
Abstract
Anoxia or ischemia causes hyperexcitability and cell death in mammalian neurons. Conversely, in painted turtle brain anoxia increases γ-amino butyric acid (GABA)ergic suppression of spontaneous electrical activity, and cell death is prevented. To examine ischemia tolerance in turtle neurons, we treated cortical sheets with an in vitro mimic of the penumbral region of stroke-afflicted mammalian brain (ischemic solution, IS). We found that during IS perfusion, neuronal membrane potential (Vm) and the GABAA receptor reversal potential depolarized to a similar steady state (–92 ± 2 to −28 ± 3 mV, and −75 ± 1 to −35 ± 3 mV, respectively), and whole-cell conductance (Gw) increased > 3-fold (from 4 ± 0.2 to 15 ± 1 nS). These neurons were electrically quiet and changes reversed after reperfusion. GABA receptor antagonism prevented the IS-mediated increase in Gw and neurons exhibited enhanced electrical excitability and rapid and irreversible rundown of Vm during reperfusion. These results suggest that inhibitory GABAergic mechanisms also suppress electrical activity in ischemic cortex. Indeed, after 4 hours of IS treatment neurons did not exhibit any apparent damage; while at 24 hours, only early indicators of apoptosis were present. We conclude that anoxia-tolerant turtle neurons are tolerant of exposure to a mammalian ischemic penumbral mimic solution.
Introduction
Most vertebrates suffer brain damage within minutes of anoxia; however, facultative anaerobes such as the Western painted turtle, Chrysemys pictα belli, survive anoxia for days to months without apparent injury (Bickler and Buck, 2007). The key to this tolerance is the maintenance of ATP supply via a coordinated reduction of ATP demand in the absence of oxygen (Bickler and Buck, 2007). In mammals, 50% to 60% of total brain ATP consumption is attributable to Na+/K+-ATPase-mediated ion pumping to maintain ionic gradients and synaptic activity (Erecinska and Silver, 1989; Hansen, 1985); while at the cellular level, action potential (AP) generation and propagation, and excitatory postsynaptic potentials account for ~81% of neuronal energy expenditure (Attwell and Laughlin, 2001). Therefore, limiting excitatory electrical activity is key to tolerating prolonged low-oxygen stress. Facultative anaerobes defend ATP supplies during anoxia by reducing the workload on ATPases through the mechanisms of ‘channel arrest’ and ‘spike arrest’ (Feng et al, 1988; Hochachka, 1986), which reduce ion leakage and neuronal excitability.
Channel arrest is a mechanism whereby membranes become less permeant to ions during anoxia, thereby reducing the requirement for compensatory ionmotive ATPase activity. In anoxic turtle brain, the conductance and expression of ion channels, in particular excitatory glutamatergic receptors, are rapidly and reversibly decreased (Bickler et al, 2000; Pamenter et al, 2008), thereby reducing ionic flux and opposing excitatory depolarization. Concomitantly, γ-amino butyric acid (GABA), the primary inhibitory neurotransmitter in adult mammalian central nervous system (Krnjevic, 1997), increases > 80-fold in anoxic turtle brain (Nilsson and Lutz, 1991), which activates a dominant conductance to Cl− ions, clamping neuronal membrane potential (Vm) near the Cl− ion reversal potential (Ecl or EGABA) and opposing excitatory depolarization via a shunt-like inhibitory mechanism (i.e., spike arrest; Pamenter et al, 2011). Together, these neuroprotective mechanisms act to decrease neuronal excitability, ionic flux, and compensatory ion pump activity, thus preserving ATP and enabling neuronal survival during prolonged anoxia (Buck et al, 1998; Hylland et al, 1997; Pamenter and Buck, 2008; Pamenter et al, 2011).
Based on these adaptations, C. picta has been championed as a model of anoxia tolerance in which to explore protective mechanisms against low-oxygen insult in brain (Bickler and Buck, 2007). However, in mammalian ischemic stroke pathologic examination, the deleterious effects of anoxia or hypoxia are compounded by impaired cerebral blood flow, which also limits nutrient delivery and slows the removal of signaling molecules, ions, and metabolically derived lactate and CO2 (Branston et al, 1974). These events enhance cytotoxicity, ionic imbalance, and acute acidification in the occluded region (the infarct core) and hypoperfused surrounding tissue (the penumbra) (Anderson et al, 1999; Yao et al, 2007). Similarly to the mammalian ischemic penumbra, turtle brain pH becomes more acidic during anoxia (Buck et al, 1998; Wasser et al, 1991); however, cerebral blood flow is increased (Bickler, 1992) and liver glycogen stores are mobilized and continuously delivered to the brain, facilitating glycolytic ATP production (Bickler and Buck, 2007). This represents a significant systemic advantage relative to ischemic stress in mammals; however, turtles are also considerably more tolerant to ischemic stress and survive > 1 hour after cardiac excision at 22°C (Belkin, 1968). Furthermore, mimicking the turtle's endogenous cell-level neuroprotective mechanisms (inhibiting glutamatergic activity—channel arrest, or enhancing GABAergic activity—spike arrest) is neuroprotective against ischemic or hypoxic injury in mammalian brain (Arundine and Tymianski, 2004; Costa et al, 2004). Therefore, the ability of anoxia-tolerant turtle brain to withstand ischemic stress is of interest to better understand how neuroprotective adaptations to low-oxygen environments may provide protection against more complex ischemic challenges.
Previous attempts to examine the resistance of anoxia-tolerant turtle neurons to ischemic stress have relied on oxygen-glucose deprivation or chemical mimics of ischemia that model the mammalian infarct core (i.e., iodoacetate and sodium cyanide to inhibit glycolysis and oxidative phosphorylation, respectively, e.g., Doll et al, 1991), where cell death occurs within minutes of insult onset. Conversely, the penumbral region is hypoperfused and therefore hypoxic, and cell death here spreads over hours to days postinsult and accounts for the majority of morbidity and mortality after stroke in mammalian models (Lo, 2008). Therefore, mechanisms of neuroprotection in this region are of considerably greater clinical relevance than in the infarct core. Recently, our laboratory developed an in vitro treatment paradigm that mimics the key ionic, hypoxic, and acidic parameters of the penumbral milieu, termed as ischemic solution (IS; Yao et al, 2007), and we asked whether anoxia-tolerant turtle cortical neurons are resistant to this clinically relevant ischemic stress. To answer this question, we examined ischemia tolerance in pyramidal neurons from intact cortical sheets because synaptic connectivity and endogenous neurotransmitter release are maintained in this preparation ex vivo (Blanton et al, 1989; Feng et al, 1988). Specifically, we treated turtle cortical sheets with artificial cerebral spinal fluid (ACSF) or IS for up to 24 hours, and examined pyramidal neuron viability and synaptic activity using the whole-cell and perforated-patch electrophysiological techniques and also molecular and biochemical viability assays.
Materials and methods
Please see Supplementary information for an expanded description of Materials and methods.
