
Abstract
We studied the acute (up to 2 hours after reperfusion) effects of localized cortical hypothermia on ischemia-induced dendritic structural damage. Moderate (31°C) and deep (22°C) hypothermia delays, but does not block the onset of dendritic blebbing or spine loss during global ischemia in mouse in vivo. Hypothermic treatment promoted more consistent recovery of dendritic structure and spines during reperfusion. These results suggest that those using therapeutic hypothermia will need to consider that it does not spare neurons from structural changes that are the result of ischemia, but hypothermia may interact with mechanisms that control the onset of damage and recovery during reperfusion.
Introduction
Of the many neuroprotective strategies under investigation for stroke, none have been as effective as hypothermia in mitigating detrimental acute and chronic effects of stroke and in improving later functional outcomes (Zhao et al, 2007). Nonetheless, while postischemic hypothermia reduces neuronal injury after global ischemia, spared neurons may still show some ultrastructural abnormalities in the days after the initial insult (Colbourne et al, 1999). Thus, we are interested in evaluating hypothermia's effects on dendrite morphology in the immediate phase of ischemia and in early reperfusion, specifically as it relates to ischemia-induced dendritic blebbing (regular swellings) and loss of spines. We hypothesized that intraischemic hypothermia may protect dendrites from ischemia-induced loss of structure. To this end, we have used in-vivo two-photon (2P) imaging to compare the induction and recovery of ischemia-induced dendritic blebbing and spine loss under normothermic (37°C, NORMO), moderate (31°C, HYPO), and deep (22°C, DEEP) hypothermic conditions in global ischemia (Murphy et al, 2008).
Materials and methods
Animals, Surgical Procedures, and Regulation of Cortical Temperature
Adult male C57BL/6 mice (2–5 months of age) expressing yellow fluorescent protein-H or green fluorescent protein-M (Feng et al, 2000) were used. Experimental protocols were approved by the University of British Columbia animal care committee and conformed to the Canadian Council on Animal Care and Use guidelines. Body temperature was maintained throughout surgical and imaging procedures at 37±0.5°C. Tracheostomies were performed in most animals (Li and Murphy, 2008) to ensure an adequate airway. In some animals that did not have the procedure, breathing was still monitored closely and experiments were terminated if the breathing or heart rate was inadequate; however, the procedure was not always necessary. In some animals, cortical surface temperature was measured using a thermocouple (IT-24P, Physitemp Instruments, Clifton, NJ, USA) placed over the cortical surface within agarose. A custom-made stainless steel head plate adhered to the skull was connected with tubing to a water pump (T/Pump TP500; Gaymar, Orchard Park, NY, USA), which circulated water and regulated cortical surface temperature to 37°C or 31°C. For DEEP experiments, a separate cooling pump was used to maintain cortical surface temperature at 22°C.
In-Vivo Two-Photon Imaging
For 2P imaging, urethane and isoflurane-anesthetized animals underwent surgical procedures to perform a 3 × 3 mm craniotomy over the somatosensory cortex. 2P imaging of apical dendrites of layer five neurons was performed as described previously (Murphy et al, 2008). 2P imaging was restricted to the first 70 μm of cortex within the forelimb somatosensory cortex or forelimb/hindlimb border as determined by intrinsic optical signal imaging or mouse atlas coordinates.
Stroke Model
Bilateral occlusion of the common carotid arteries (CCAO, 4 to 10 minutes in duration) as in Murphy et al (2008) was used to induce global forebrain ischemia under urethane anesthesia in mice assigned to the following groups: NORMO+ure, HYPO+ure, DEEP+ure, and a second normothermic group receiving a shorter period of occlusion (2 to 3 minutes), SHORT+ure. We also performed these experiments under isoflurane anesthesia that is more typical for stroke recovery experiments in NORMO+iso and DEEP+iso groups. In some cases, the duration of ischemia was shortened if we observed a depression of the heart rate (accounting for the range of occlusion durations). Ischemia was confirmed by monitoring: a slowing of surface blood flow through video microscopy; ischemic depolarization by DC electroencephalogram (EEG, unfiltered); AC EEG (filtered 0.1 to 10 Hz) depression; and 2P imaging of Texas-Red dextran (70 kDa; Invitrogen, Eugene, OR, USA)-labeled blood. To determine whether animals were maintained in a stable state during imaging procedures some mice had physiological parameters monitored using a Starr Life Sciences (Oakmont, PA, USA) Mouse-Ox pulse oximeter.
