
Abstract
An angiogenic factor, vascular endothelial growth factor (VEGF), might be associated with the blood-brain barrier (BBB) disruption after focal cerebral ischemia; however, it remains unknown whether hemorrhagic transformation (HT) after tissue plasminogen activator (tPA) treatment is related to the activation of VEGF signaling pathway in BBB. Here, we hypothesized that inhibition of VEGF signaling pathway can attenuate HT after tPA treatment. Rats subjected to thromboembolic focal cerebral ischemia were assigned to a permanent ischemia group and groups treated with tPA at 1 or 4 hours after ischemia. Anti-VEGF neutralizing antibody or control antibody was administered simultaneously with tPA. At 24 hours after ischemia, we evaluated the effects of the antibody on the VEGF expression, matrix metalloproteinase-9 (MMP-9) activation, degradation of BBB components, and HT. Delayed tPA treatment at 4 hours after ischemia promoted expression of VEGF in BBB, MMP-9 activation, degradation of BBB components, and HT. Compared with tPA and control antibody, combination treatment with tPA and the anti-VEGF neutralizing antibody significantly attenuated VEGF expression in BBB, MMP-9 activation, degradation of BBB components, and HT. It also improved motor outcome and mortality. Inhibition of VEGF signaling pathway may be a promising therapeutic strategy for attenuating HT after tPA treatment.
Introduction
The thrombolytic agent tissue plasminogen activator (tPA) is the only therapeutic agent approved by the US Food and Drug Administration for the treatment of patients with acute ischemic stroke (The National Institute of Neurological Disorders and Stroke rt-PA stroke Study Group, 1995). Its use remains limited to patients who are treated within 4.5 hours of symptom onset (Hacke et al, 2008). The benefits of tPA thrombolysis are heavily dependent on time to treatment, and use of tPA may be associated with hemorrhagic transformation (HT), especially when tPA is administered beyond the therapeutic time window. Several animal studies on focal cerebral ischemia have shown that the development of HT after tPA treatment is associated with ischemic endothelial injury (Wang et al, 2004) and subsequent blood-brain barrier (BBB) disruption (Yepes et al, 2003; Su et al, 2008). Identification of therapeutic target molecules that are involved in endothelial injury and BBB disruption associated with tPA treatment is, therefore, of great importance.
Studies on experimental animal models or patients with acute ischemic stroke showed that matrix metalloproteinase-9 (MMP-9) has important functions in tPA-associated BBB disruption (Sumii and Lo, 2002). Tissue plasminogen activator might activate MMP-9 in cultured astrocytes (Lee et al, 2007) and increases ischemic neuronal injury by elevating the level of MMP-9 in mice (Wang et al, 1998). It is considered that MMP-9 causes BBB disruption via the dysregulated proteolytic degradation of BBB components such as endothelial barrier antigen (EBA) and basal lamina type IV collagen (Gursoy-Ozdemir et al, 2004). Consequently, it is necessary to determine effective MMP-9 inhibitors, which can be applied in a clinical setting.
Several studies have shown that an angiogenic factor, vascular endothelial growth factor (VEGF), can activate MMP-9 (Bergers et al, 2000; Valable et al, 2005), and that it is one of the potent mediators of vascular permeability (Ferrara et al, 2003). In cerebral ischemia, expressions of VEGF are increased in the blood vessels, neurons, and other cells (Zhang et al, 2002a). Topical application of VEGF on the ischemic cortex or intravenous administration of VEGF to rats 1 hour after ischemic stroke exacerbated BBB disruption (Zhang et al, 2000; Chi et al, 2005). In addition, several studies using different modalities to inhibit VEGF signaling pathway have shown its potential utility in reducing edema and vessel injury (van Bruggen et al, 1999; Chi et al, 2007; Foster et al, 2009). Although these findings suggest that VEGF is responsible for the BBB disruption after focal cerebral ischemia, it remains unknown whether HT after tPA treatment is related to the enhanced activation of VEGF signaling pathway in BBB. Here, we hypothesized that inhibition of VEGF signaling pathway can attenuate MMP-9 activation and HT after tPA treatment.