This study was approved by the University of California San Diego Institutional Animal Care and Use Committee and the University of Toronto Animal Care Committee. Cortical slice dissection and patch-clamp recording methods are described elsewhere (Pamenter et al, 2011). Briefly, cortical sheets were dissected and placed in a bath with flow-through perfusion system. Neurons were perfused with (1) ACSF (in mmol/L: K+ 2.6, Na+ 107, Cl− 113, Ca2+ 1.2, Mg2+ 1.0, NaH2PO4 2.0, NaHCO3 26.5, glucose 10, imidazole 5 (280 to 290 mOsm, pH 7.4, 21% O2, 5% CO2, balance N2); Pamenter et al, 2011) at 4°C, (2) ACSF at 24°C, or (3) an ischemic penumbral perfusate mimic (IS, in mmol/L: K+ 64, Na+ 51, Cl− 77.5, Ca2+ 0.13, Mg2+ 1.5, glucose 3.0, glutamate 0.1 (315 mOsM, pH 6.5, 1.5% O2, 15% CO2, balance N2); Yao et al, 2007) at 24°C; alone or with pharmacological modifiers as specified in Results section. In some experiments, cortical sheets were treated with ACSF containing high [K+] or with IS containing normal [K+] (2.6 mmol/L). Electrical activity was recorded for up to 2 hours from pyramidal neurons using an Axon multiclamp 700B amplifier and Clampex 10 software (Molecular Devices, Sunnyvale, CA, USA). For perforated-patch experiments, electrodes were back-filled with intracellular cerebral spinal fluid containing 25 μg/mL gramicidin.
Cell viability was assessed using propidium iodide (PI) exclusion, annexin V expression, DNA ‘comet-tail’, and ATP-luciferase assays. The PI and annexin V fluorescence were examined using an Olympus FV1000 scanning confocal microscope, with 572 nm (TRITC) and 488 nm (FITC) laser lines (Olympus, San Diego, CA, USA).
Statistical Analysis
Data were analyzed using a two-tailed Student's t-test or one-way analysis of variance, followed by Dunnet's post test. Significances were indicated if P < 0.05 assuming two groups had an equal variance.
Results
Cortical Neurons Are Reversibly Depolarized During Ischemic Solution Perfusion
In normoxia, neurons exhibited a steady resting membrane potential (Vm) of −90.3 ± 1.1 mV in recordings of up to 2 hours (n = 10, Figures 1A and 1B(i)), and voltage ramps elicited APs at a threshold (APth) of −51.3 ± 1.0 mV (Figures 1C and 1D), values which are similar to previous measurements from normoxic turtle cortex (Pamenter et al, 2011). Conversely, upon IS perfusion neurons rapidly depolarized ~60 to 70 mV to a new steady state [Vm = −22.9 ± 1.35 mV after 20 minutes and −28.4 ± 2.3 mV after 60 minutes IS perfusion, respectively; n = 26, Figures 1A and 1B(ii, iii)). Notably, neurons did not exhibit excitatory events during the initial depolarization and were electrically quiet during the entire period of IS treatment, such that APs could not be evoked (n = 26, Figures 1C and 1D). These changes were reversed by IS washout with normoxic ACSF. Vm hyperpolarized to −68.6 ± 9.3 mV after 20 minutes reperfusion, and at 40 minutes was not significantly different from pre-IS baseline controls (–85.4 ± 1.5 mV). Neuronal excitability also returned after reperfusion, as evidenced by the firing of APs in response to stimuli (recovery APth = −45.5 ± 3.4 mV, Figures 1C and 1D).
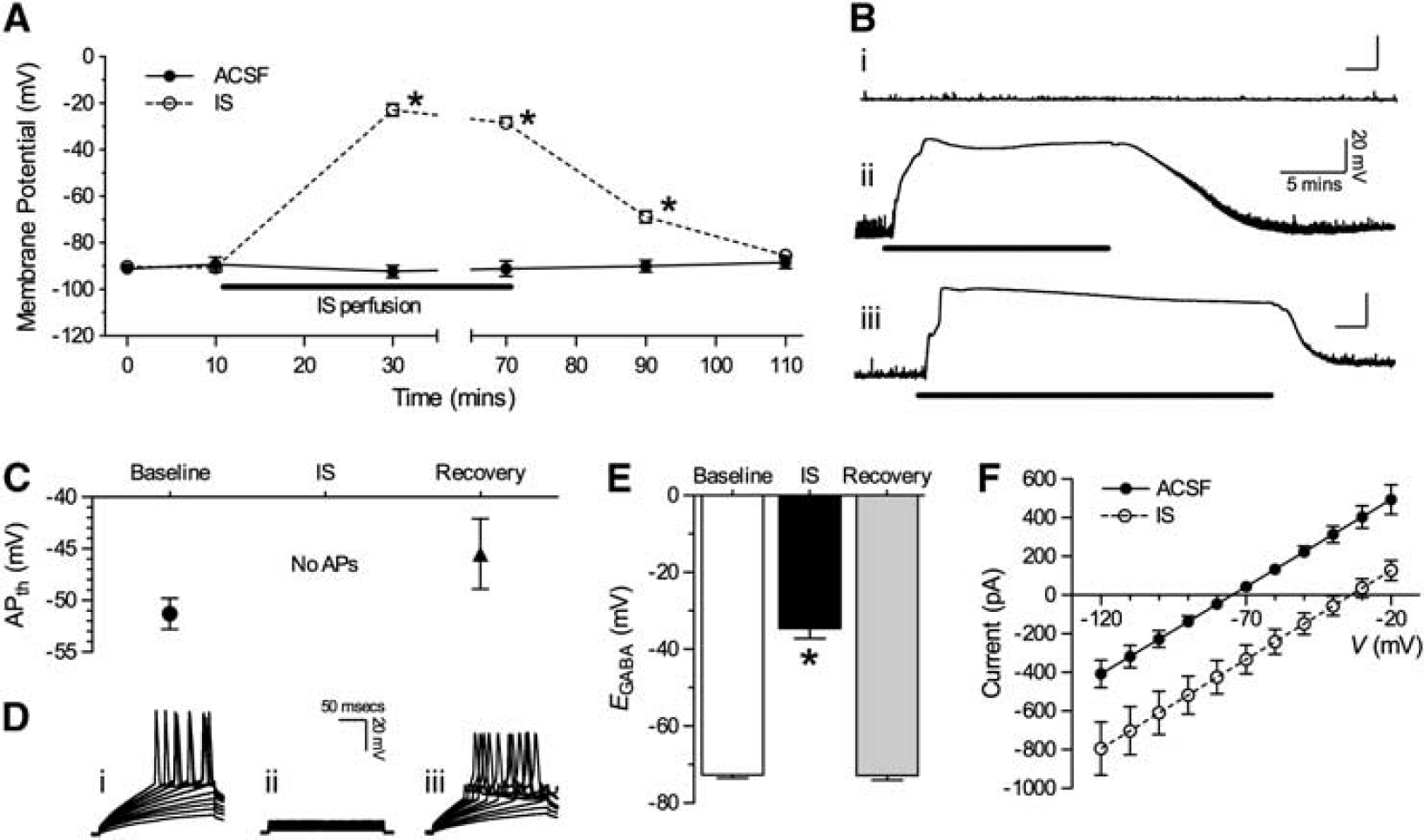
IS-treated cortical neuronal membrane potential (Vm) depolarizes to EGABA. (
In ischemic mammal brain, neuronal depolarization is deleterious and leads to seizure-like events and irreversible anoxic depolarization within minutes of insult onset; and therefore, the lack of similar electrical hyperexcitation in IS-treated turtle neurons, despite a rapid and marked Vm depolarization, suggests that inhibitory (i.e., spike arrest) mechanisms are strongly recruited during IS treatment and reperfusion in this model. In anoxic turtle neurons, Vm is ‘clamped’ at EGABA, which is determined by the distribution of Cl− across the plasma membrane (Ecl); however, the IS used in our experiments incorporates deleterious ionic imbalances that are characteristic of the penumbral milieu, which would alter this distribution. Therefore, to assess a potential role in the IS-mediated depolarization of Vm, we measured EGABA to determine the new set point of Cl− distribution during IS. In normoxia, EGABA was −72.7 ± 0.8 mV and depolarized relative to control Vm (n = 20, Figures 1E and 1F); whereas during IS perfusion EGABA depolarized to −31.6 ± 2.6 mV and was not significantly different from Vm in IS at the same time point. After normoxic reperfusion, EGABA returned to baseline levels (–72.9 ± 1.2 mV). This depolarization represents a significant increase in intercellular chloride ([Cl−]i). According to the Nernst potential equation [Cl−]i in ACSF is ~6.3 mmol/L (extracellular chloride ([Cl−]o) = 113 mmol/L), whereas after the transition to IS, [Cl−]i increases to ~22.2 mmol/L ([Cl−]o = 77.5 mmol/L).
