
Abstract
Disruption of the blood–brain barrier (BBB) has an important part in cellular damage in neurological diseases, including acute and chronic cerebral ischemia, brain trauma, multiple sclerosis, brain tumors, and brain infections. The neurovascular unit (NVU) forms the interface between the blood and brain tissues. During an injury, the cascade of molecular events ends in the final common pathway for BBB disruption by free radicals and proteases, which attack membranes and degrade the tight junction proteins in endothelial cells. Free radicals of oxygen and nitrogen and the proteases, matrix metalloproteinases and cyclooxgyenases, are important in the early and delayed BBB disruption as the neuroinflammatory response progresses. Opening of the BBB occurs in neurodegenerative diseases and contributes to the cognitive changes. In addition to the importance of the NVU in acute injury, angiogenesis contributes to the recovery process. The challenges to treatment of the brain diseases involve not only facilitating drug entry into the brain, but also understanding the timing of the molecular cascades to block the early NVU injury without interfering with recovery. This review will describe the molecular and cellular events associated with NVU disruption and potential strategies directed toward restoring its integrity.
Keywords
Introduction
Cerebral blood vessels form the major interface between the blood and brain tissues, providing the basis for the immunological sequestration of the brain tissues and the prevention of perturbation of the neuronal microenvironment by fluctuations in the systemic circulation. They are dynamic, highly metabolic structures that resemble epithelial membranes in their high electrical resistance and ability to form fluids through the action of an ATPase pumping mechanism. Capillaries are critical for delivering the essential nutrients and maintaining a constant supply of oxygen. Much of our knowledge of the physiology of the blood–brain barrier (BBB) was discovered before the modern era of neuroimaging and molecular biology and remains the foundation of clinical practice. However, discoveries in the past 25 years have added a new dimension to our understanding of the what is now referred to as the neuro(glio)vascular unit (NVU).
Disruption of the BBB occurs in many neurological disorders (Table 1). In some situations, extrinsic systemic factors, such as infection and autoimmune processes, affect blood vessels, initiating the damage, while in others, including cerebral ischemia, the blood vessels are damaged secondary to the injury by the activation of intrinsic cellular mechanisms that are referred to collectively as neuroinflammation. While the cerebral vasculature provides an important protective role in maintaining homeostasis essential to neuronal function, the BBB also prevents the entry of drugs, making treatment of disorders of the central nervous system more difficult than in the systemic circulation where circulating blood and entrained molecules access tissues via fenestrated capillaries.
Neurological disorders with disruption of the neurovascular unit

Early physiological studies in animals showed the high electrical resistance of the cerebral capillary tight junctions and revealed the complex transport mechanisms involved in the movement of glucose and essential amino acids into the brain. However, the recent emphasis in BBB studies is on the molecular biology of the blood vessels and surrounding cells and the applications to human disease. Isolation and cloning of the proteins that form the tight junctions between the cerebral endothelial cells was a major breakthrough in understanding normal BBB function. With those molecular biology tools, it was possible to unravel the mechanisms of injury due to proteases and free radicals. This involved identification of the important roles played by cyclooxygenases (COXs), matrix metalloproteinases (MMPs), free radicals of nitrogen and oxygen, hypoxia-inducible factor-1α (HIF-1α), and the family of aquaporins (AQPs). Finally, discoveries in immunology on the role of selectins, integrins, and other adhesion molecules in the trafficking of white blood cells across endothelial cells have informed thinking about immunological processes involved in pathological states. This review will focus on the applications of these new ideas to the study of the NVU in neurological disorders. Several excellent reviews should be consulted for information on the basic science of the NVU (Iadecola and Nedergaard, 2007; Neuwelt et al, 2011; Zlokovic, 2010).
Cellular and molecular basis of vascular permeability
Tight junction proteins, occludin and claudin, were identified and cloned and shown to be the major proteins that self-assemble into ‘zip-locked’ structures, forming the physical barrier in the endothelial cell clefts (Furuse et al, 1998; Hirase et al, 1997). Occludins are a family of transmembrane proteins with four transmembrane domains, two extracellular loops, and two intracellular domains. They interact directly with intracellular proteins, zonula occludens, which are connected to actin. Claudins are transmembrane proteins that are the main constituent of the tight junctions; the claudin family has a number of members. Junctional adhesion molecules have a single transmembrane domain, an extracellular domain containing two Ig-like motifs, and a cytoplasmic tail (see for review Hawkins and Davis, 2005).
Surrounding the abluminal surface of the endothelial cell is a basal lamina composed of type IV collagen, fibronectin, heparan sulfate, and laminin, which functions as a charge and molecular weight barrier and interacts in complex ways with integrins to regulate permeability and cellular transport across the BBB (Milner et al, 2008). Pericytes are macrophage-like cells with smooth muscle properties that are embedded in the basal lamina around the blood vessels (Dore-Duffy, 2008). They are an important component of the NVU, which regulate permeability by release of vasoactive substances; since they are next to the vessels they have immediate access to both the basal lamina and the endothelial cells. Pericytes are recruited to developing blood vessels and contribute to the formation of tight junctions (Daneman et al, 2010). Another interesting study of pericytes showed that they decrease with age, paralleling an increase in BBB permeability (Bell et al, 2010).
Proteases and the neurovascular unit
The final common pathway for BBB disruption involves neutral proteases and free radicals that are induced by the inflammatory response to the injury. Proteases regulate a large number of molecular events at the cell surface and inside the cell (Rivera et al, 2010). They activate growth factors to promote normal development and along with tissue inhibitors to metalloproteinases-3 (TIMP-3) control the death receptors on the cell surface that are involved in apoptosis (Cunningham et al, 2005). Under normal conditions, proteases remodel the extracellular matrix and participate in vasculogenesis, angiogenesis, and neurogenesis. While they participate in the damage to the tissues in the early stages of an injury, they later become important contributors to the repair process. Cyclooxygenases are another important family of inflammatory enzymes. Cyclooxygenase-1 is a constitutive enzyme, while COX-2 is inducible and contributes to BBB damage as part of a secondary inflammatory response from 24 to 72 hours after the initial insult (Nagayama et al, 1999).
