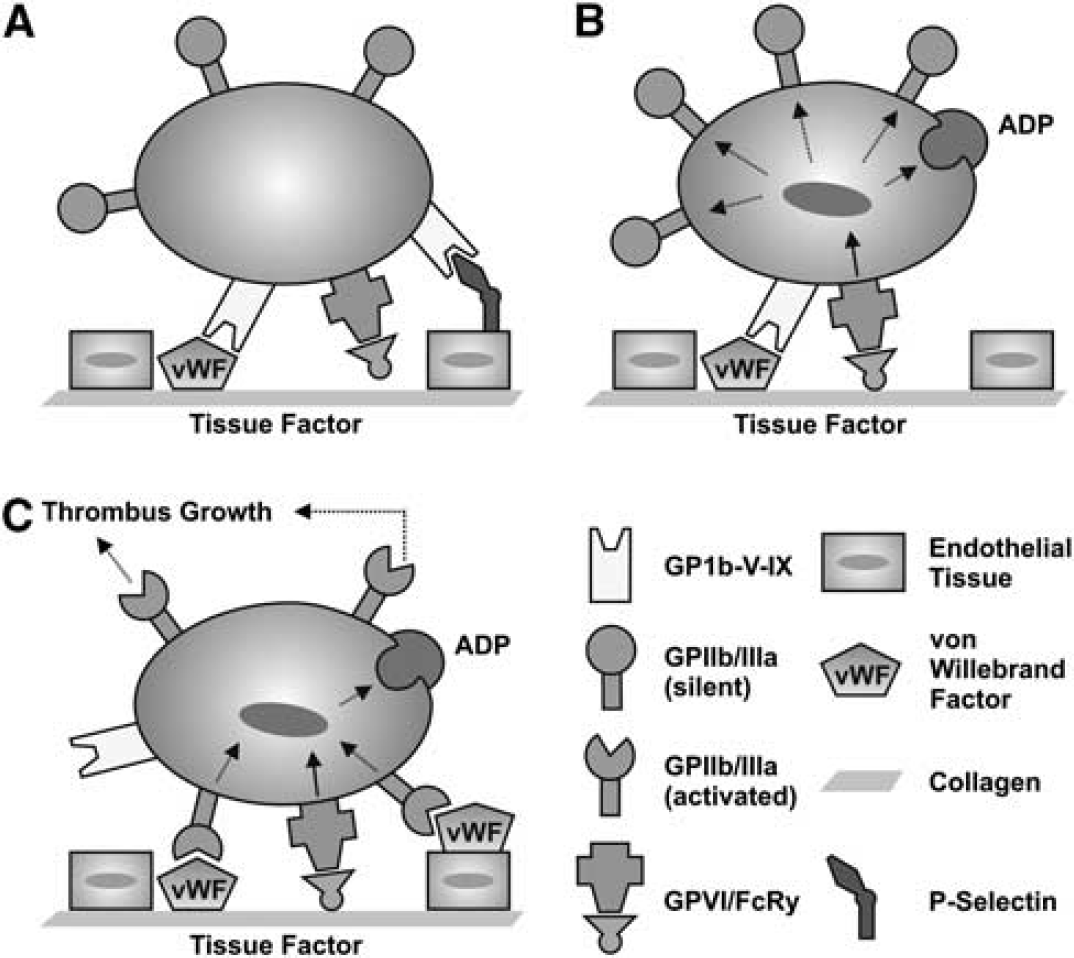
Distinct steps of platelet adhesion, activation, and aggregation at the activated endothelium. (
In the last few years, extensive effort has been made to investigate whether this classical view of arterial thrombus formation is also valid in (ischemic) brain vasculature. These studies have revealed interesting insights that could lead to new treatment strategies in stroke management.