Neuronal Membrane Conductance is Greatly Increased During Ischemic Solution Treatment
Spike arrest in turtle cortical neurons is mediated by an increase in GABA-sensitive Cl− conductance (Gcl)i which resets Vm to EGABA (Pamenter et al, 2011). Therefore, since neuronal Vm and EGABA depolarized to similar levels (approximately −30 mV) during IS perfusion but neurons did not exhibit electrical excitation we hypothesized that a similar increase in GCl occurs during ischemia, and that this increase underlies the depolarization of Vm to EGABA. To test this hypothesis, we examined changes in whole-cell conductance (Gw) during IS perfusion. At rest, neuronal Gw was 3.9 ± 0.2 nS, but increased >3-fold to 14.6 ± 1.3 nS during IS perfusion (n = 31, Figures 2A to 2C); and Gw recovered to pre-IS control levels on reperfusion (4.8 ± 0.5 nS). We have previously shown that 2 mmol/L GABA administration induces similar-magnitude changes to Gw in turtle neurons (Pamenter et al, 2011); therefore, in separate experiments we treated cortical sheets with GABA during an ACSF to IS transition with recovery. In these experiments, Gw in neurons pretreated for 15 seconds with 2 mmol/L GABA was 15.1 ± 1.7 nS in normoxic ACSF and 17.1 ± 2.4 nS in IS (n = 14, Figure 2A). These conductance states were not significantly different from GABA-free IS-treated samples.
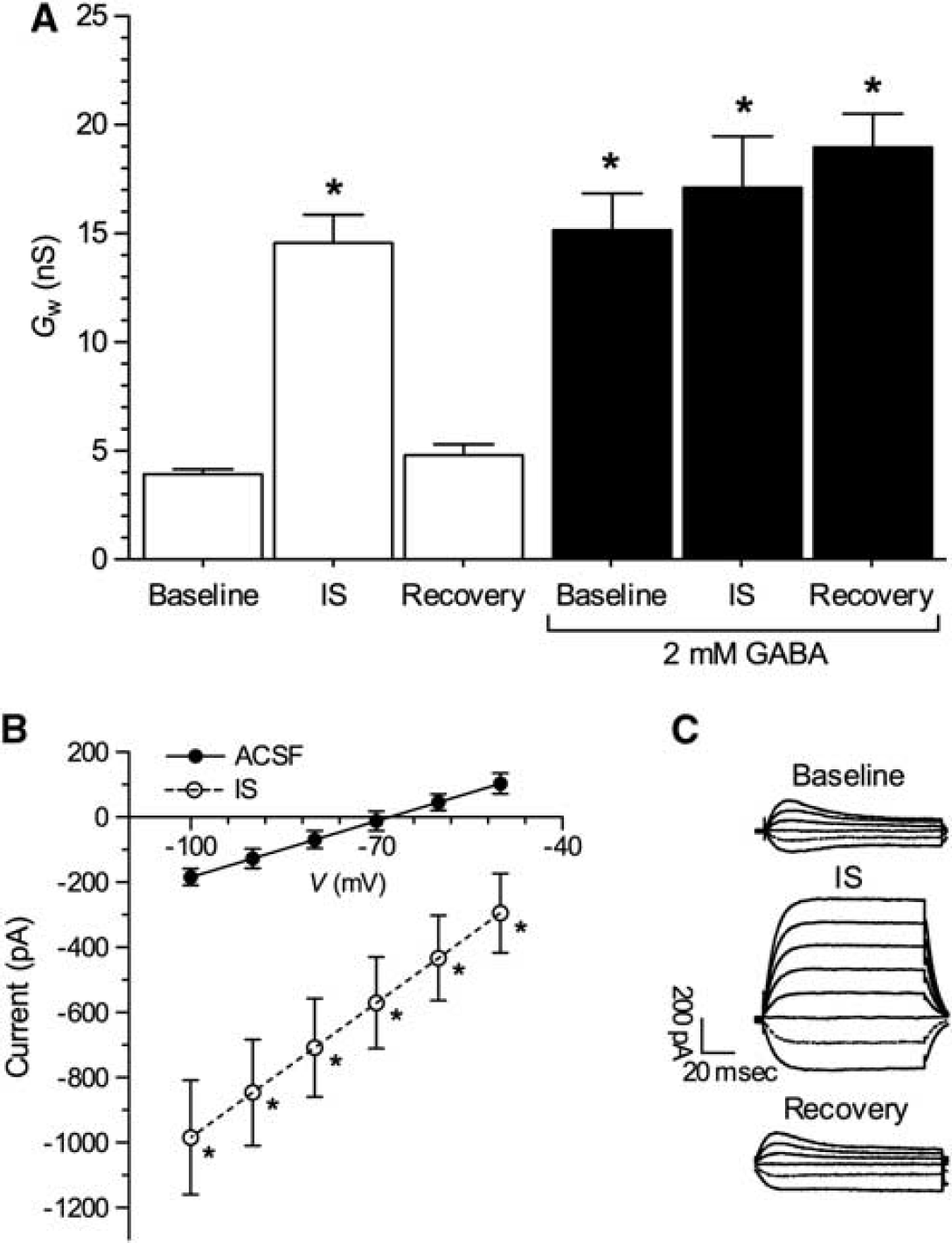
Ischemic solution (IS) and γ-amino butyric acid (GABA) increase whole-cell conductance (Gw). (
Next, we examined the effect of preventing the inhibitory action of spike arrest mechanisms during IS perfusion by blocking GABAA and GABAB receptors with picrotoxin or gabazine (GZ), and CGP55845 (CGP), respectively, because we have previously shown that both GABA receptor subtypes contribute to spike arrest mechanisms in anoxic turtle cortex (Pamenter et al, 2011). In these experiments, neurons exhibited electrical excitability during the transition to IS perfusion (n = 5, Figure 3A, segment a) and also after normoxic reperfusion (Figure 3A, segment b). In all experiments, Vm was more depolarized during IS perfusion with GABA blockers than with IS alone (–15.7 ± 0.6 versus −28.4 ± 2.3 mV, Figure 3B); and after normoxic reperfusion increased spontaneous APs were observed and Vm did not recover, whereas in neurons treated with IS alone Vm returned to baseline levels (–19.4 ± 1.5 and −82.0 ± 2.0 mV after 30 minutes reperfusion, respectively; Figures 3A and 3B). Importantly, the IS-mediated increase in Gw was prevented by GABA receptor antagonism [Gw was 4.5 ± 0.8 and 6.3 ± 1.5 nS before, and during IS + GZ + CGP perfusion, respectively, Figures 3C and 3D).