Materials and methods
A Rat Thromboembolic Model
As per the protocol approved by the Niigata University Administrative Panel on Laboratory Animal Care, male Sprague-Dawley rats weighing 300 to 390 g were subjected to thromboembolic focal cerebral ischemia (Busch et al, 1997). At 1 or 4 hours after middle cerebral artery occlusion, the tPA (alteplase, a gift from Mitsubishi Tanabe Pharma Co., Osaka, Osaka, Japan) was administered via the femoral vein. A catheter was inserted into the tail artery of the rats for blood pressure measurement and blood gas analysis. The level of cerebral cortical blood flow (CCBF) was monitored by laser Doppler flowmetry as described previously (Okubo et al, 2007). Briefly, a Plexiglass tube was mounted onto the skull to aid placement. Cerebral cortical blood flow at 30 minutes before ischemia and 30 minutes and 24 hours after ischemia were measured after the CCBF stabilized. Rats in which the residual CCBF at 30 minutes after ischemia did not decrease below 50% of the preischemic baseline value were excluded (9 of the total of 137 rats were excluded). Tissue plasminogen activator was administered at 1 or 4 hours after ischemia because previous studies using a rat embolic stroke model showed that the administration of tPA 1 hour after ischemia reduced infarct volume and improved functional outcome, whereas its administration 4 hours after ischemia caused BBB damage (Jiang et al, 2000; Zhang et al, 2002b). Rats subjected to acute focal cerebral ischemia were randomly allocated to the following three groups: a permanent ischemia group without tPA treatment and groups treated with tPA (10 mg/kg) at 1 hour after ischemia (tPA 1-hour group) or at 4 hours after ischemia (tPA 4-hour group). At 24 hours after ischemia, deeply anesthetized rats were transcardially perfused with phosphate-buffered saline. Unfixed 3 mm coronal brain slices were incubated in 2% 2,3,5-triphenyltetrazolium chloride, and the infarct and edema volumes were calculated as described previously (Swanson et al, 1990). Hemoglobin concentrations of the lysates of whole ischemic hemisphere were measured by a spectrophotometric assay (Asahi et al, 2000). Outcomes at 24 hours after ischemia were scored using the 5-point motor function scale, as described previously (Andersen et al, 1999), in a nonblinded manner: 0 = no motor deficit, 1 = flexion of the forelimb contralateral to the ischemic hemisphere, 2 = reduced resistance against push toward the paretic side, 3 = spontaneous circling toward the paretic side, and 4 = death.
Immunohistochemistry
Deeply anesthetized rats were perfused intracardially with 4% paraformaldehyde at 24 hours after ischemia. The brains were removed and embedded in paraffin wax. Serial sections (4 µm thick) were cut from the paraffin blocks. We stained paraffin-embedded sections using antibodies against VEGF and EBA (Supplementary Table 1). The immunoreactive products were visualized using the ABC Vectastain kit (Vector, Burlingame, CA, USA) and diaminobenzidine as the chromogen. We also stained freefloating sections using antibodies listed in Supplementary Table 1 as described previously (Kanazawa et al, 2011). Three-dimensional reconstructions, z-sections collected at 0.23 µm z intervals, were created and analyzed using IMARIS imaging software (BitplaneAG, Zurich, Switzerland). To evaluate immune complex deposition in the liver, spleens, and kidneys, we immunostained these organs with Alexa Flour 568-conjugated anti-rabbit IgG antibody at 24 hours after ischemia.
Physiological parameters after ischemia
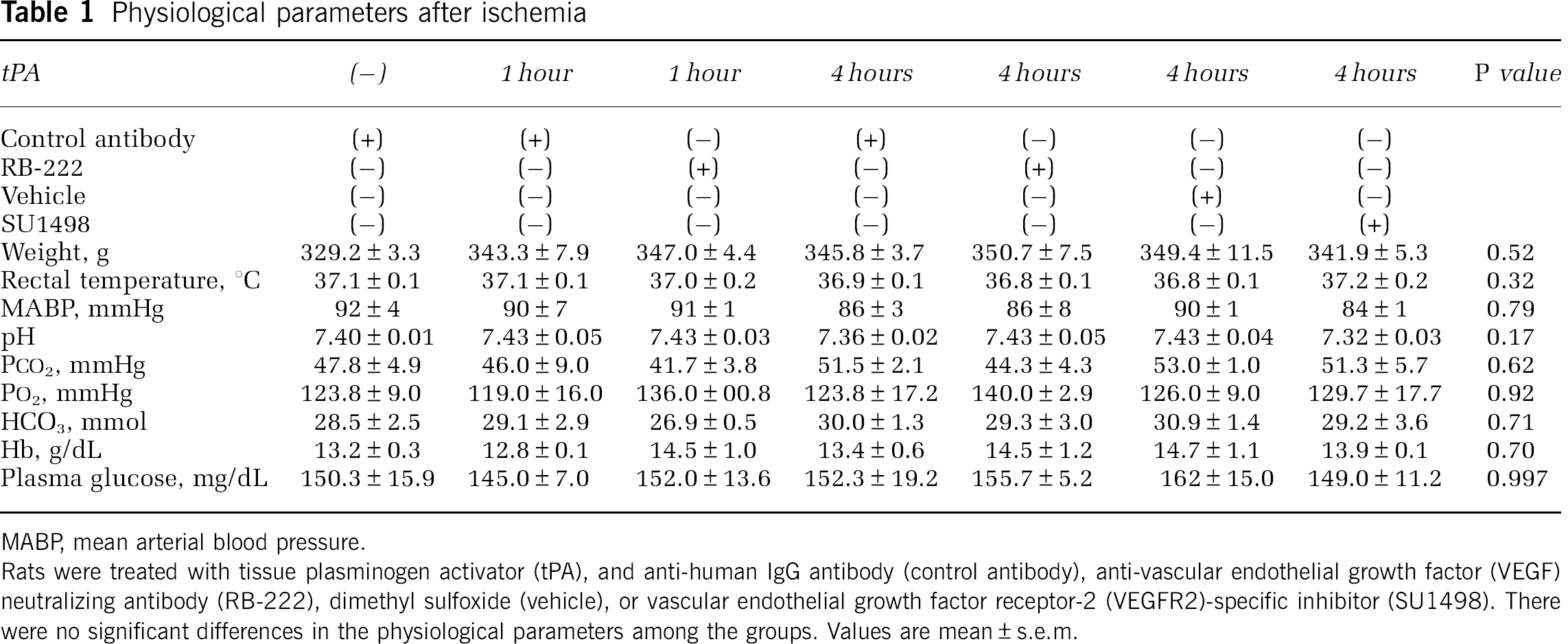
MABP, mean arterial blood pressure.