Blocking the GPIb–VWF axis
Because of its absolute importance for platelet adhesion under high shear conditions, the GPIb–VWF axis has become an attractive target for the development of new antithrombotic agents. During the last decade, the antithrombotic efficacy of GPIb–VWF inhibition has been well established using various experimental arterial thrombosis models (reviewed in De Meyer et al (2012a)). However, the role of GPIb–VWF during cerebral ischemia remained elusive until recent studies showed a clear involvement of the GPIb–VWF axis in stroke development. First, we showed in mice that inhibition of early platelet adhesion by anti-GPIb
As described above, binding of GPIb to VWF is essential for platelet adhesion. von Willebrand factor is a multimeric GP produced in megakaryocytes and endothelial cells only. Lack of functional VWF results in the common bleeding disorder von Willebrand disease (De Meyer et al, 2009). The crucial role of VWF in cerebral ischemia/reperfusion injury was revealed by two independent studies showing that VWF-deficient mice develop significantly smaller strokes compared with control mice (Kleinschnitz et al, 2009; Zhao et al, 2009). This protection was reversed when VWF-deficient mice were reconstituted with VWF via hydrodynamic gene transfer (Kleinschnitz et al, 2009; De Meyer et al, 2010). To gain more insight into the specific roles of the different VWF domains, we transferred different VWF mutants into VWF-deficient mice. These experiments showed that binding of VWF to collagen and GPIb
Recently, population-based studies have shown that enhanced VWF serum levels are an independent risk factor for ischemic stroke (Bongers et al, 2006; Wieberdink et al, 2010). Moreover, different VWF polymorphisms associated with an increased risk of stroke have been identified (Dai et al, 2001; van Schie et al, 2011). Not all of these are associated with higher VWF levels, suggesting that other mechanisms, such as increased VWF activity, could contribute to the risk of stroke as well.
Activity of VWF is regulated by an enzyme called ‘a disintegrin-like and metalloprotease with thrombospondin repeats 13’ (ADAMTS13). Activity of VWF is strongly correlated with multimer size, and ultralarge VWF multimers can even spontaneously form microthrombi. To prevent spontaneous thrombus formation, ultralarge VWF is cleaved into smaller, less reactive molecules by ADAMTS13. Lack of ADAMTS13 activity results in thrombotic thrombocytopenic purpura (Moschcowitz syndrome), characterized by microthrombi that occlude the vascular beds of many organs, including the brain (Lindblom et al, 2009; Sevy et al, 2011). Interestingly, ADAMTS13-deficient mice develop significantly larger strokes than wild-type mice (Fujioka et al, 2010; Zhao et al, 2009), further confirming the notion of VWF as a central player in thrombus formation in cerebral vessels after stroke. Conversely, treatment of wild-type mice with ADAMTS13 led to smaller infarcts, without sensitizing to brain hemorrhage (Zhao et al, 2009). This indicates that, next to inhibiting the GPIb–VWF interaction, also cleavage of VWF itself may become a potential new strategy in stroke treatment.
Despite the promising results in experimental stroke, the translational impact of GPIb–VWF inhibition (or ADAMTS13 treatment) is still limited. Broader time windows after the onset of stroke and other stroke models than the mouse tMCAO model have to be tested to fully proof the translational character of these findings. Nevertheless, a relevance of platelet GPIb for human stroke might indeed exist. Several
GPVI blockade
Platelet GPVI is crucial for stable platelet adhesion on exposed collagen because it induces strong signaling for platelet activation (Figure 2). Glycoprotein VI is a type 1 transmembrane receptor from the immunoglobulin superfamily and is expressed exclusively on platelets (Nieswandt and Watson, 2003). Blockade of GPVI with specific antibodies very effectively prevented thrombus formation in different