Neuroimaging of the neurovascular unit
Quantification of BBB permeability by magnetic resonance imaging (MRI) was achieved by adapting to MRI, the graphical autoradiographic method developed for use in animals by Patlak et al (1983). The accuracy of the MRI measurements was shown in animal studies directly comparing permeability constants obtained from autoradiograms with MRIs from the same animal (Ewing et al, 2003). Dynamic contrast-enhanced MRI (DCEMRI) method can be used in studies of humans, and it is possible to quantify abnormal BBB permeability in chronic diseases (Figure 1) (Taheri et al, 2011).
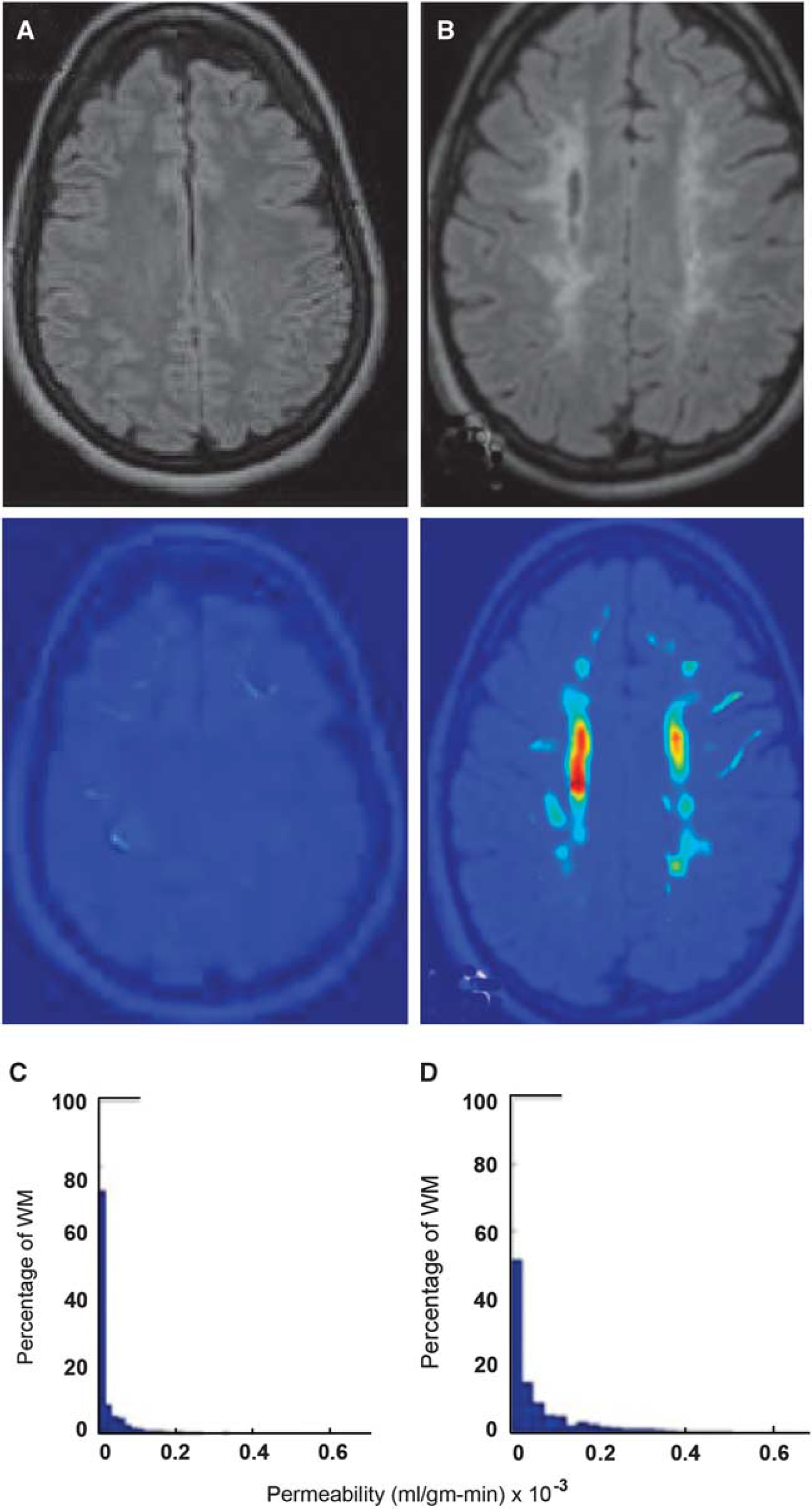
Density distribution of permeability values for white matter (WM) voxels of a control and a vascular cognitive impairment patient. (
Measurements of BBB permeability can also be performed during computed tomography (CT) angiography using the Patlak graphical method. Computed tomography has the advantage over MRI in that the studies can be performed more rapidly, allowing measurements to be made in acute stroke before treatment with tPA (tissue plasminogen activator). In one study, CT showed disruption of the BBB in the early stages of stroke, which was proposed as an aid in deciding when it was safe to give tPA, which has a risk of inducing hemorrhage (Hom et al, 2011). Another CT study showed that increased permeability indicated a higher risk for hemorrhagic transformation and suggested that it could be used to select patients for hemicraniectomy (Figure 2) (Bektas et al, 2010). The disadvantage of the CT method is the relatively large amount of radiation delivered to the patient, which is not generally a problem in the elderly patient, who is getting a CT angiogram as part of the diagnostic work-up, but would be one in younger individuals. In addition, the risk of a life-threatening allergic reaction to the contrast agent is higher with CT contrast than MRI.