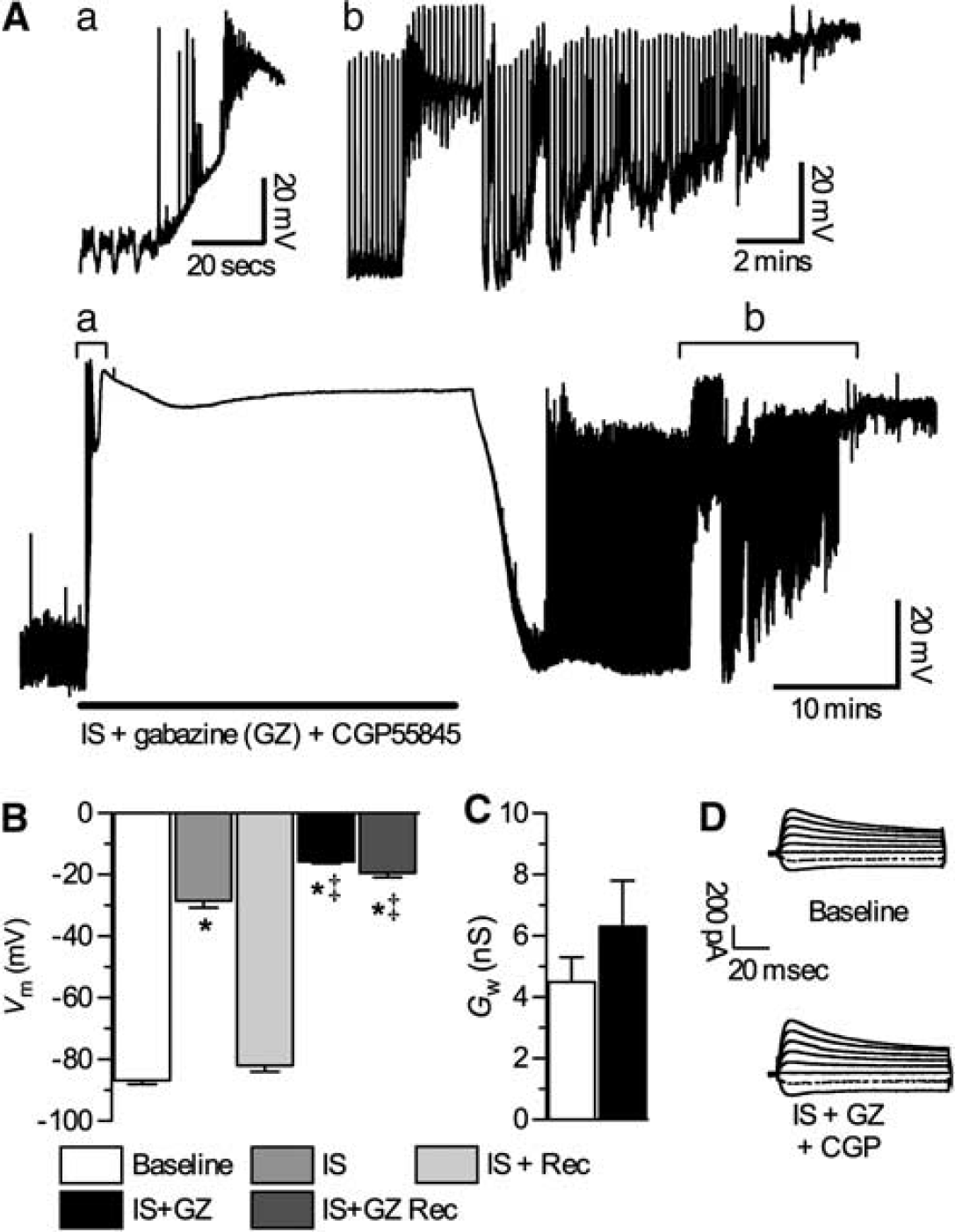
GABA receptor antagonism during ischemic solution (IS) causes electrical hyperexcitation and Vm depolarization after normoxic reperfusion. (
High External [K+] Increases GABAergic Inhibitory Tone
Neuronal membrane potential is determined primarily by the K+ gradient (Hodgkin and Huxley, 1952). Similarly, EGABA is largely determined by this gradient through the activity of K+/Cl− cotransporters, such that increases in extracellular [K+] are expected to depolarize EGABA (Rivera et al, 1999; Thompson and Gahwiler, 1989). External [Cl−] is reduced from 113 to 77.5 mmol/L in IS relative to ACSF, which would account for an ~10 mV depolarizing shift in EGABA (Nernst potential equation); however during IS perfusion, EGABA was much more depolarized (by ~30 to 40 mV), indicating that the internal [Cl−] is altered by IS perfusion in addition to changes to external [Cl−]. Since K+ is greatly increased in the mammalian ischemic penumbra and in our penumbral mimic solution, we next examined the impact of altered K+ on neuronal electrical activity and GABAergic inhibition in normoxia and during IS treatment in turtle brain. First, we treated cortical sheets with a high-K+ ACSF solution containing 64 mmol/L K+ (to match the K+ composition of IS). In these experiments, neurons rapidly depolarized ~75 mV and Vm was −10.3 ± 2.3 mV after 30 minutes perfusion (n = 9, Figures 4A and 4B). EGABA was similarly depolarized by high-K+ ACSF to −44.6 ± 2.2 mV (n = 5, raw data not shown), neurons were electrically quiet during this depolarization (Table 1), and Vm returned to baseline levels after 30 minutes reperfusion with normal ACSF. Similarly, Gw reversibly increased from 4.6 ± 0.7 to 12.3 ± 1.5 nS when neurons were treated with high-K+ ACSF (n = 5, Figures 4C and 4D), a conductance state that is consistent with that of normoxic neurons during GABA application.