Platelet signaling
The promising results regarding GPIb and GPVI inhibition entailed follow-up studies on pathways downstream of these receptors. One fundamental step in platelet activation is the rapid rise of intracellular calcium concentration (Feske, 2007; Grosse et al, 2007). Following binding of VWF, collagen, or adenosine diphosphate to their respective receptors on the platelet surface (see above), phospholipase C hydrolyzes phosphatidylinositol-4,5-bisphosphate to form inositol-1,4,5-trisphosphate and diacyl-glycerol. Inositol-1,4,5-trisphosphate then triggers the release of Ca2+ from the endoplasmic reticulum. The intracellular Ca2+ sensor stromal interaction molecule 1 (Stim 1) is an important activator of calcium release-activated calcium channel protein 1, a calcium selective ion channel in the plasma membrane encoded by the
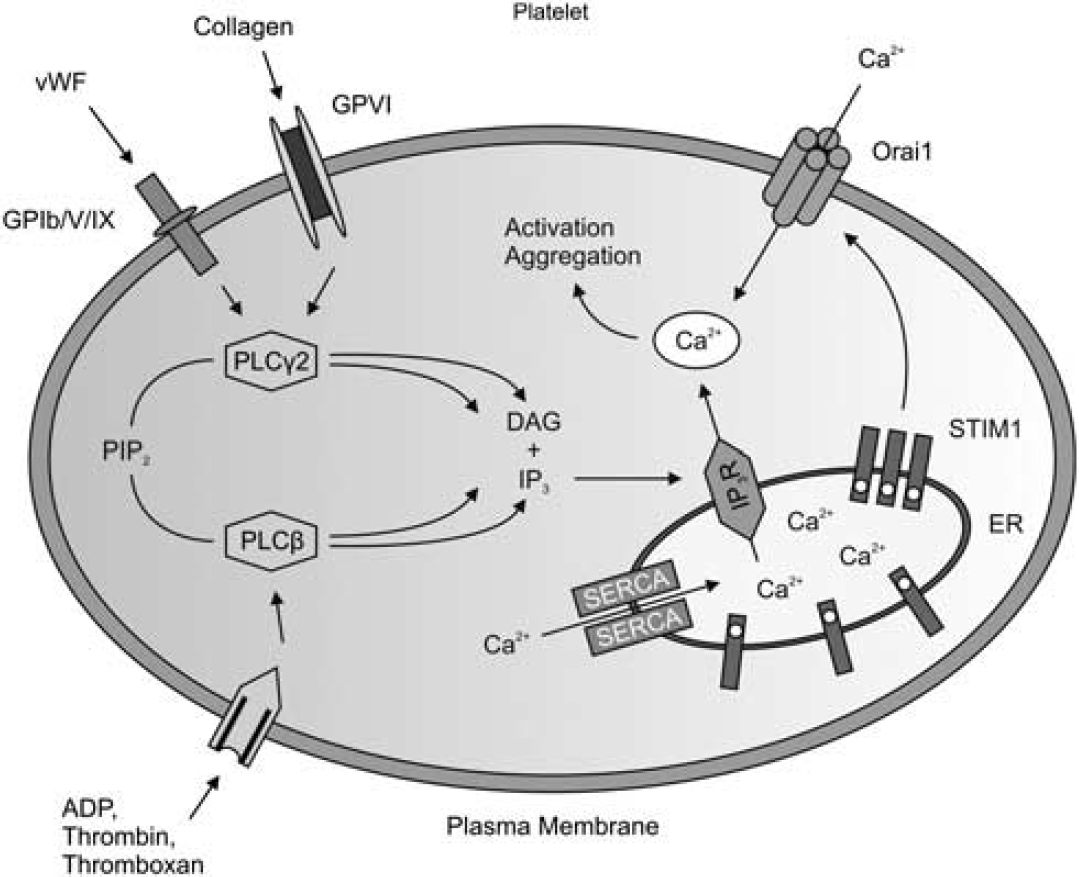
Simplified diagram of Ca2+ homeostasis in platelets. On activation of several receptors by ligands such as adenosine diphosphate (ADP), von Willebrand factor (VWF) or collagen, different phospholipase (PL)C isoforms hydrolyze phosphatidilinositol-4,5-bisphosphate (PIP2) to inositol-1,4,5-trisphosphate (IP3) and diacyl-glycerol (DAG). Inositol-1,4,5-trisphosphate leads to a Ca2+ release from intracellular stores (endoplasmic reticulum, ER). On Ca2+ release STIM1 activates the Ca2+ channel Orai1 in the plasma membrane that mediates Ca2+ influx from the extracellular space. Sarcoplasmic/endoplasmic reticulum Ca2+ ATPases (SERCAs) are involved in the counteracting mechanisms (adapted from Varga-Szabo et al, 2009). IP3R, IP3-receptor; STIM1, stromal interaction molecule 1.
Another interesting molecule in platelets is phospholipase D1, which acts downstream of GPIb
GPIIb/IIIa blockade
The GPIIb/IIIa pathway is crucial for mediating irreversible platelet aggregation and subsequent thrombus growth (Figure 2). Consequently, pharmacological inhibition of this final pathway of platelet activation has been a favored antithrombotic strategy (Armstrong and Peter, 2012). Surprisingly however, application of anti-GPIIb/IIIa Fab did not alter stroke size or functional outcome in the murine tMCAO model (Kleinschnitz et al, 2007). Instead, the therapy resulted in a massive rise in intracerebral hemorrhages and mortality. Similarly, reconstituting
Other studies using different GPIIb/IIIa antagonists reported an improved functional outcome after experimental stroke, but these were also accompanied by a dose-dependent increase in intracranial bleeding (Choudhri et al, 1999). Similarly, bleeding problems were also observed during the AbEST-II phase III clinical trial, studying the efficacy of the GPIIb/IIIa antagonist Abciximab in patients with acute ischemic stroke (Adams et al, 2008). The study had to be stopped prematurely due to a significant increase in bleeding complications and a lack of efficacy in the treated group. In the Safety of Tirofiban in Acute Ischemic Stroke (SaTIS) trial, tirofiban reduced overall 6-month mortality, but failed to improve neurological outcome and disability (Siebler et al, 2011). Another trial compared the efficacy of tirofiban and ASA in a 6 hours time window after stroke, but did not find superiority of tirofiban over ASA in improving short-term neurological course and reducing long-term disability (Torgano et al, 2010). Thus, GPIIb/IIIa inhibitors seem to have a low-therapeutic range, at least in ischemic stroke. If GPIIb/IIIa antagonists fall below the lower therapeutic margin, activation of platelets can result; if they go above the upper therapeutic margin, the risk for bleeding complications rises (Bhatt and Topol, 2003). A better understanding of the molecular pathways of GPIIb/IIIa during stroke and of the pharmacology of GPIIb/IIIa antagonists will be necessary for substantial improvement in this regard.