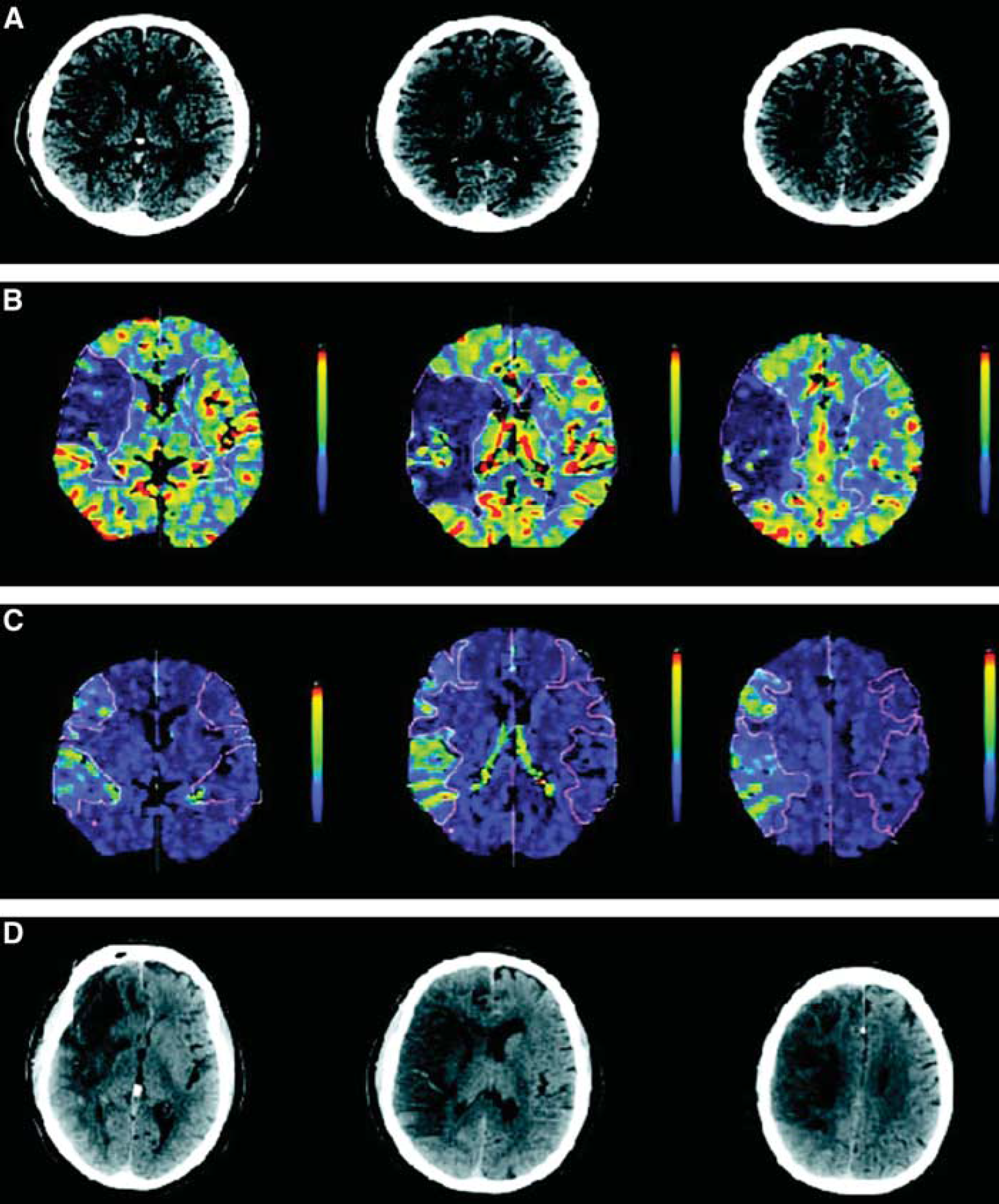
A 75-year-old right-handed man who presented with sudden onset of left hemiparesis and dysarthria. (
Disruption of the blood–brain barrier in ischemia/hypoxia and hemorrhage
Cerebral ischemia with reperfusion in animals leads to a biphasic disruption of the BBB (Kuroiwa et al, 1985). The initial opening is reversible and associated with activation of MMP-2 by membrane-type MMP (MMP-14); the activated MMP-2 attacks the tight junction proteins (Yang et al, 2007). Hypoxia-inducible factor-1α has an essential role in cellular and systemic oxygen homeostasis by regulating the expression of genes important in glycolysis, erythropoiesis, angiogenesis, and catecholamine metabolism (Semenza, 2010). Hypoxia-inducible factor-1α-deficient mice were protected from hypoxia-induced cell death, suggesting that decreasing the level of HIF-1α can be neuroprotective (Helton et al, 2005). Under conditions of low oxygen, HIF-1α accumulation leads to the expression of the fur gene and transcription of the protein, Furin, which activates MMP-14, resulting in the activation of the constitutive enzyme, MMP-2 (Figure 3). Because the brain has large amounts of MMP-2 in the inactive form, the rate-limiting step for conversion to the active form is the activation of MMP-14. These reactions occur within hours after the ischemia, but because the proMMP-2 is tethered to the membrane by MMP-14, the reactions are constrained to the area of activation. During recovery, HIF induces vascular endothelial growth factor and transforming growth factor-β; both are important in neurogenesis and angiogenesis. Hypoxia-inducible factor-1α is elevated in acute ischemia and reverts to low levels after the acute insult. In more chronic situations, such as intermittent hypoxia, HIF-1α may remain elevated for longer periods (Yuan et al, 2008). How this affects the activation of MMP-2 and other MMPs remains to be resolved.
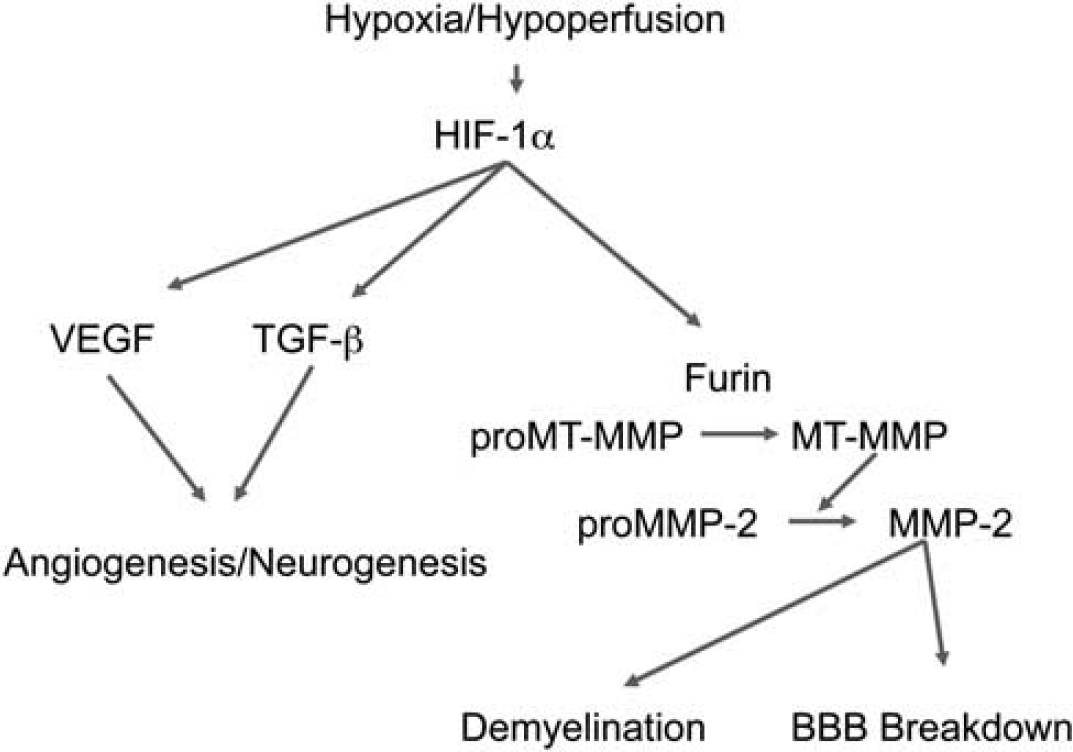
Hypoxic hypoperfusion in acute and chronic ischemia induces hypoxia-inducible factor-1α (HIF-1α), which induces the fur gene to transcribe the convertase, furin. Activation of proMMP-14 (membrane-type MMP) is necessary for the activation of proMMP-2, which attacks the tight junction proteins and basal lamina opening the blood–brain barrier (BBB). As a consequence of the activation of the MMPs, myelin is broken down. MMP, matrix metalloproteinases.