Rats were treated with tissue plasminogen activator (tPA), and anti-human IgG antibody (control antibody), anti-vascular endothelial growth factor (VEGF) neutralizing antibody (RB-222), dimethyl sulfoxide (vehicle), or vascular endothelial growth factor receptor-2 (VEGFR2)-specific inhibitor (SU1498). There were no significant differences in the physiological parameters among the groups. Values are mean ± s.e.m.
Immunoblotting
Those rats that survived for 24 hours after ischemia were euthanized with an overdose of halothane, followed by intracardiac perfusion with cold normal saline. The brains were removed, and the cortical tissue from the whole middle cerebral artery territory was dissected. The corresponding regions from sham-operated rats were also dissected as controls. The brain tissues were homogenized in a glass homogenizer (Wheaton, Millville, NJ, USA) in 7 volumes of cold cell lysis buffer (#9803; Cell Signaling Technology, Beverly, MA, USA; containing 1% Triton X-100, 1 mmol/L Na3VO4), protease inhibitor (P8340; Sigma-Aldrich, St Louis, MO, USA), and phosphatase inhibitor cocktails 1 and 2 (P2850 and P5726; Sigma-Aldrich); centrifuged; and the supernatants were collected, as described previously (Shimohata et al, 2007). For Trisglycine SDS-PAGE, 50 µg of sample was mixed with Laemmli sample buffer (161-0737; Bio-Rad, Hercules, CA, USA) and boiled for 5 minutes. After transfer to PVDF membrane, the blots were probed with primary antibodies against VEGF and type IV collagen (Supplementary Table 1), followed by incubation with a secondary horseradish peroxidase-conjugated antibody (1:10,000). We used the anti-VEGF antibody, which has been shown to detect VEGF165 and VEGF121 isoforms in rats (Soh et al, 1997; Chodobski et al, 2003; Zisch et al, 2003). Signals were detected by enhanced chemiluminescence (Millipore, Billerica, MA, USA) and semiquantified by densitometry. The membranes were stripped and probed with an anti-β-actin antibody to confirm even loading of proteins.
Active Matrix Metalloproteinase-9 Assay
The Fluorimetric SensoLyte™ 490 (AnaSpec Corp., San Jose, CA, USA) was used to quantify the specific enzymatic activity of active MMP-9 by using a fluorescence resonance energy transfer peptide containing a fluorescent donor and quenching acceptor (Lin et al, 2008). We measured MMP-9 activities of the samples according to the manufacturer's instructions. Briefly, the cortical tissues were homogenized in a glass homogenizer in 7 volumes of cold assay buffer. The homogenates were centrifuged at 10000 g for 15 minutes at 4°C, and the supernatants were collected. The protein concentrations of the samples were determined using a bicinchoninic acid protein assay kit. Samples (75 µg) were placed in a 96-well plate containing 50 µL of assay buffer. The proteolytic activities of MMP-9 were quantified using the fluorescence resonance energy transfer peptide and expressed as a change in fluorescence intensity at excitation of 355 nm/emission of 460 nm.
Quantification of Endothelial Barrier Antigen-Positive Vessels
To determine the number of EBA-positive vessels, tissue sections were immunostained with the EBA antibody (Supplementary Table 1) and counted as described previously (Fagan et al, 2003), with some modifications. Briefly, 6 randomly chosen nonoverlapping high-power fields (0.27 mm2, × 400) at the level of the anterior commissure of the sham-operated rats or ischemic cortex in the middle cerebral artery territory were examined at 24 hours after ischemia. Double labeling with the anti-EBA antibody and Alexa fluoro 488-conjugated lectin (Zhang et al, 2009) was also performed to investigate the reason for the loss of vessels after ischemia. These analyses were conducted blindly to the treatment information.