Effects of extracellular [K+] and GABA on APth
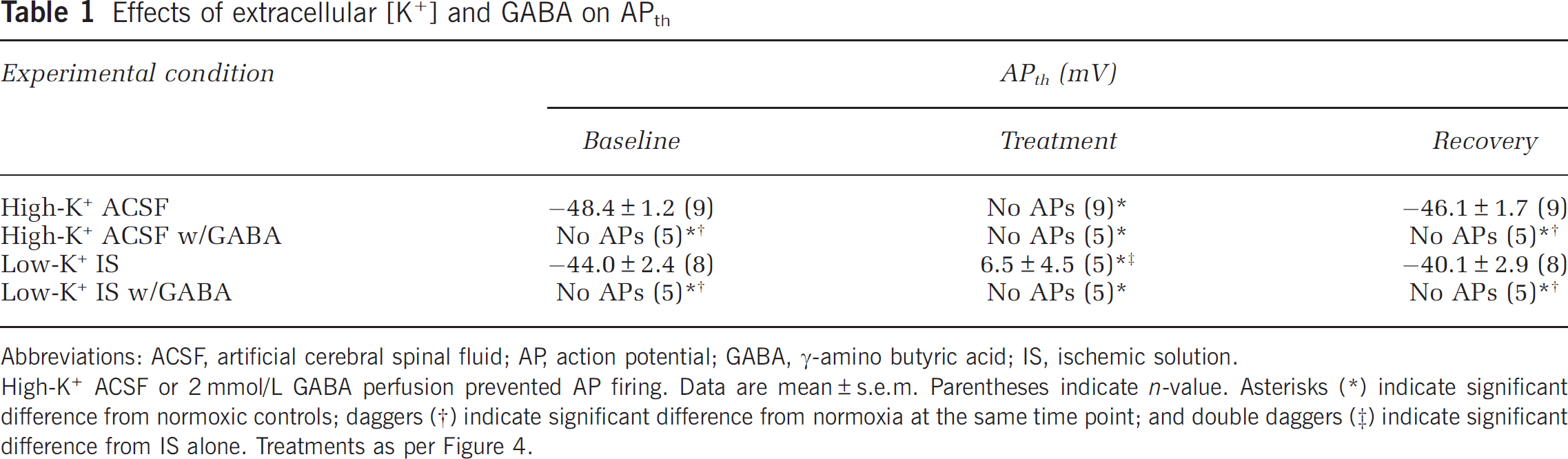
Abbreviations: ACSF, artificial cerebral spinal fluid; AP, action potential; GABA, γ-amino butyric acid; IS, ischemic solution.
High-K ACSF or 2 mmol/L GABA perfusion prevented AP firing. Data are mean±s.e.m. Parentheses indicate n-value. Asterisks
indicate significant difference from normoxic controls; daggers
indicate significant difference from normoxia at the same time point; and double daggers
indicate significant difference from IS alone. Treatments as per Figure 4.
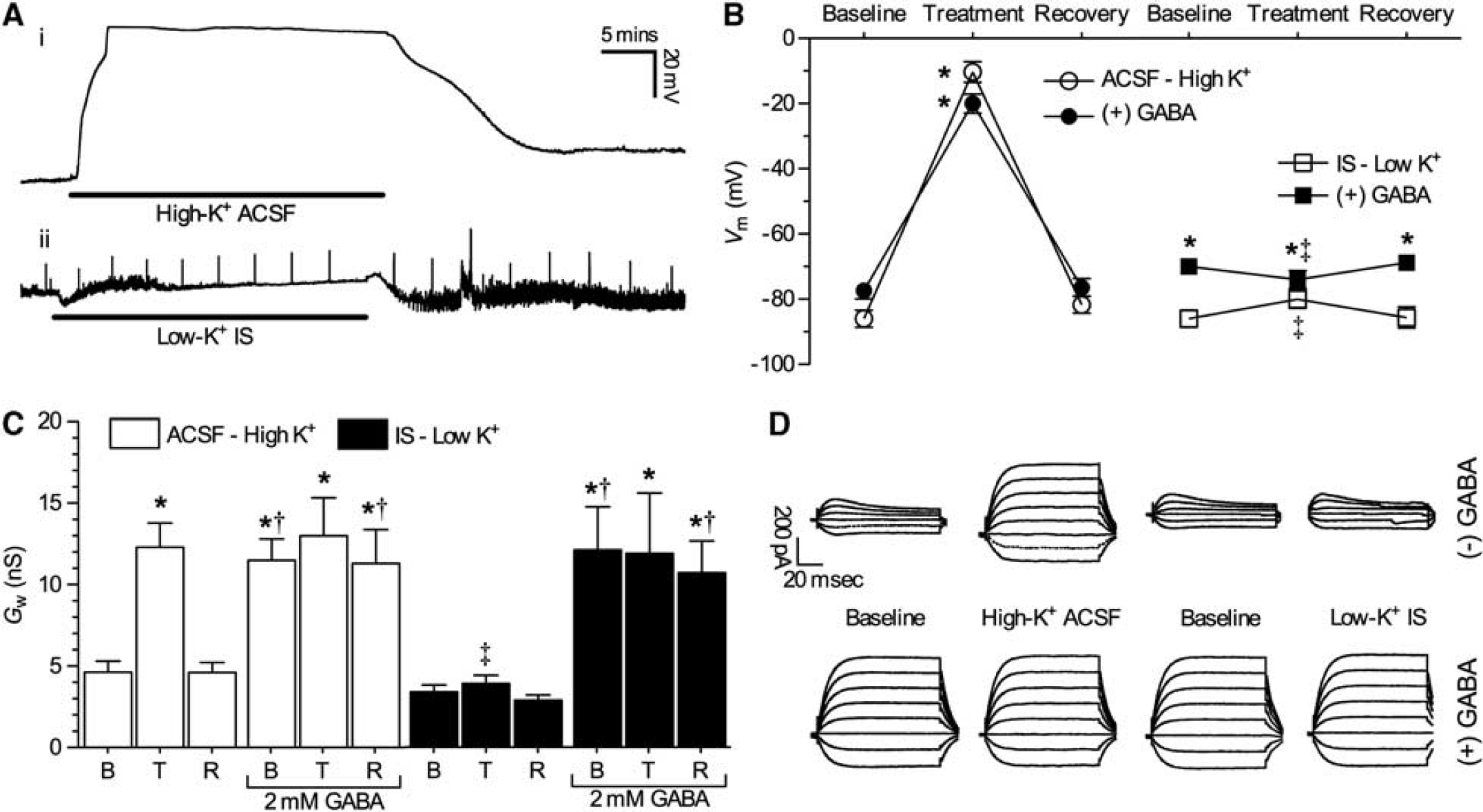
Extracellular K+ underlies the ischemic solution (IS)-mediated Vm depolarization and increase in Gw. (
To examine the role of GABA in high-K+-mediated changes, we next applied 2 mmol/L GABA during an ACSF to high-K+ ACSF transition with recovery. In this analysis, GABA-treated neurons were slightly depolarized with regard to untreated neurons (–78.6 ± 2.5 mV, n = 5, Figure 4B), and reversibly depolarized to −20.0 ± 2.3 mV after 30 minutes perfusion of high-K+ ACSF with GABA application. Gw was increased by GABA treatment alone (13.3 ± 2.3 nS, n = 5 for each, Figures 4C and 4D) and the subsequent switch to high-K+ ACSF with GABA had no additional effects on Gw (13.0 ± 2.3 nS). In addition, APs could not be evoked from neurons treated with high-K+ ACSF alone or in the presence of GABA (n = 9 and 5, respectively, Table 1). Taken together, these results indicate that inhibitory GABAergic activation (i.e., GABA release), and the depolarizing shift in EGABA occurs downstream of elevated extracellular K+ accumulation since GABA perfusion alone increases Gw but does not markedly depolarize neurons, whereas high-K+ ACSF perfusion alone increases Gw to the same degree as GABA perfusion and also depolarizes neurons, while simultaneously preventing electrical excitability.