The second opening of the BBB occurs 24 to 48 hours after reperfusion depending on the length of ischemia; the longer the ischemia, the earlier and more disruptive the BBB opening (Rosenberg et al, 1998). This second phase is mediated by the inducible MMPs, MMP-3 and MMP-9, which are induced by cytokines during inflammation. Cyclooxygenase-2 is also an important mediator of the second, more destructive phase of BBB damage. Because the inducible MMPs and COX-2 are free in the extracellular space, their action is more destructive and the second opening of the BBB is more damaging.
Cytokines induce the expression of inflammatory MMPs and COX-2 through the action of nuclear-factor-κB and the activator protein-1 gene transcription sites. This opening of the BBB is due to intrinsic cellular activity that is most likely initiated by activation of pericytes, microglia, and astrocytes by the hypoxia. Once the BBB is damaged, however, neutrophils and monocytes enter the brain, bringing with them another source of MMPs and toxic blood products that amplify the injury.
Aquaporins are pore-forming molecules located in astrocytic end feet that facilitate the passage of water molecules across the BBB. Deletion of AQP4 reduces edema in models in which cytotoxic edema is the pathophysiological mechanism. However, in conditions in which vasogenic edema is significant, AQP4 deletion exacerbated the brain edema. This suggests that the AQP4 functions as a passive pore, allowing water to follow pressure gradients to remove extracellular fluid and resolve vasogenic edema (Zador et al, 2007).
Oxidative stress damages endothelial cells of the BBB and contributes to vasogenic edema. The superoxide radical (O2−.) has been identified as the primary reactive oxygen species involved in increased vascular permeability and edema formation in global and focal cerebral ischemia, cold-induced brain injury, and brain tumors. Scavenging O2−. radicals using recombinant superoxide dismutase (SOD) or polyethylene glycol-SOD reduces ischemia-induced BBB injury and vasogenic edema. Superoxide dismutase 1-overexpressing mice also have reduced activation of MMP-9 by reactive oxygen species, which may be involved in early BBB disruption and progressive striatal damage induced by the mitochondrial excitotoxin, 3-nitropropionic acid (Kim et al, 2003).
An indirect indication of the leakiness of blood vessels in the acute stage of stroke has been the qualitative visualization of Gd-DTPA. Several hours after injection of Gd-DTPA at admission, fluid-attenuated inversion recovery images showed disruption of the BBB, which appeared as enhancement in sulci over the infarct area. This was termed, hyperintense acute reperfusion marker, and was found in one-third of ischemic stroke patients; those with the sign had a higher risk of hemorrhagic transformation and worse clinical outcome (Henning et al, 2008).
Damage to the BBB is a factor in the growth of intracerebral hemorrhage, which can occur in the first 24 hours after the onset of the bleed (Brott et al, 1997). Agents that increase the potential for clot formation and probably act by closing the BBB have been used in treatment. Factor VII, which promotes clotting, reduced growth of the hematoma in the initial trial, which could not be confirmed in a second study (Mayer et al, 2008). The extent of hemorrhagic transformation and intraparenchymal hemorrhage after stroke correlated with MMP-9 elevation in the blood (Montaner et al, 2001).
Blood–brain barrier disruption in multiple sclerosis
Multiple sclerosis (MS) is a disease of young adults that begins with inflammation in the venules. This is an extrinsic BBB disruption pattern where the initial injury occurs to the blood vessels, allowing T and B cells to cross the BBB (Miller et al, 2003). Evidence from the experimental animal model, EAE (experimental allergic encephalomyelitis), which is caused by the injection of myelin basic protein into the footpad, suggests that it is an autoimmune disease. The initial pathology in EAE is disruption of the BBB with exudation of fibrin, which causes an inflammatory reaction with demyelination (Paterson, 1976). Although EAE is a monophasic illness as opposed to MS and the offending antigen remains unknown, it is the model used to study the immunology of MS and to test treatments.
Current concepts of pathophysiology of MS consider it a heterogeneous disease (Lucchinetti et al, 2000). Recent data suggest that in a subset of MS patients, the brain lesions show profound similarities to tissue alterations found in vascular diseases, suggesting that a hypoxia-like metabolic injury is occurring in the inflammatory brain lesions. Both, vascular pathology as well as metabolic disturbances may be responsible for apoptosis of oligodendrocytes (Henderson et al, 2009; Lassmann, 2003).