Therapeutic Intervention
The sample size was calculated to detect a difference between the placebo group and treatment group with 80% power (with a two-tailed α level of 0.05). We administered rabbit anti-VEGF antibody (RB-222, Lab Vision-Neomarkers, Fremont, CA, USA), which has been shown to neutralize VEGF in rat brains (Kimura et al, 2005), or rabbit anti-human IgG antibody as control (R5G10-048, OEM Concepts, Toms River, NJ, USA) to the rats. To investigate the effects of RB-222 on VEGF, tPA and either 30 or 100 µg RB-222 was administered for 30 minutes at 4 hours after ischemia. Moreover, tPA was infused in combination with either 100 µg of RB-222 or control antibody at 1 or 4 hours after ischemia for 30 minutes. We also intraperitoneally administered VEGF-receptor-2 (VEGFR2)-specific inhibitor, SU1498 (572888, Calbiochem, San Diego, CA, USA) (Warner-Schmidt and Duman, 2007) dissolved in dimethyl sulfoxide (20 mg/kg in 1 mL/kg dimethyl sulfoxide) or 1 mL/kg dimethyl sulfoxide as vehicle to the rats after the tPA infusion at 4 hours after ischemia, because a previous study showed that intraperitoneal injection of this amount of SU1498 effectively blocked VEGF in vivo (Weis et al, 2004). Although SU1498 has additional effects such as blocking the receptors for the epidermal growth factor and platelet-derived growth factor, its inhibitory effects on these receptors are known to be very weak compared with those on VEGFR2 (Strawn et al, 1996).
Statistical Analysis
All the data are presented as mean ± s.e.m. Differences in the parameters were analyzed using one-way analysis of variance followed by Tukey's post hoc test, the Kruskal-Wallis test followed by post hoc Mann-Whitney U tests, or unpaired t-test. Two-way analysis of variance was used to determine the effects of RB-222 on the optical densities of protein bands and MMP-9 activities. Differences in the frequencies were assessed with Fisher's exact test. All the tests were considered statistically significant at P values < 0.05.
Results
Hemorrhagic Transformation After the Delayed Tissue Plasminogen Activator Treatment in a Rat Thromboembolic Model
We evaluated the rat thromboembolic model, which could allow us to investigate the pathogenic mechanisms of HT after the delayed tPA treatment. There were no differences in the physiological parameters after ischemia (Table 1). In the tPA 1-hour group, tPA treatment reduced the infarct and edema volumes compared with those in the permanent ischemia group (P = 0.019 and P =0.034, respectively) without increasing the hemoglobin concentration of the lysate (Figures 1C–1E) and improved motor scale at 24 hours after ischemia compared with that in the permanent ischemia group (P < 0.001) (Figure 1F). However, in the tPA 4-hour group, the infarct and edema volumes were not reduced compared with those in the permanent ischemia group in spite of the recovery of CCBF (Figures 1A, 1C, and 1D). Macroscopic HT was observed only in the brains of the tPA 4-hour group and was observed in > 80% of rats in this group (Figure 1B, arrow). In accordance with this result, the hemoglobin concentration of the lysate of the cortex from the tPA 4-hour group was higher than that from the permanent ischemia group and tPA 1-hour group (P < 0.001 and P < 0.001, respectively) (Figure 1E). Tissue plasminogen activator 4-hour group showed worsening of motor function at 24 hours after ischemia compared with tPA 1-hour group (P < 0.001) (Figure 1F). In addition, the mortality rate (percentage of grade 4) of tPA 4 hour group (59.0%) was markedly higher than that of the permanent ischemia group (17.4%) (P = 0.006). Rats in the tPA 4-hour group produced HT and showed increased mortality rate, suggesting that the therapeutic time window of this model was < 4 hours after ischemia.

Rat thromboembolic model produces hemorrhagic transformation (HT) and shows increased mortality rate after delayed 4 hours treatment of tissue plasminogen activator (tPA). (
Delayed Tissue Plasminogen Activator Treatment at 4 hours After Ischemia Promotes the Expression of VEGF165 and VEGF121 in Blood-Brain Barrier
While VEGF was undetectable in the brains from sham-operated rats, its expression was observed in the periinfarct area of the brains from the permanent ischemia group at 24 hours after ischemia (Figure 2A), especially in the arterioles (Figure 2B). Vascular endothelial growth factor expression was more prominent in arterioles of the tPA 4-hour group than in the permanent ischemia group or the tPA 1-hour group (Figure 2B), while it was expressed in a limited number of capillaries (Supplementary Figure 1). Vascular endothelial growth factor expression was observed in endothelial cells, pericytes, and astrocytic foot processes (Figures 2C and 2D), which constitute BBB. In all the ischemic groups, VEGF expression was also observed in the neurons and astrocytic cell bodies, but not in microglia (Supplementary Figure 2), while it was not observed in the brains from sham-operated rats (data not shown).