This relationship was indirectly confirmed in experiments examining the effect of a modified IS perfusate containing low [K+] (2.6 mmol/L). In these experiments, neurons treated with low-K+ IS did not exhibit significant depolarization of Vm, Gw did not increase, and potentials could be evoked with stimulus injection (n = 8, Figures 4A to 4D, Table 1). In addition, EGABA was not depolarized relative to controls (n = 5, −75.0 ± 0.6 mV, raw data not shown). Conversely, application of GABA in low-K+ IS restored the large increase in Gw observed in control IS experiments and ablated stimulus-evoked potentials, but did not restore the depolarization of Vm, indicating that EGABA was not markedly depolarized by low-K+ IS perfusion. Therefore, increased GABA release and postsynaptic Gw, along with the IS-mediated shift in EGABA, are regulated by high-K+ in the unmodified IS perfusate such that the change in Vm and electrical inhibition is due to increased presynaptic release of GABA, while the change in EGABA is due to postsynaptic alterations of neuronal K+ gradients with a smaller contribution from a reduced driving force on Cl− due to lower [Cl−]o.
Turtle Neurons Tolerate Prolonged Ischemic Solution Treatment
Finally, we examined the ability of turtle cortical sheets to survive prolonged IS treatment. It is not feasible to assay long-term cell viability using electrophysiological approaches; therefore, we used a variety of biochemical and molecular assays to examine the effects of prolonged IS perfusion (0.5 to 24 hours) on turtle cortical sheet viability. Necrotic cell death is generally characterized by rapid permeability barrier (plasma membrane) rupture in the absence of apoptotic markers (Galluzzi et al, 2007); therefore, we first examined membrane viability using a PI exclusion assay in live cortical sheets bathed in ACSF or IS treated at 24°C [n = 3 for each, Figure 5A). The PI fluorescence was assessed at 0.5, 1, 2, 4, 8, 12, and 24 hours, and at each of these time points PI fluorescence was unchanged in either treatment group relative to controls treated in ACSF at 4°C. In other experiments, PI fluorescence was assessed in combination with the translocation of the apoptotic marker annexin V to the outside of neuronal plasma membranes. After 4 hours of IS treatment, we did not observe significant PI or annexin V fluorescence in either ACSF or IS perfused cortical sheets treated at 24°C relative to controls treated with ACSF at 4°C (n = 5 for each, Figures 5B and 5C). Similarly, after 24 hours treatment neurons in all groups continued to exclude PI; however, the extent of annexin V fluorescence in IS-treated samples increased ~6-fold relative to controls, indicating the occurrence of apoptosis in IS-treated cortical sheets. In parallel experiments, cortical sheets were treated with the proapoptotic agent staurosporine in ACSF. Relative to controls staurosporine increased annexin V fluorescence ~11- and 15-fold at 4 and 24 hours, respectively (n = 3, Figures 5B and 5C).
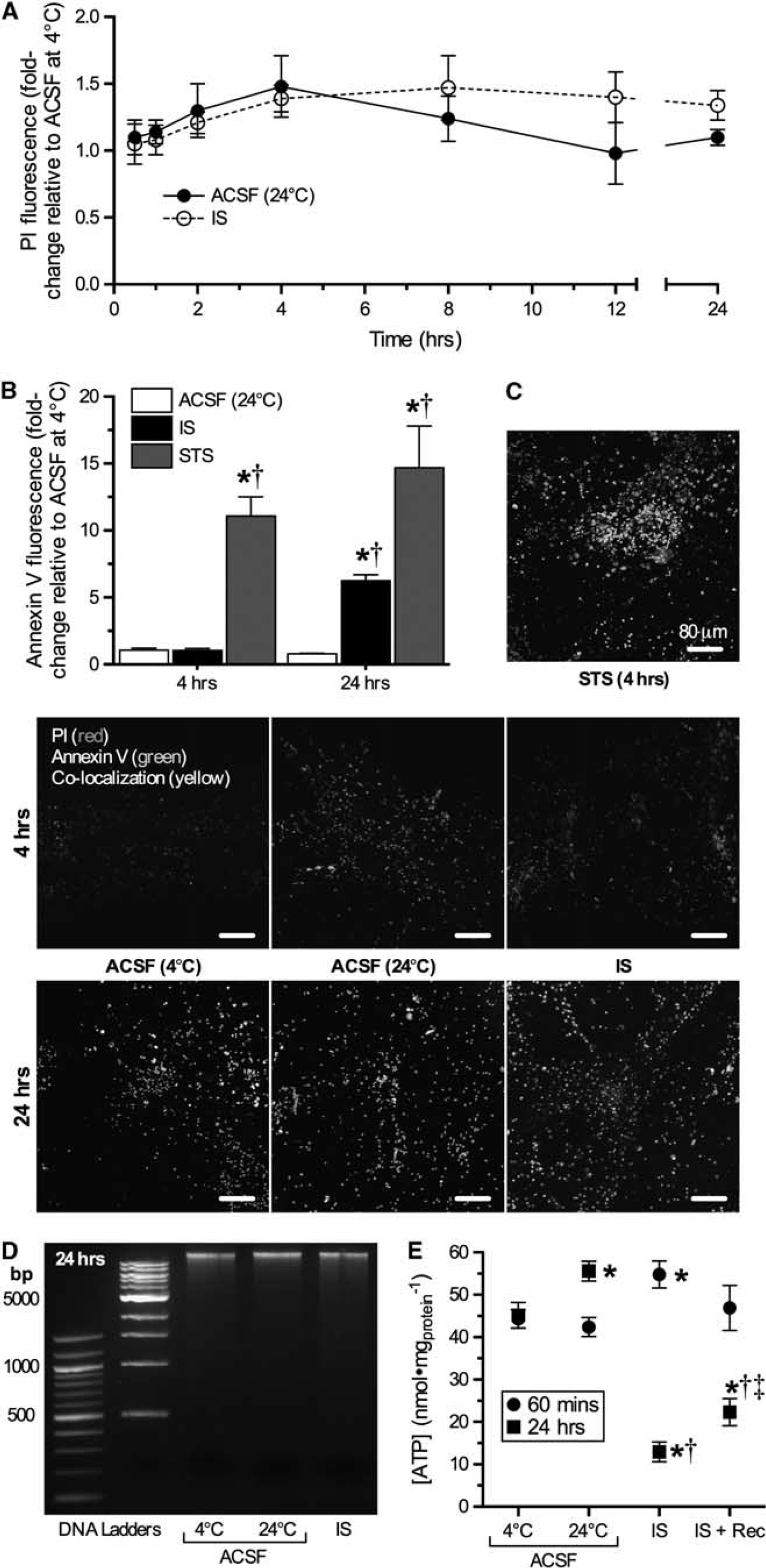
Ischemic solution (IS) perfused cortical sheets retain DNA and plasma membrane integrity, but express an early apoptotic marker at 24 hours. (
To further examine cell viability at 24 hours, we examined DNA fragmentation using a DNA gel electrophoresis assay, since laddered oligonucleosomal DNA fragmentation is a hallmark of advanced apoptosis (Galluzzi et al, 2007). In this analysis, we did not observe fragmentation in ACSF- or IS-treated samples (n = 3 for each, Figure 5D), suggesting that the induction of apoptosis in IS-treated cortical sheets at 24 hours is at a very early stage. Finally, we also assayed total cellular [ATP] at 1 and 24 hours and found that [ATP] was unchanged after 1 hour of IS treatment, but decreased ~75% in IS-treated samples at 24 hours (n = 3 to 5, Figure 5E). Nonetheless in samples that were treated with IS and then allowed to recover in normoxic ACSF for 1 hour, [ATP] began to recover, suggesting that samples treated with this paradigm remain viable and recover from prolonged IS exposure.
Discussion