Early pathologists championed the concept of vascular lesions being an important component of the MS attack. Dawson (1916) showed that inflammatory cells clustered around veins in the center of the demyelinated plaque. Putnam and Adler (1937) carefully reconstructed serial brain sections from MS patients, and showed that fibrin deposits surrounded the inflamed vessels. Magnetic resonance imaging has confirmed these early pathological studies. Correlation of venous pathology with MS lesions is possible with MR venography, which takes advantage of the difference between oxygenated and deoxygenated blood to visualize venous blood. In most MS lesions, venograms show a vein in the center of the lesions. This pattern is not specific for MS since a similar pattern with venule involvement occurs with hypoxic ischemic white matter lesions. However, in contrast to MS lesions, ischemic white matter lesions showed no consistent relationship to the shape and location of the veins (Tan et al, 2000). A recent study using a 7-T MRI showed that T2∗-weighted images can reliably distinguish all patients with clinically definite MS. Small venules in the center of lesions are visualized using T2∗-weighted MRI because of the paramagnetic effect of deoxyhemoglobin. Those patients have >40% of the MS lesions in a perivenous location, while in those without clinical MS, <40% of the lesions appeared perivenous (Figure 4) (Tallantyre et al, 2011).
The 7-T T2∗-weighted magnitude images were viewed in orthogonal planes. For each lesion, the presence or absence of a central vein was noted. The proportion of perivenous lesions in individual patients with multiple sclerosis (MS) (mean 80%, range 53% to 100%) was consistently much higher than in individual subjects without MS (mean 16%, range 0% to 34%; (
Blood–brain barrier disruption in amyotrophic lateral sclerosis
Blood–brain barrier disruption occurs in mice with genetic defects in the SOD gene, which is model of the familial form of amyotrophic lateral sclerosis. A subset of patients with familial and sporadic amyotrophic lateral sclerosis have mutations in the gene encoding Cu, Zn SOD (SOD1). Transgenic mice that have a glycine 93 to alanine 93 (G93A) mutation develop an amyotrophic lateral sclerosis-like syndrome. Disruption of BBB and blood–spinal cord barrier was seen in areas of motor neuron degeneration in G93A mice at both early and late stages of disease, and capillary ultrastructure revealed that endothelial cell membrane and/or basement membrane damage occurred, and was followed by vascular leakage (Garbuzova-Davis et al, 2007). Superoxide dismutase 1 mutant mice with disrupted blood–spinal cord barrier had reduced levels of the tight junction proteins zonula occludens 1, occludin and claudin-5 between endothelial cells, which caused microhemorrhages and release of neurotoxic hemoglobin-derived products. These changes in the blood vessels, which reduced microcirculation and caused hypoperfusion, were present before motor neuron degeneration and the neurovascular inflammatory response occurred (Zhong et al, 2008).
Blood–brain barrier dysfunction in vascular cognitive impairment