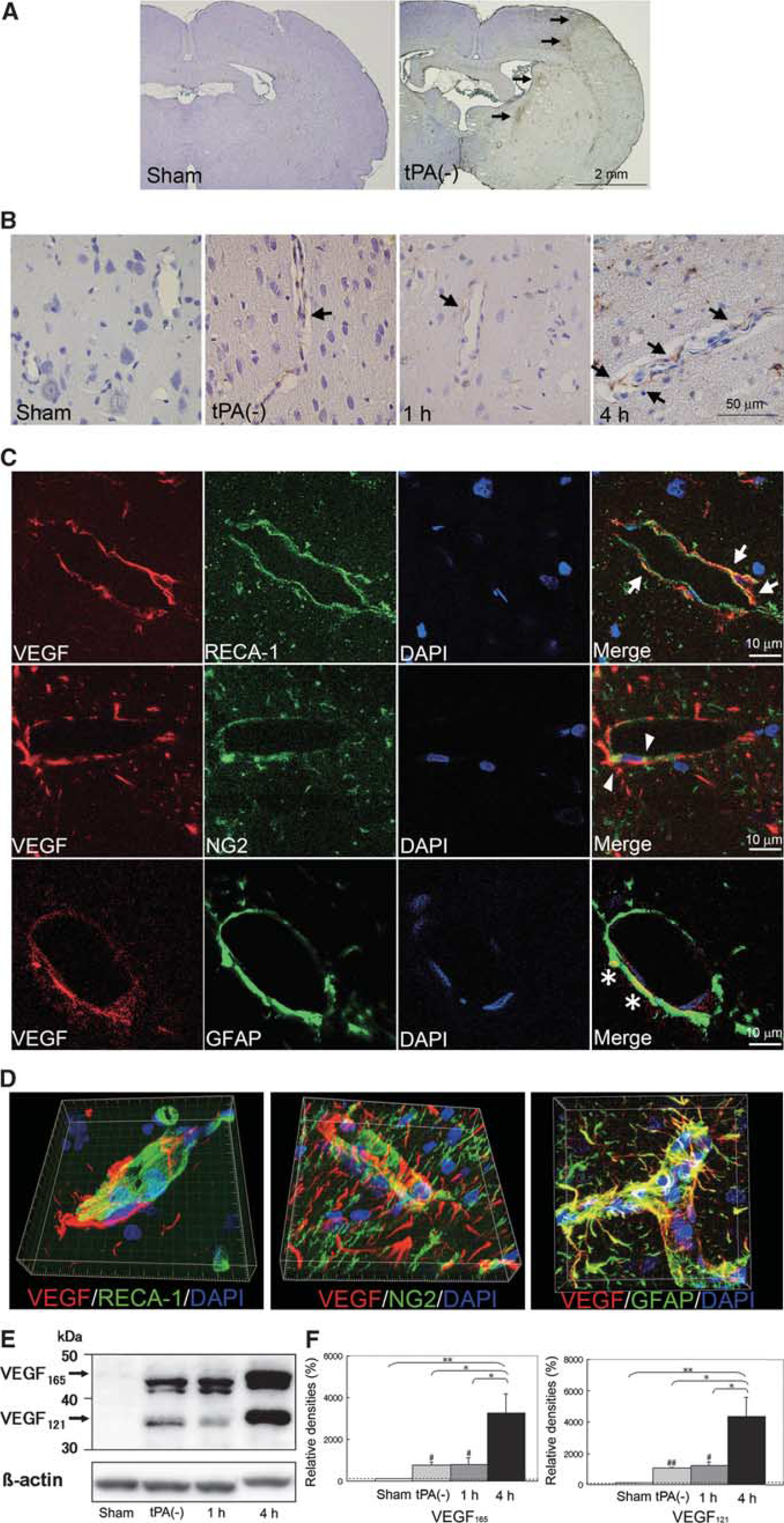
Tissue plasminogen activator (tPA) treatment at 4 hours after ischemia promotes expression of vascular endothelial growth factor (VEGF) in the blood-brain barrier (BBB). (
On the basis of the molecular weights determined by immunoblotting, we speculated that VEGF165 (46 kDa) and VEGF121 (38 kDa), which are the secretory isoforms of VEGF, were induced after ischemia (Figure 2E). The levels of both the isoforms in the tPA 4-hour group were higher than those in the permanent ischemia group (P = 0.030 and P =0.026, respectively) or the tPA 1-hour group (P = 0.032 and P = 0.035, respectively) (Figures 2E and 2F). There were significant differences in the expression level of VEGF165 and VEGF121 between the sham-operated group and the permanent ischemic group (P = 0.015 and P = 0.004, respectively) as well as between the sham-operated group and the tPA 1-hour group (P = 0.047 and P = 0.027, respectively).