We show that in addition to being anoxia-tolerant, Western painted turtle cortical neurons are tolerant to a high-K+ ischemic penumbral mimic solution. This conclusion is supported by our observations that turtle neurons: (1) depolarize but are electrically quiescent during 1 hour of IS perfusion and recover fully after normoxic reperfusion; (2) defend [ATP] in this time period, and [ATP] recovers from ischemic lows after 24 hours treatment; and (3) maintain plasma membrane and DNA integrity through 24 hours of IS treatment. Although turtles likely do not experience ischemia naturally, this tolerance is nonetheless remarkable and the mechanisms that mediate this survival may be used to protect tissue in ischemia-sensitive mammals. An important component of this tolerance during the transition to IS treatment and reperfusion is an increase in inhibitory GABAergic signaling. GABA is released in anoxic turtle brain (Nilsson and Lutz, 1991) and upon binding to postsynaptic GABAA receptors, activates Gcl to a degree sufficient to dominate Gw, and thus clamp Vm at EGABA (Pamenter et al, 2011). As a result, excitatory inputs are neutralized by a ready flux of opposing charge mediated by Cl−, which dampens excitatory depolarization via a shunt-like mechanism. Here, we report a similar phenotype in IS-treated cortex: Vm depolarizes to EGABA; Gw increases >3-fold and neurons are electrically silent despite extreme depolarization, while APs cannot be elicited with stimulus injection.
The mechanism underlying these postsynaptic changes is likely mediated by GABA receptor activation since (1) GABA perfusion in normoxic ACSF increases neuronal Gw to the same degree as IS and APs cannot be elicited; while (2) cotreatment of IS plus GABA does not result in further elevations of Gw; and (3) antagonism of neuronal GABAA + B receptors during IS prevents the IS-mediated increase in Gw, and neurons become hyperexcited and Vm runs down within minutes of reperfusion. Therefore, since IS and GABA have similar but not additive effects on Gw and neuronal excitability, and since GABAergic inhibition during IS treatment prevents the increase in Gw and induces excitotoxicity characteristic of ischemic mammalian neurons, we conclude that enhanced GABAergic inhibition prevents electrical excitability and at a minimum, underlies early-stage IS tolerance in this model.
GABAergic inhibition and the large depolarizing shift in EGABA are mediated by the high [K+] of our IS. In mammals, a high [K+] perfusate induces extensive neuronal hyperexcitation by shifting the K+ gradient such that nervous cells depolarize and become more electrically active within 4 to 5 minutes of perfusion (Zhou et al, 2010). In mammalian slice models, this excitation leads to depolarization of both presynaptic and postsynaptic neurons, which causes extensive neurotransmitter release, excitotoxicity, and spreading depression (Gardner-Medwin, 1981). Conversely, in turtle brain high-K+ ACSF causes extensive postsynaptic depolarization, but electrical activity is suppressed and Vm recovers after reperfusion with normal ACSF. GABAergic increases in postsynaptic Gw are activated by high-K+ and do not occur in neurons treated with low-K+ IS, indicating that presynaptic GABA release is greatly increased by high-K+. Our observation that turtle neurons are electrically silent in high-K+ experiments despite significant depolarization is notable as this indicates that inhibitory tone in turtle brain greatly outweighs excitatory tone, as in these experiments the high [K+] will have chronically activated excitatory glutamatergic signaling in addition to inhibitory GABAergic signaling. This may represent a significant adaptive advantage in turtle brain relative to mammal brain. Conversely, our observation that turtle neurons treated with low-K+ IS do not exhibit increases in Gw also indicates that this change is mediated by activation of postsynaptic GABA receptors due to high-K+-induced presynaptic GABA release. The lack of electrical hyperexcitability in neurons treated with low-K+ IS indicates that the remaining deleterious alterations in this solution are not overly damaging to turtle neurons, or are protected against by alternative mechanisms since the inhibitory GABAergic shunt that underlies SA does not appear to be active under these conditions.