Several lines of evidence suggest that the BBB is abnormal in a subset of patients with small vessel disease secondary to hypertension and diabetes. Early studies of cerebrospinal fluid (CSF) documented increased albumin, which is an indicator of a disrupted BBB (Skoog et al, 1998; Wallin et al, 1990). Autopsy studies in patients with vascular cognitive impairment showed increases in HIF-1α in the affected white matter supporting a role of hypoxia in the process that leads to death of the oligodendrocytes and white matter gliosis (Fernando et al, 2006). Autopsy studies of patients with Binswanger's disease showed the presence of serum proteins in the brain (Akiguchi et al, 1998). Several etiologies are proposed to explain the changes in the white matter. Silent strokes are suspected in many cases, and may be the initiating event particularly when hypertension is damaging the blood vessels (Vermeer et al, 2007). An alternative mechanism is disruption of the NVU with vasogenic edema (Feigin and Popoff, 1963). Inflammation as a possible cause of the BBB damage has been proposed because of the presence of MMP-2 in reactive astrocytes and MMP-3 in microglia/macrophages (Rosenberg et al, 2001).
Vascular cognitive impairment has multiple etiologies, which can be separated into large and small vessel disease. Multiple strokes from large vessel thromboses or emboli affect primarily the cortex, while small vessel disease is more commonly found in lacunar strokes and in subcortical ischemic vascular disease or Binswanger's disease. Demyelination in Binswanger's disease can be extensive and growth of the white matter damage occurs slowly over time sparing the subcortical U-fibers. This pattern of demyelination is more consistent with an inflammatory response secondary to blood vessels damaged by hypertension, diabetes, and hereditary disorders. Protease-secreting macrophages in the form of pericytes around blood vessels and recruited microglia/macrophages release the proteases that attack the BBB and break down myelin.
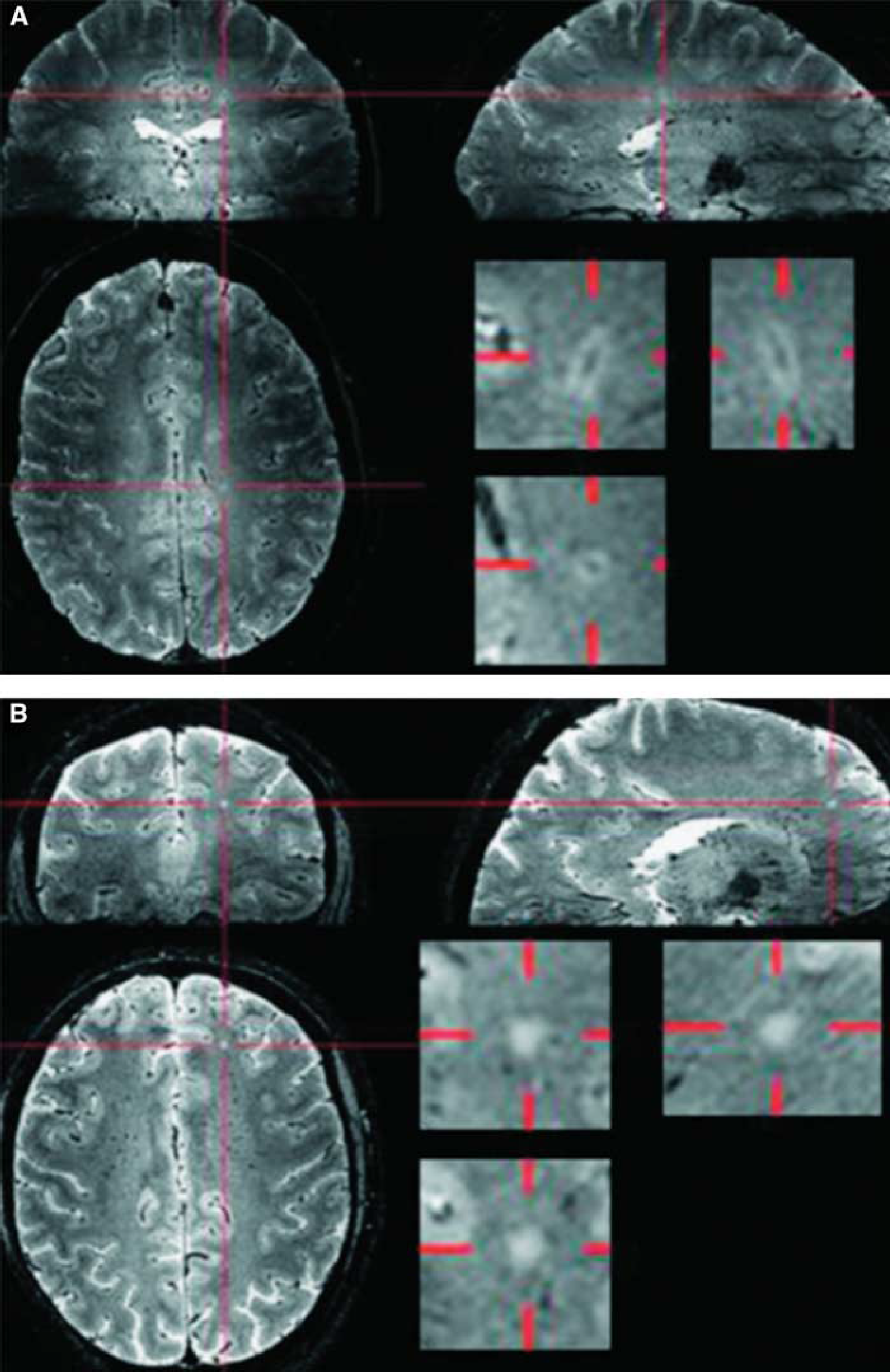
Recent studies using the DCEMRI method showed BBB disruption in white matter hyperintensities in vascular cognitive impairment patients that are inside the areas of white matter hyperintensities. Disruption of the BBB suggests that vasogenic edema with secondary hypoxia in the white matter may be a contributing factor in the growth of the subcortical lesions (Taheri et al, 2011). Serial MRI scans in patients with Binswanger's disease shows growth of the lesion size rather than discreet stroke-like events (Figure 5) (unpublished data).
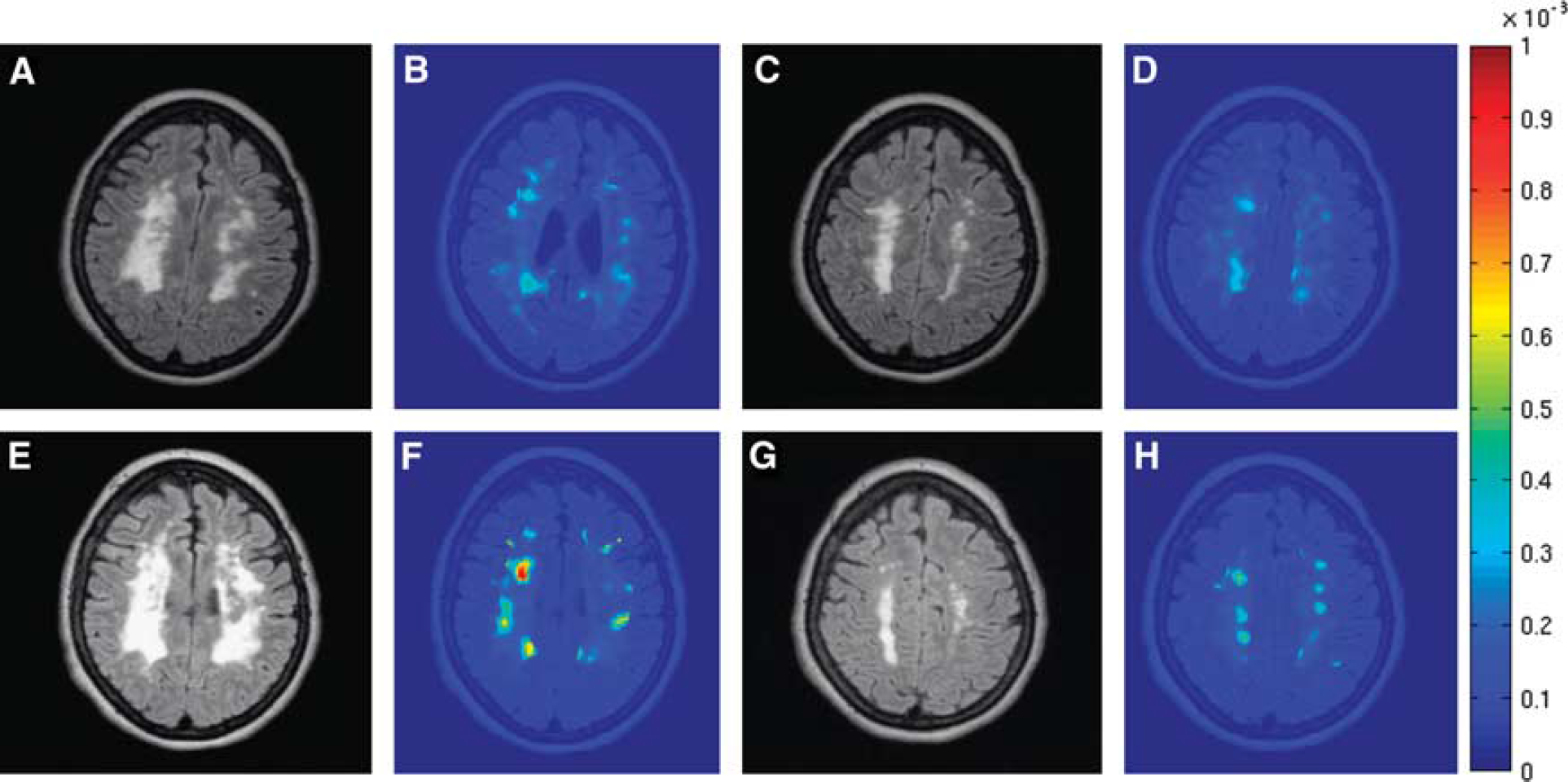
Fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) and dynamic contrast-enhanced MRIs (DCEMRIs) from a patient with Binswanger's disease. Initial MRIs (
Blood–brain barrier dysfunction in Alzheimer's disease
Several lines of evidence primarily from studies in animals implicate an abnormality of the BBB in the pathogenesis of Alzheimer's disease, but only a few qualitative studies of the BBB are available in humans. Pathological studies show that most patients with Alzheimer's disease have some form of vascular disease (Gold et al, 2007; Schneider et al, 2007; Snowdon et al, 1997). There is accumulation of amyloid in the form of plaques, which suggest that the rate of amyloid clearance across blood vessels may be less than the rate of production (Bateman et al, 2009). A number of factors determine the rate of amyloid clearance. Low-density lipoprotein receptor-related protein-1 (LRP1), a member of the low-density lipoprotein receptor family, has a major role in the cellular transport of amyloid across the BBB as well as many other roles (Zlokovic, 2010). Recent evidence indicates that LRP1 regulates the brain and systemic clearance of Alzheimer's disease amyloid β-peptides (Aβ). The cell surface LRP1 at the BBB binds Aβ from the brain interstitial fluid facilitating transcytosis across the blood vessels into the blood. Circulating soluble LRP1 normally binds 70% to 90% of plasma Aβ preventing free Aβ access to the brain, and when the LRP1 is unable to bind the amyloid protein, levels in the blood can increase.