Antithrombotic therapy without bleeding: a realistic option in stroke therapy?
An important characteristic of an ideal antithrombotic drug is that it should not increase the risk of bleeding. This is particularly important in the brain where a small intracranial hemorrhage already can have devastating consequences. As described above, several new experimental strategies seem promising in this regard, including inhibition of factors IX, X and XII, GPIb–VWF, and GPVI. Interestingly, in several models of arterial thrombosis, inhibition of the collagen–VWF–GPIb axis showed a broader therapeutic window free of bleeding events when compared with GPIIb/IIIa blockers (reviewed in De Meyer et al (2006)). Whether this holds true for stroke remains to be established. Direct comparisons of novel experimental stroke therapies are still lacking, but will be necessary to find the most promising candidates. In this respect, it will be interesting to see whether targeting molecules of which deficiency is not associated with a severe human bleeding diathesis (e.g., FXII, GPVI) will be safer in terms of hemorrhagic risk than inhibiting factors that are essential for normal hemostasis (e.g., FIX, VWF, GPIb). For example, inhibition of GPVI seemed to be safer and more effective than inhibition of GPIIb/IIIa (Kleinschnitz et al, 2006). These comparisons in various models of stroke will also help to segregate the various strategies in relation to prevention or treatment of stroke (primary prevention, thrombolysis, secondary prevention, and so on).
Nevertheless, it should be stressed that absence of bleeding in experimental models of stroke will not necessarily mean bleeding-free protocols in humans. Similarly, there is little evidence that experimental bleeding times in animals closely reflect hemorrhagic events in humans. Thus, while initial experimental stroke studies may indicate a beneficial effect on bleeding risk, close computed tomography or MRI monitoring of potential hemorrhagic transformation in further translational studies are required to show reduced bleeding risks in patients. Finally, absence of intracranial hemorrhage does not exclude that hemostasis could be impaired in other vascular beds, as it has been suggested that the hemostatic system in the central nervous system is different from that in other organ systems (Rosenberg and Aird, 1999).
Summary and perspective
During recent years, an increasing amount of experimental stroke studies has greatly improved our understanding of various coagulation and/or thrombotic pathways that mediate stroke progression. Although we should be aware of translational pitfalls, use of these animal models has revealed several novel targets that potentially could improve stroke therapy and/or prevention. Such innovations are badly needed considering that a plethora of attempts to implement novel thrombolytics or platelet inhibitors have spectacularly failed in the past. In terms of translation to human therapy, it is encouraging that several of these new compounds are currently enrolled in late preclinical and even clinical studies. Examples are rHA-Infestin-4 (Esmon, 2010; Jackson, 2011), nanobodies or aptamers inhibiting GPIb–VWF (reviewed in De Meyer et al (2012a)) or humanized anti-VWF antibodies that inhibit VWF-mediated platelet adhesion (Fontayne et al, 2006; De Meyer et al, 2006; Staelens et al, 2006). Larger translational studies and proof-of-principle clinical trials are now needed to further validate these novel approaches in stroke management, either confirming their safety and efficacy or showing them falling victim to the frequently cited ‘translational roadblock’ in stroke research.
Footnotes
Acknowledgements
SFDM is a postdoctoral fellow of the Fonds voor Wetenschappelijk Onderzoek Vlaanderen.
Disclosure/conflict of interest
CK received financial support from CSL Behring GmbH, Marburg, Germany, for studying novel FXIIa inhibitors in ischemic stroke. PK and SFDM have no conflicts of interests.