Activation of Matrix Metalloproteinase-9 and Degradation of Blood-Brain Barrier Components via Vascular Endothelial Growth Factor Signaling Pathway After Tissue Plasminogen Activator Treatment
To directly determine the MMP-9 activities after ischemia, we measured the proteolytic activities of MMP-9 using the fluorescence resonance energy transfer peptide. We found that the MMP-9 activities at 24 hours after ischemia increased, especially in the tPA 4-hour group (Figure 3A). The levels of MMP-9 activities in the tPA 4-hour group were higher than those in the sham-operated group and tPA 1-hour group (P = 0.011 and P =0.031, respectively). Significant differences were also noted in the MMP-9 activity between the sham-operated group and the permanent ischemic group (P = 0.028) as well as between the sham-operated group and the tPA 1-hour group (P = 0.037). We also confirmed the degradation of BBB components such as EBA (Figures 3B and 3C) and type IV collagen (Figures 3D and 3E) in the ischemic region, especially in the tPA 4-hour group. Double labeling with the anti-EBA antibody and Alexa fluoro 488-conjugated lectin revealed that the loss of EBA expression was not due to the complete loss of vessels but due to the loss of EBA expression on the vessels (Supplementary Figure 3). This finding is suggestive of potentially leaky vessels after delayed tPA treatment.

Tissue plasminogen activator (tPA) treatment at 4 hours after ischemia promotes degradation of blood-brain barrier (BBB) components. (
We next examined the effect of intravenous administration of an anti-VEGF antibody, RB-222, on the expressions of VEGF, MMP-9 activities, and degradation of BBB components. Immunoblotting analysis revealed that 100 µg RB-222, but not 30 µg RB-222, inhibited VEGF expression compared with that in the group administered the control antibody (Supplementary Figure 4). Therefore, we selected 100 µg RB-222 for combination treatment with tPA in the subsequent experiments. Immunohistochemical analyses revealed that RB-222 administered at 4 hours after ischemia dramatically reduced VEGF expression in the arterioles and astrocytes at 24 hours after ischemia in the tPA 4-hour group (Figure 4A; Supplementary Figure 5). Immunoblotting confirmed that administration of RB-222 reduced the levels of both VEGF165 and VEGF121 in both the tPA 1-hour (P = 0.003 and P = 0.002, respectively) and tPA 4-hour groups (P = 0.003 and P = 0.002, respectively) compared with that of the group treated with the control antibody (Figures 4B–4D). Immunoblotting analysis showed that RB-222 continued to inhibit VEGF expression for 2 weeks (data not shown). RB-222 attenuated MMP-9 activities in both the tPA 1- and 4-hour groups (P = 0.033 and P = 0.033, respectively) (Figure 4E). RB-222 also attenuated the degradation of EBA (Figures 5A and 5B) and type IV collagen (Figures 5C and 5D) after ischemia. In addition, SU1498 attenuated the degradation of EBA compared with vehicle (Supplementary Figure 6).