Image and Statistical Analyses
For the final analysis, we included only animals that had a confirmed reduction of blood flow during occlusion as seen on 2P images of Dextran-labeled plasma. We also confirmed that all animals had recovery of blood flow with reperfusion. Detection and quantification of the fraction of blebbed dendrites was performed using a MATLAB-based software program, which compared and averaged 5 to 7 μm maximal intensity z-projections over 20 μm of cortex. This software program had been modified from the original version where analyses of 20 μm maximal intensity z-projections were used (Chen et al, 2011). Dendritic spines were counted in the same sections of cortex using blinded manual analysis. An one-way analysis of variance (ANOVA) followed by Bonferroni's post-tests were used to compare differences in blebbed dendrites and spines across time in experimental groups. Significance was set at P<0.05. F-tests were used to compare variance in the percentage of blebbed dendrites and spines.
Results
In this study, we investigated the effect of localized cortical hypothermia on ischemia-induced structural damage and structural recovery in reperfusion. We verified in some animals that the average cortical surface temperature during experiments obtained using our warming and cooling method (Figures 1A—C) was consistent with normothermia (36.5±0.2°C, n=5), moderate hypothermia (31.1±0.2°C, n=4), and deep hypothermia (21.8±0.8°C, n=5) (Supplementary Figure 1). While temperature measurements at different cortical depths were not directly obtained, we did observe the reported (Nakashima and Todd, 1996) changes in ischemic depolarization latency (Table 1) suggesting cooling is effective. For all animals, successful CCAO preceded the onset of ischemic depolarization (Table 1) and was typically associated with an ∼90% or greater suppression of the spontaneous EEG power (Figure 1C).
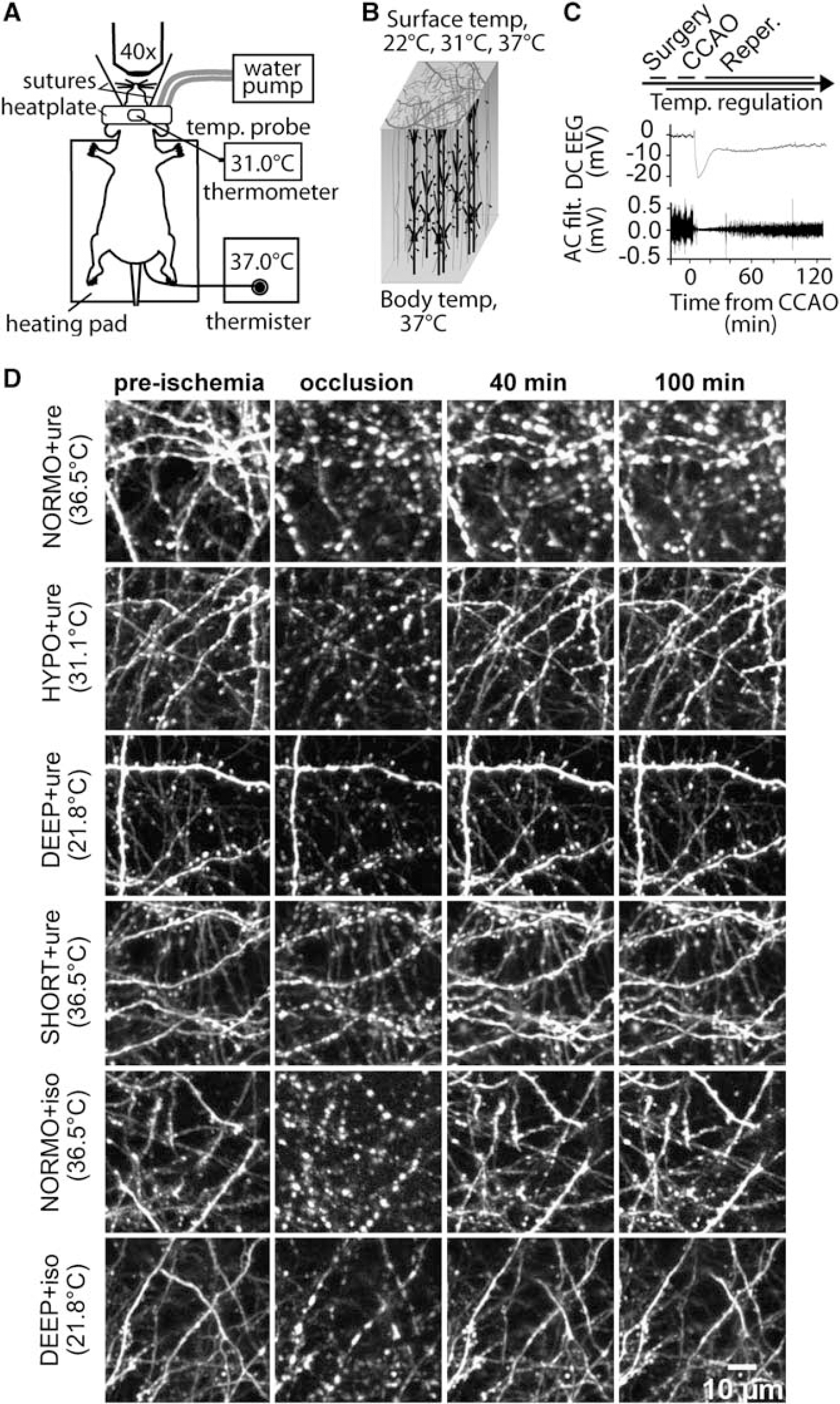
Local cortical surface cooling does not prevent the onset of ischemia-induced dendritic blebbing. (
Summary of ischemic depolarization amplitudes, rates, and latencies observed under NORMO (36.5°C), NORMO short (36.5°C), HYPO (31.1°C), and DEEP (21.8°C) conditions
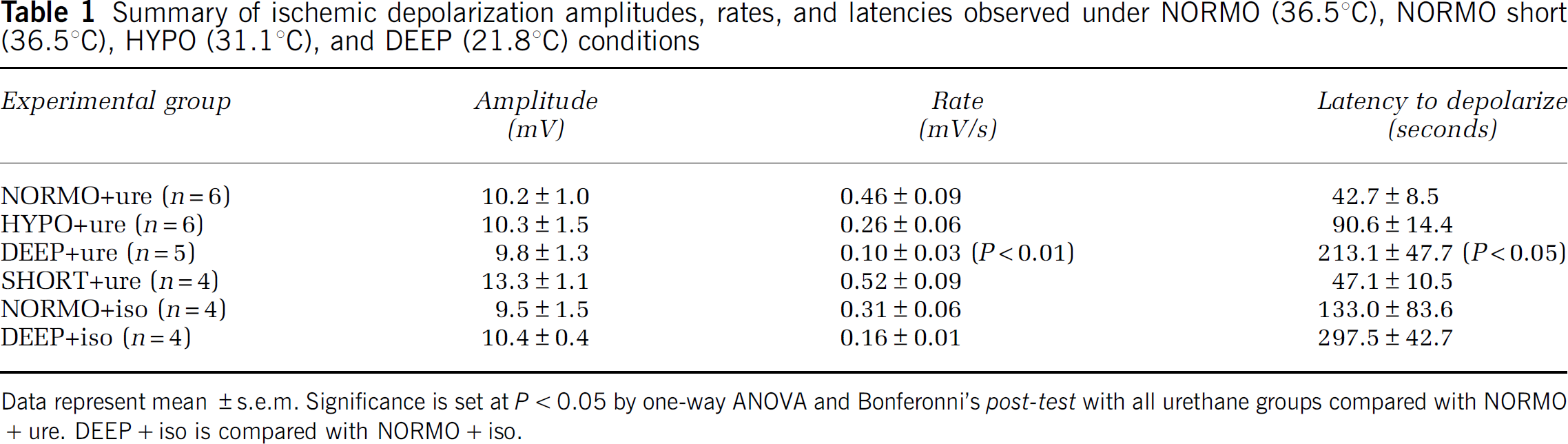
Data represent mean ±s.e.m. Significance is set at P<0.05 by one-way ANOVA and Bonferonni's post-test with all urethane groups compared with NORMO+ure. DEEP+iso is compared with NORMO+iso.