Neurotoxicity is due to accumulation of Aβ1−42, which is linked to the apoE4 allele through the action of LRP1. Binding of apoE4 to Aβ1−42 slows the clearance of Aβ1−42 from the brain compared with the other alleles, apoE2 and apoE3. Thus, apoE isoforms differentially regulate Aβ1−42 clearance from the brain, and this might contribute to the effects of apoE genotype on the disease process in both individuals with Alzheimer's disease and animal models of the disease (Deane et al, 2008). The cell surface LRP1 in the liver mediates systemic clearance of soluble, sLRP1–Aβ, complexes and free Aβ, ultimately eliminating Aβ from the body. In addition, the kidney removes sLRP1–Aβ complexes and Aβ (Zlokovic et al, 2010). Since amyloid proteins are removed across the blood vessels, disease of the blood vessels could reduce clearance, leading to accumulation. While the importance of vascular disease in acceleration of Alzheimer's disease is established, the exact role of the blood vessels is unclear.
Amyloid angiopathy occurs commonly in Alzheimer's disease as shown by Aβ in small arteries, arterioles, and capillaries (Vinters et al, 1988). In a study in transgenic mice with genes for the familial form of Alzheimer's disease, amylodogenesis was found to mediate BBB disruption and leakiness through promoting neoangiogenesis and hypervascularity by causing a redistribution of tight junction proteins (Figure 6) (Biron et al, 2011).
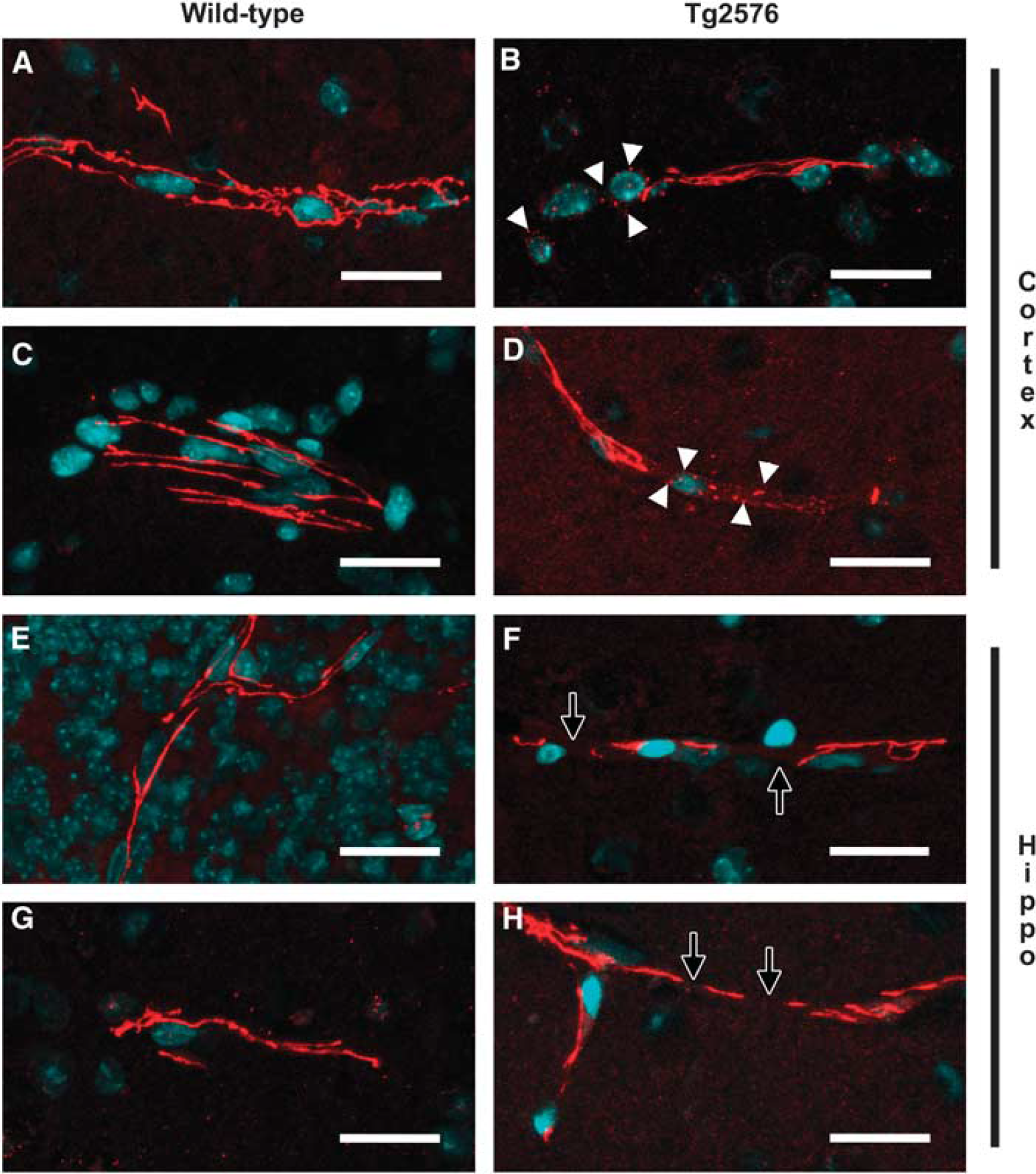
Representative confocal micrographs of cerebral blood vessels from aged Tg2576 and wild-type mice immunolabeled for either occludin or zonula occludens (ZO)-1 (red) and counterstained for DNA (blue) with TOTO-3. Blood vessels, imaged in the neocortex and hippocampus, which exhibited strong, continuous, and linear occludin (
Blood–brain barrier in bacterial meningitis, neuropathic pain, and brain trauma
Bacterial meningitis causes disruption of the BBB with exudation of white blood cells and proteins into the subarachnoid space. Infectious agents can enter the brain from several sources, including infected sinuses and through hematogenous spread. When the bacteria enter the brain tissue either through the Virchow–Robin spaces or directly across the capillaries, they cause a cerebritis, which can evolve into an abscess. Proteases are important in the disruption of the subarachnoid blood vessels with high levels of MMPs detected in the CSF, particularly in fungal meningitis (Leppert et al, 2001). Antibiotics attack the bacteria releasing proteins from the bacterial walls that can cause an inflammatory response. In children, this can lead to hearing loss. Cytokines are important in this inflammatory response. Steroids are given to reduce the inflammation and reduce the damage to the eighth nerve. Matrix metalloproteinase inhibitors are also effective in reducing the opening of the BBB and controlling the inflammatory response (Meli et al, 2006).