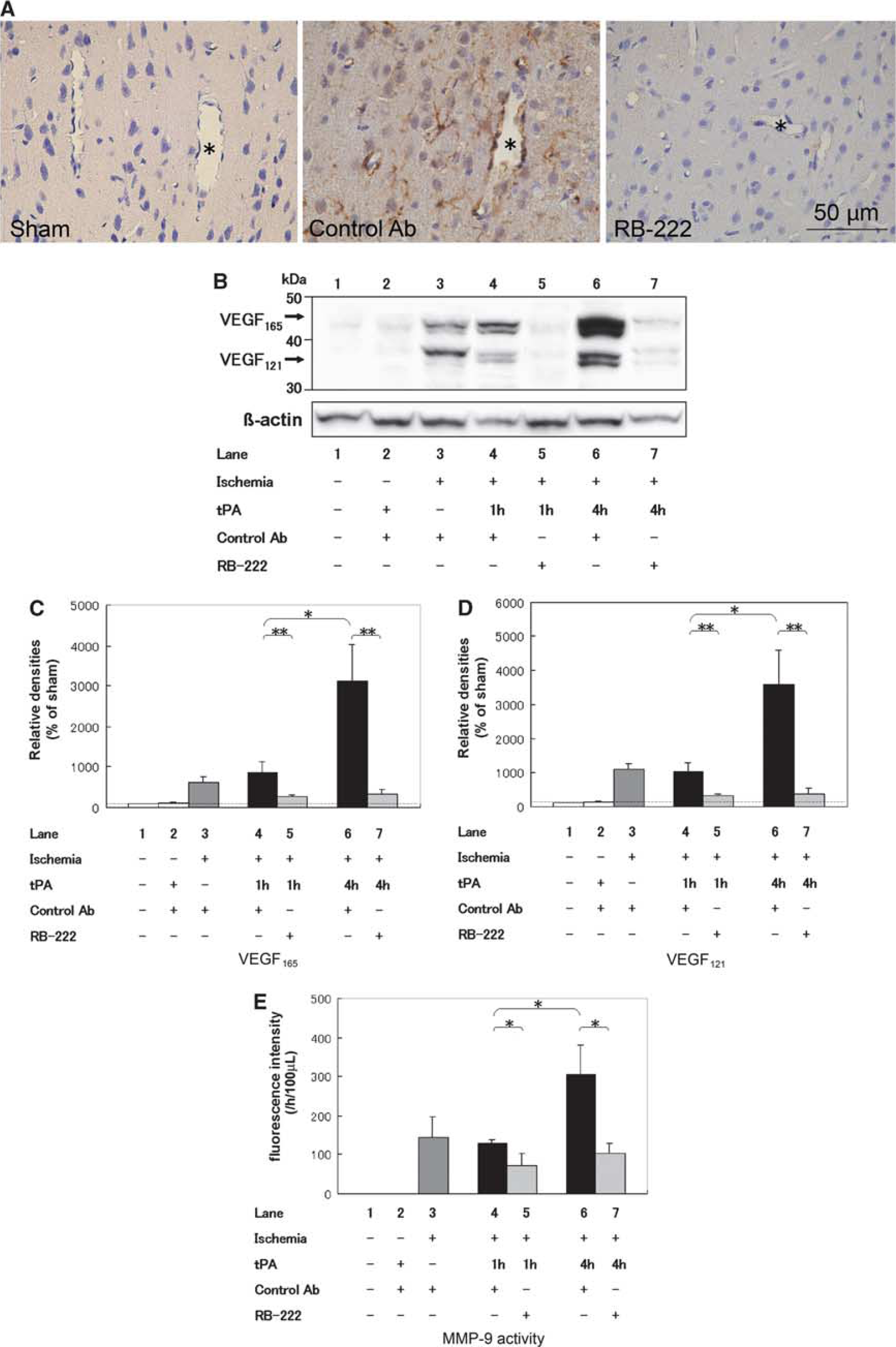
RB-222 attenuates vascular endothelial growth factor (VEGF) expression and matrix metalloproteinase-9 (MMP-9) activation after tissue plasminogen activator (tPA) treatment. (

RB-222 attenuates the degradation of blood-brain barrier (BBB) components after tissue plasminogen activator (tPA) treatment. (

RB-222 or vascular endothelial growth factor receptor-2 (VEGFR2) inhibitor, SU1498 attenuates hemorrhagic transformation (HT) after tissue plasminogen activator (tPA) treatment. (
Inhibition of Vascular Endothelial Growth Factor Signaling Pathway Suppresses Hemorrhagic Transformation Associated with Tissue Plasminogen Activator Treatment
To investigate the effect of RB-222 on BBB disruption and outcomes after tPA treatment, we evaluated infarct and edema volumes, hemoglobin concentrations of the lysate of the ischemic hemisphere, and motor scales at 24 hours after ischemia between the rats treated with tPA in combination with RB-222 or control antibody at 4 hours after ischemia. The rats treated with tPA and RB-222 showed reduced hemoglobin concentration compared with those treated with tPA and control antibody (P = 0.013), although they did not show reduced infarct or edema volumes (Figures 6A–6C). Furthermore, the rats treated with tPA and RB-222 showed improved motor scales compared with those of the groups treated with tPA and control antibody (P = 0.001) or those treated with control antibody alone (P = 0.038) (Figure 6D). Additionally, at 24 hours after ischemia, the mortality rate of the tPA 4-hour group treated with RB-222 was lower than that of the tPA 4-hour group treated with control antibody alone (P = 0.007). Pathological examination of the rats treated with tPA and RB-222 revealed no inflammatory/immune responses such as increased number of microglia/macrophages in the brain as well as no immune complex deposition in their livers, spleens, and kidneys (data not shown).
Additionally, the group treated with tPA and SU1498 at 4 hours after ischemia showed reduced hemoglobin concentration compared with that of the group treated with tPA and vehicle (P = 0.005) (Figure 6G), although the former group did not show reduced infarct and edema volumes (Figures 6E and 6F) and improvement of motor outcome (Figure 6 H) at 24 hours after ischemia compared with that treated with tPA and vehicle.