In-vivo 2P imaging of green fluorescent protein or yellow fluorescent protein expressing layer 5 apical dendrites was used to assess ischemia-induced dendritic structural damage and recovery with urethane or isoflurane anesthesia in mice. Under our experimental conditions, baseline dendritic structure was apparently not altered with cooling in that we did not observe hypothermia-induced structural alterations either in the form of beading or spine loss, indicating that localized hypothermia to 22°C does not induce any detrimental structural effects (data not shown). We report that under all treatment conditions, dendrites blebbed with occlusion (Figures 1D, 2A and 2B). The time to bleb (mean±s.e.m.) under NORMO+ure was 87.0±13.7 seconds (Figure 2C). Compared with NORMO+ure, the onset to bleb was significantly longer under DEEP+ure at 348.5±26.9 seconds (P<0.001), as well as between HYPO+ure and DEEP+ure (P<0.001) (Figure 2C). We also found that dendrites blebbed with occlusion under isoflurane anesthesia and dendritic blebbing was more prominent under DEEP+iso than NORMO+iso (Figures 1D and 2B). Similarly to urethane-anesthetized mice, the time to bleb was inversely related to cortical surface temperature under isoflurane (Figure 2C). Animals which showed a longer latency to depolarize also tended to show delayed onset to dendritic blebbing (Supplementary Figure 2). By one-way ANOVA, no statistically significant differences were observed in the latency between ischemic depolarization and dendritic blebbing onset for all groups. There was no relationship between average area (μm2) of blebs at occlusion and cortical surface temperature under urethane or isoflurane anesthesia (Supplementary Figure 3).
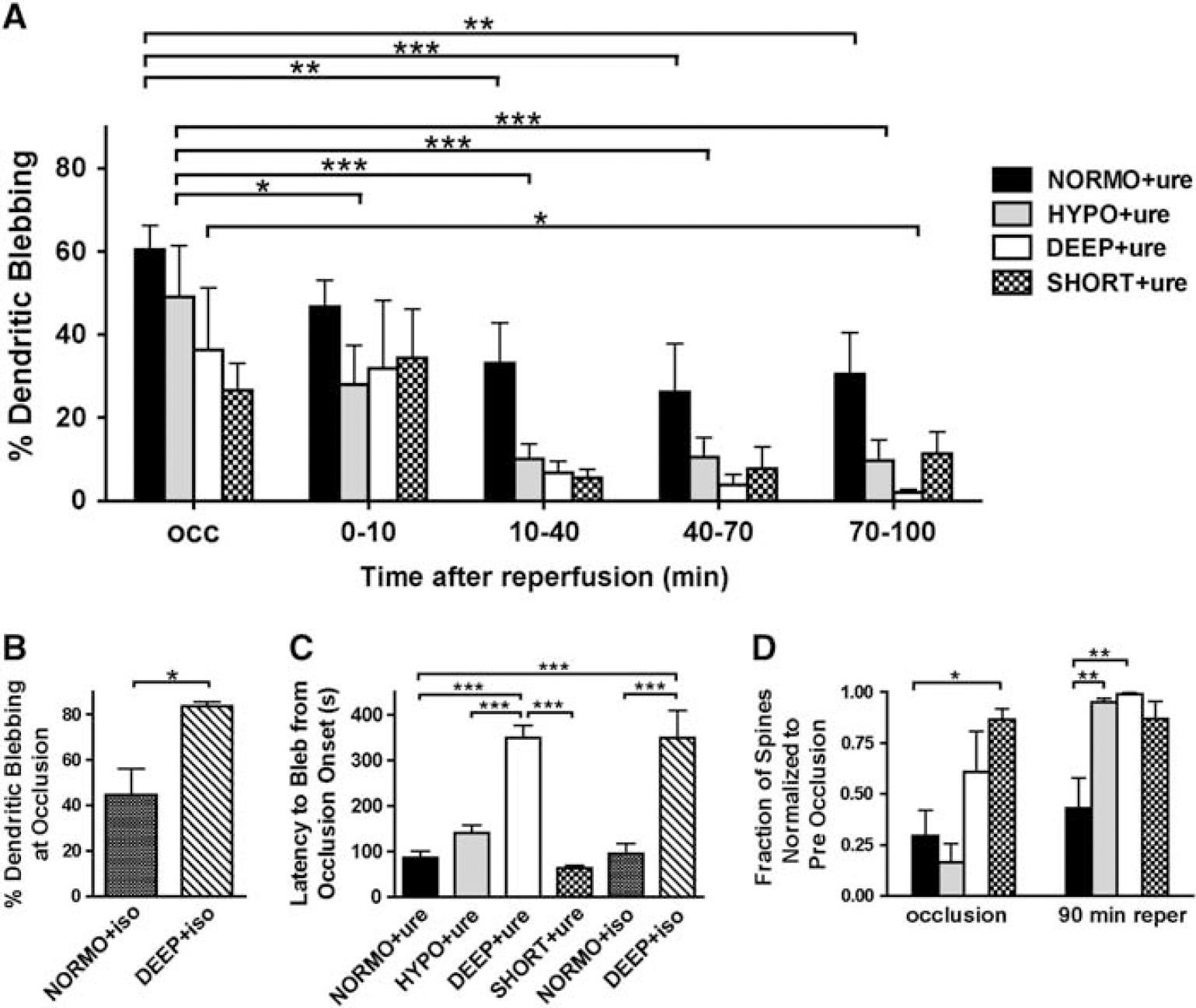
Quantification of dendritic damage under normothermia, moderate hypothermia, and deep hypothermia. (