In addition to regulating GABA release during IS perfusion, high-K+ also predominately mediates the depolarizing shift in EGABA observed in IS-treated neurons. EGABA is the reversal potential of GABAA receptors, which are primarily permeant to Cl− ions. EGABA therefore is determined by the Cl− gradient, which is in turn determined by the activity of K+/Cl− cotransporters (Rivera et al, 1999). Therefore, increasing external K+ decreases the driving force on Cl−, and depolarizes EGABA. This relationship is supported by our observations that high-K+ alone in ACSF was sufficient to depolarize EGABA in normoxia and shift Vm toward this depolarized value, and that low-K+ IS did not induce a shift in Vm or EGABA, even when postsynaptic GABAA receptors were chronically activated in the presence of 2 mmol/L GABA. Conversely, in anoxic turtle cortex GABA release is drastically increased, but EGABA is not depolarized and extracellular [K+] does not change significantly during prolonged anoxia, suggesting that alternative mechanisms are used to activate endogenous inhibitory GABAergic neuroprotective mechanisms in anoxic turtle brain (Jackson and Heisler, 1983; Nilsson and Lutz, 1991; Pamenter et al, 2011).
Traditionally researchers have primarily used oxygen-glucose deprivation or chemical ischemia to mimic stroke in vitro. Although these paradigms accurately model the metabolic consequence of impaired cerebral blood flow, they do not incorporate other deleterious aspects of the ischemic milieu (Lo, 2008; Yao et al, 2007). The ischemic paradigm used in our experiments similarly mimics the metabolic consequences of reduced cerebral blood flow (low glucose, hypoxia), but also incorporates severe ionic derangements, acidification (~pH 6.4), and excitatory neurotransmitter accumulation (glutamate) characteristic of ischemic mammalian penumbral tissue (Hansen and Nedergaard, 1988; Siesjo, 1992; Yao et al, 2007). Indeed, many of these alterations alone are sufficient to induce depolarization or death in mammalian neurons. For example, the increase in external [K+] from 2.6 (in ACSF) to 64 mmol/L (in IS) is sufficient to induce spreading depression in cortex (Zhou et al, 2010), whereas holding external pH at 6.4 for 6 hours causes 50% neuronal and glial cell mortality in forebrain (Nedergaard et al, 1991). Therefore when combined, these alterations present a highly challenging stress, and the ability of turtle cortex to withstand IS treatment for up to 24 hours with minimal apparent damage is remarkable.
Unlike turtle neurons, mammalian cells are intolerant to IS treatment. In hippocampal slice models and in cell cultures, mammalian neurons become electrically excited and rapidly depolarize within minutes of IS onset at 24°C or 37°C, and Vm does not recover after normoxic reperfusion (Yao and Haddad, unpublished observations). Furthermore, after a 24-hour IS treatment ~90% of neurons take up PI and > 90% of cellular lactate dehydrogenase is released (Yao et al, 2007), while [ATP] is depleted > 85%, and DNA exhibits extensive oligonucleosomal fragmentation and TUNEL (terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling)-positive staining characteristic of late-stage apoptosis (Galluzzi et al, 2007; Pamenter et al, 2010, 2012). Similar results have been reported in a wide variety of in vivo and in vitro models of the ischemic mammalian penumbra, where in general ischemic stress induces: electrical hyperexcitation and extreme Vm depolarization within minutes; ionic and neuro transmitter derangements, [ATP] depletion, and necrotic cell rupture or extensive activation of apoptotic mechanisms within < 1 to 4 hours; and extensive or total neuronal cell death within 6 to 24 hours (Broughton et al, 2009; Hansen and Nedergaard, 1988; Nedergaard and Hansen, 1993; Siesjo, 1992).
Comparatively, patch-clamped turtle cortical neurons tolerate at least 60 minutes of IS treatment (i.e., at least 20 to 30 × longer than mammalian neurons) and Vm and synaptic function fully recover after reperfusion. Necrosis does not occur through 24 hours of IS treatment, as indicated by maintained vital dye exclusion, and DNA does not exhibit fragmentation typical of either necrosis or apoptotic cell degradation at 24 hours (Galluzzi et al, 2007). However, we observed a significant increase in the translocation of annexin V, an early indicator of apoptosis (Galluzzi et al, 2007), after 24 hours, but not 4 hours of IS treatment. This result suggests that cortical sheets are likely reaching the limit of their tolerance to the stress at this point and are beginning to undergo programmed cell death. Nonetheless, the recovery of [ATP] after 1-hour reperfusion after 24 hours IS treatment suggests that tissue remains viable and recovers even after prolonged stress. There is evidence that cells damaged by ischemic stress are replaced via neuronal regeneration in this model (Kesaraju and Milton, 2009), and it is tempting to speculate that in vivo, damaged cells marked for apoptosis would be replaced by new neurons, potentially allowing this organism to tolerate prolonged brain ischemia and recover without long-term detriment, even if some cells are damaged by the initial stress. If this were the case, then the observed annexin V staining at 24 hours may actually be adaptive in turtle brain and indicative of maintenance-related local apoptosis to mark damaged cells for removal and eventual replacement with new neurons, instead of an indication of the onset of widespread apoptosis.
Experimental temperature differences may contribute to the enhanced ischemia tolerance of turtle brain in the present study relative to previously published studies in mammal brain. Mammalian experiments are typically conducted between 24°C and 37°C, and hypothermia slows metabolism and extends ischemic or hypoxic survival time (Lampe and Becker, 2011). Conversely, turtles survive months of anoxia at 4°C, and typically tolerate 2 to 3 days of anoxia at room temperature, and just hours at 37°C (Bickler and Buck, 2007). Nonetheless, turtles have consistently displayed a remarkable degree of anoxia and ischemia tolerance relative to mammals across a wide range of experimental temperatures (Belkin, 1968; Bickler, 1992; Bickler and Buck, 2007; Doll et al, 1991) and our present results provide further support for this relationship. For example, at temperatures of 22°C to 24°C turtle brain sheets tolerate > 3 hours of anoxic perfusion and > 1 hour of ischemic perfusion, whereas rat brain slices last < 40 minutes in anoxia and < 5 minutes in ischemia (Doll et al, 1991; Pamenter et al, 2010, present study). Similarly at the whole-organism level, turtles tolerate > 1 hour of ischemia after cardiac excision, while mammals die within minutes of blood flow cessation (Belkin, 1968; Bickler and Buck, 2007).
In summary, we have shown that turtle cortical neurons tolerate a highly deleterious ischemic stress for up to 24 hours. Electrophysiological studies indicate that one key mechanism underlying this tolerance is a large-scale activation of inhibitory GABAergic Gcl, which clamps Vm at EGABA and prevents excitatory events and cell death during IS onset. A similar mechanism underlies spike arrest in the anoxic turtle cortex, but the degree to which inhibitory GABAergic mechanisms are activated in IS-treated neurons is several-fold greater than that observed previously in anoxic experiments (Pamenter et al, 2011), likely because GABAergic inhibition here is activated by greatly increased extracellular K+, whereas in the anoxic cortex GABA release is activated by an unknown mechanism that is likely K+ independent. Nonetheless, these results illustrate that naturally evolved adaptations to survival in anoxic environments can also protect against clinically relevant paradigms of ischemic stress derived from mammalian studies. This study highlights the value of a comparative model in which to elucidate medically relevant protective mechanisms against anoxic and ischemic insults in brain.
Disclosure/conflict of interest
The authors declare no conflict of interest.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.