Pain induces a reaction in the central nervous system that disrupts the BBB. Injection of λ-carrageenan into the rat hind paw produced after 3 hours a marked change in the relative amounts of occludin isoforms and resulted in an increase in BBB permeability from the peripheral inflammatory pain (McCaffrey et al, 2008).
Traumatic brain injury causes the disruption of the BBB that is mediated by MMP-9 and AQP4 (Higashida et al, 2011). In the early stages of the injury, the mechanical disruption of the vessels causes intracerebral bleeding, which sets up an inflammatory response to the blood that continues to damage the BBB. Recruitment of macrophages and activation of the microglia results in the release of free radicals and proteases. Ventricular CSF has increased levels of MMP-3 and MMP-9, which may be important in BBB opening and hemorrhage secondary to the brain injury in patients (Grossetete et al, 2009).
Treatment in blood–brain barrier disruption and future directions
Treatments that restore BBB integrity are important in diseases that involve BBB disruption. Many agents have been shown to reduce BBB opening in MS. Matrix metalloproteinases 9 is elevated in the CSF of MS patients during an acute exacerbation. High-dose methylprednisolone reduces the MMP-9 levels in the CSF by blocking the activator protein-1 site in the MMP-9 gene, which results in closure of the BBB (Rosenberg et al, 1996). However, the effects are transient and repeated treatments with high-dose steroids have too many complications to be used routinely. A more sustained reduction of BBB damage occurs with immunomodulatory drugs, such as interferon-β and glatiramer acetate (Filippi et al, 2011; Kala et al, 2011).
The antiinflammatory tetracycline derivative, minocycline, is an inhibitor of MMPs with multiple actions, including antiinflammatory effects, and is well tolerated in low doses over long periods of time (Matsukawa et al, 2009). Minocycline reduced the BBB disruption in a pilot study of patients with relapsing-remitting MS over 24 months of open-label treatment. Despite a moderately high pretreatment relapse rate in patients in the study before treatment, no relapses occurred between months 6 and 24. The activity of MMP-9 was decreased by treatment (Yong et al, 2007). Patients taking 200 mg of minocycline for 5 days within 24 hours of an ischemic stroke showed an improvement in functional state and stroke severity over a period of 3 months compared with patients receiving placebo (Lampl et al, 2007). It has been used in the treatment of acute stroke to reduce early disruption of the BBB and extend the therapeutic window for treatment with tPA (Fagan et al, 2010). Larger, placebo-controlled trials will be needed to confirm efficacy of Minocycline in MS and stroke. Several MMP inhibitors have been tested and shown to reduce the opening of the BBB in stroke in animals, including BB-94, BB-1101, and GM6001 along with selective COX-2 inhibitors. However, none of these agents is suitable for clinical studies, and new classes of MMP and COX inhibitors are needed (Hu et al, 2007).
Clearance of drugs by efflux from the brain into the blood is facilitated by a series of enzymes, the most important of which is the multidrug resistance transporter (Mdr)-1. These ATP-binding cassette carriers carry toxic substances back into the blood, but also remove drugs important in therapy of a number of neurological disorders. During cerebral ischemia, the Mdr-1 transporter, P-glycoprotein, is upregulated. This enhances the removal of lipophilic drugs used to treat stroke. Blockade of the P-glycoprotein after stroke raised the levels of drugs that are transported out of the brain by the transporter (Spudich et al, 2006).
An important aspect of the design of treatment protocols to reduce BBB disruption in MS and acute and chronic stroke is the dual nature of the molecules targeted for treatment. Many of those molecules that participate in the death of cells in the early stages of the injury also have a critical role in the recovery period. An example is the benefit derived from treatment with MMP inhibitors in the early stages of injury may be lost if the same enzymes are blocked in the later stages when they are used in angiogensis and neurogenesis (Zhao et al, 2006). Understanding the timing of the expression of each of the agents to be blocked and their actions at each point of the injury cycle is necessary in planning the use of inhibitors. In the future, as the multiple cascades are better understood, treatments will be tailored to start when the damaging effects of that agent are maximal and to be stopped as the beneficial effects are beginning.
In conclusion, we have described some of the major advances in understanding the function of the NVU that go beyond the early physiological studies and add a molecular dimension. Unraveling the proteins that comprise the tight junctions provided tools to observe the effects of proteases on the tight junctions following acute stroke. Agents that block the BBB disruption can protect the brain from the adverse effects of tPA, extending the therapeutic window. The combined effect of agents that act early, including MMP inhibitors and those that protect the delayed opening of the BBB, such as the COX-2 inhibitors, need to be tested. The recent studies of chronic effects of hypoperfusion in humans and animals demonstrate a role for MMPs in both the disruption of the BBB and the breakdown of myelin, which may contribute to the death of oligodendrocytes. Defining the molecular mechanisms underlying damage to the vasculature provides important information on which to base further trials of novel therapies to protect the BBB.
Footnotes
Disclosure/conflict of interest
The author declares no conflict of interest.