Due to technical challenges of obtaining complete reperfusion, we were not able to chart the recovery of dendritic blebbing during reperfusion for all isoflurane animals, but in the animals which we were able to monitor fully, we observed recovery from dendritic blebbing along similar timescales as under urethane anesthesia (Figure 1D). Analysis of blebbed dendrites in urethane-anesthetized mice showed recovery of blebbed dendrites during reperfusion compared with occlusion (Figures 1D and 2A). A two-way ANOVA indicated a significant effect of time and temperature and individual time points were further assessed using one-way ANOVAs. The percentage of blebbed dendrites under NORMO+ure was reduced from 60.4±5.9% at occlusion to 26.2±11.6% (P<0.001) at 40 to 70 minutes of reperfusion and 30.4±10.0% (P<0.01) by 70 to 100 minutes of reperfusion. No statistically significant reductions in blebbing were observed under SHORT+ure; however, this could be attributed to less extensive blebbing at occlusion and a relatively smaller number of animals lowering statistical power. Under HYPO+ure, dendrites blebbed to 49.1±12.4% at occlusion and in contrast to NORMO+ure, recovery in blebbing was more rapid during reperfusion with significant reductions in blebbing apparent by 0 to 10 minutes (P<0.05) of reperfusion. Under HYPO+ure, blebbing was reduced to 10.5±4.8% (P<0.001) by 40 to 70 minutes of reperfusion and to 9.7±5.0% (P<0.001) by 70 to 100 minutes of reperfusion. Dendrites also blebbed under DEEP+ure at occlusion to 36.3±14.9% and this blebbing was reduced to 2.0±0.8% (P<0.05) by 70 to 100 minutes of reperfusion. Statistical tests for variance showed that the variability in blebbed dendrites during reperfusion was reduced for HYPO+ure and DEEP+ure as compared with NORMO+ure, indicating that structural recovery was more uniform at lower cortical temperatures. For HYPO+ure, an F-test for variance was significant (P<0.05) at 10 to 40 minutes of reperfusion and for DEEP+ure, significance was observed at 10 to 40 minutes (P<0.05), 40 to 70 minutes (P<0.05), and 70 to 100 minutes (P<0.001) of reperfusion.