Discussion
We investigated the effects of tPA thrombolysis on VEGF expression in BBB using the rat thromboembolic model. After ischemia, VEGF was mostly associated with arterioles. This association is interesting because a previous study suggests the importance of arterioles in BBB function (Petito and Levy, 1980). In contrast, VEGF was undetectable in BBB of the cerebral cortex of sham-operated rats, which was consistent with the previous reports that detectable amounts of VEGF were not observed in rat BBB under physiological conditions (Kovács et al, 1996). Vascular endothelial growth factor was expressed in BBB after ischemia, and the isoforms of VEGF were estimated to be VEGF165 and VEGF121. The mechanism by which the delayed 4-hour treatment induced higher VEGF expression in BBB remains to be elucidated. The increased level of VEGF (Figures 2 and 4) might be explained by the increased production of VEGF in the brain, as we used perfused rat brains. As VEGF expression was not induced by tPA treatment alone in our model, we speculated that ischemia/reperfusion injury caused by tPA treatment promoted VEGF secretion from surviving cells in and around the ischemic regions. We also speculated that VEGF was secreted by endothelial cells, astrocytes, and neurons for the following reasons: VEGF has been shown to be secreted by these cells subjected to hypoxia in vitro (Namiki et al, 1995; Ijichi et al, 1995; Ogunshola et al, 2002), and VEGF expression was also observed in these cells after ischemia in our model.
To test the hypothesis that activation of VEGF signaling pathway in BBB promoted HT after tPA treatment, we investigated the effects of RB-222 on VEGF expression, MMP-9 activities, BBB component degradation, and HT. RB-222 efficiently eliminated both the secretory isoforms of VEGF after ischemia and suppressed MMP-9 activation, degradation of EBA and type IV collagen, and induction of HT, although it was not sufficient to suppress brain edema. Taken together, these findings showed that the activation of VEGF signaling pathway in BBB promoted HT, and that anti-VEGF antibody can attenuate HT via inhibition of MMP-9 activity. The effect of RB-222 on HT might be further supported by the pharmacological effects of humanized anti-VEGF antibody, bevacizumab, which suppresses hemorrhage in ophthalmologic diseases, including diabetic retinopathy (Ishikawa et al, 2009) or neovascular glaucoma (Wakabayashi et al, 2008). In addition, anti-VEGF antibody can be administered intravenously, enabling the combination treatment with tPA. These findings raise the possibility that the anti-VEGF antibody is a candidate for adjunctive thrombolytic therapy, which could extend the therapeutic time window of tPA via suppression of HT.
It is interesting that both RB-222 and SU1498 with tPA reduced hemoglobin concentration but did not decrease infarct or edema volumes. We considered that RB-222 could enter the brain, because VEGF expression in the brain decreased after intravenous administration of the anti-VEGF antibody, as revealed by the immunoblotting and immunohistochemical analyses. Inhibition of VEGF probably does not provide a neuroprotective effect, and factors other than VEGF may also be involved in the increased vascular permeability (Yepes et al, 2003; Su et al, 2008). We speculated that the improvement of outcome by RB-222 treatment was caused by the inhibition of hemorrhage. In clinical practice, inhibition of VEGF may enable prevention of HT, which may allow the extension of the therapeutic time window for tPA.
To confirm the involvement of VEGF signaling pathway in HT, we investigated whether HT after the tPA treatment was suppressed by SU1498, which is a small molecule inhibitor that freely traverses the membrane and binds the ATP-binding pocket of VEGFR2, preventing phosphorylation and downstream signaling (Warner-Schmidt and Duman, 2007). A recent study showed that SU1498 prevented the disruption of in vitro model of BBB after oxygen-glucose deprivation (Al Ahmad et al, 2009). In the present study, SU1498 attenuated HT after tPA treatment, although it did not significantly improve motor outcome or mortality in the ischemic rats. This finding might be explained by assuming that SU1498 less effectively suppresses VEGF signaling pathway compared with anti-VEGF antibody or that VEGF/VEGF-receptor-1 signaling pathway also exerts an influence on the outcome (Hiratsuka et al, 2002).
The present study has limitations. First, the long-term effects of VEGF inhibition after ischemia remain to be elucidated. Because the administration of VEGF after subacute phase of ischemia has been shown to promote angiogenesis, neurogenesis, and favorable outcome in the rat suture occlusion model (Sun et al, 2003), VEGF is considered to have biphasic roles in ischemic stroke pathophysiology. Recently, the biphasic roles of molecular targets for the treatment of ischemia during a different phase are receiving increasing attention (Lo, 2008); for example, the N-methyl-
In conclusion, HT after tPA treatment may be related to the activation of the VEGF signaling pathway in the BBB. Inhibition of VEGF signaling pathway may be a promising therapeutic strategy for attenuating HT after tPA treatment.
Footnotes
Acknowledgements
The authors thank Dr Seiji Okubo and Toshiki Inaba (Division of Neurology, Second Department of Internal Medicine, Nippon Medical School) for their technical advice, Dr Riuko Ohashi and Professor Makoto Naito (Department of Cellular Function, Division of Cellular and Molecular Pathology, Niigata University) for their support in the confocal microscopy studies, and Dr Kenshi Terajima (Department of Medical Informatics, Niigata University Medical and Dental Hospital, Niigata University) for expert statistical advice.
The authors declare no conflict of interest.