In urethane-anesthetized animals, we investigated the effect of hypothermia on dendritic spine number by examining the same spines before, during, and after ischemia (Figure 2D). By one-way ANOVA, we did not find any significant difference in normalized spine number at occlusion between NORMO+ure, HYPO+ure, and DEEP+ure; however, there was a significantly reduced number of spines between NORMO+ure and SHORT+ure (Figure 2D). Compared with the NORMO+ure group, spines significantly reemerged by 90 minutes of reperfusion under HYPO+ure (P<0.01) and DEEP+ure (P<0.01) (Figure 2D). Tests for variance also indicated that the variability in spine number at 90 minutes of reperfusion was reduced for HYPO+ure (F-test, P<0.001) and DEEP+ure (F-test, P<0.0001) as compared with NORMO+ure, suggesting more uniform recovery at lower cortical temperatures.
Discussion
This study investigated the possible protective effect of local hypothermia on ischemia-induced loss of dendritic structure using in-vivo imaging. Contrary to our hypothesis, we found that treatment with hypothermia did not prevent the onset of ischemia-induced dendritic blebbing or loss of spines as compared with normothermia, but hypothermic treatment promoted more consistent recovery during reperfusion, a mechanism that may contribute to the neuroprotective effects of hypothermia. While a global ischemia model was used herein, in contrast to the more commonly studied focal models which differ in their extent and selectivity of affected brain regions (Colbourne et al, 1997), our results provide additional support for the beneficial role of therapeutic hypothermia for cerebral protection in cardiac arrest (Bernard et al, 2002). This study expands on previous work where mild or extreme hypothermia effects on dendritic structure have been investigated in vitro in the presence (Obeidat et al, 2000) or absence of anoxia (Kirov et al, 2004) and studies showing at the ultrastructural level that effects of ischemia were not necessarily spared under hypothermia (Colbourne et al, 1999).
Although we found that dendritic structure was still affected during ischemia under hypothermia, there was a trend toward less overall dendritic blebbing and spine loss (urethane anesthesia), suggesting some general benefits of hypothermia for dendrites during stroke. Greater than 90% recovery of dendritic spines after reperfusion has been reported previously (Murphy et al, 2008), but it is important to note that these experiments were performed under conditions where the local cortical temperature was unregulated and may have been similar to the HYPO conditions of this study.
Because this study investigated only acute effects of hypothermia, we did not evaluate any relationship between acute structural changes within apical dendrites of layer 5B neurons and cell death. Indeed, short timescale neuroprotection does not necessarily prevent delayed cell death or damage to synapses from occurring over longer timescales (Iyirhiaro et al, 2008). In global ischemia, we found little evidence for cortical cell death in the first hour after reperfusion with 5 to 8 minutes of occlusion (Murphy et al, 2008), as this typically occurs in the days after insult (Olsson et al, 2003; Yonekura et al, 2004). Nonetheless, given that hypothermic treatment can block apoptotic cell death (Zhao et al, 2007), we would expect that hypothermic neurons with recovered dendritic structure may also be protected from later apoptosis.
In summary, we found that ischemia-induced dendritic blebbing could not be prevented even with deep hypothermia treatment and may be an obligate effect of energy failure and impaired ionic homeostasis. Thus, mechanisms involved in such structural changes are likely still intact during hypothermia; however, their kinetics may be altered as we have showed a temperature dependency for dendritic blebbing onset latency. While our results show structural recovery under hypothermic conditions after reperfusion, the possibility of delayed neuronal death of structurally recovered neurons still exists.
Footnotes
Acknowledgements
The authors would like to thank Cindy Jiang and Pumin Wang for surgical assistance; Alexander Goroshov, Jamie Boyd, and Jeff LeDue for technical assistance; and Albrecht Sigler for assistance with figures.
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.