
Abstract
The deficiency in the mitochondrial aspartate/glutamate transporter Aralar/AGC1 results in a loss of the malate-aspartate NADH shuttle in the brain neurons, hypomyelination, and additional defects in the brain metabolism. We studied the development of cortico/hippocampal local field potential (LFP) in Aralar/AGC1 knockout (KO) mice. Laminar profiles of LFP, evoked potentials, and unit activity were recorded under anesthesia in young (P15 to P22) Aralar-KO and control mice as well as control adults. While LFP power increased 3 to 7 times in both cortex and hippocampus of control animals during P15 to P22, the Aralar-KO specimens hardly progressed. The divergence was more pronounced in the CA3/hilus region. In parallel, spontaneous multiunit activity declined severely in KO mice. Postnatal growth of hippocampal-evoked potentials was delayed in KO mice, and indicated abnormal synaptic and spike electrogenesis and reduced output at P20 to P22. The lack of LFP development in KO mice was accompanied by the gradual appearance of epileptic activity in the CA3/hilus region that evolved to status epilepticus. Strikingly, CA3 bursts were poorly conducted to the CA1 field. We conclude that disturbed substrate supply to neuronal mitochondria impairs development of cortico—hippocampal LFPs. Aberrant neuronal electrogenesis and reduced neuron output may explain circuit dysfunction and phenotype deficiencies.
Keywords
Introduction
During postnatal brain development, many processes come to a final mature condition within a perfectly orchestrated temporal succession of events, for which cellular machinery should be ready. It has already come out evident that alterations in neurodevelopment may lead to brain pathology. There is also evidence that early electrical activity in the brain is key for development and refinement of functional networks (Khazipov and Luhmann, 2006). Activity-dependent modulation of metabolic pathways may thus determine the correct development at an early stage. We here report on the postnatal alterations of brain electrical activity in mice deficient in Aralar/AGC1, a mitochondrial transporter of aspartate—glutamate present mainly in neurons (del Arco and Satrústegui, 1998; Ramos et al, 2003; Berkich et al, 2007; Xu et al, 2007) that is critically involved in neuroglial metabolic concert (Pardo et al, 2011).
While disruption of neuroglial metabolic coupling in adult brain leads to failure of ion homeostasis, neurotransmission, and cell survival (Largo et al, 1996), little is known in the early postnatal period, when many functions are going through a refinement process and in many cases are differently handled as in the adult. Aralar deficiency leads to a loss of respiration on malate plus glutamate, a shutdown of the malate-aspartate NADH shuttle, and in alterations in neuronal aspartate levels and pyruvate to lactate ratios (Jalil et al, 2005; Pardo et al, 2011) Globally, the aralar-deficient brain shows hypomyelination and a drop of N-acetylaspartate and a progressive failure to synthesize glutamine, suggesting that glutamatergic neurotransmission may be compromised in the older animals (Pardo et al, 2011). However, the functional state of neurons and circuits in these animals is still unknown. Aralar-knockout (KO) mice show motor deficits, tremor, seizures, and premature death (Jalil et al, 2005). A patient with an homozygous loss of function mutation in the gene encoding AGC1 has also low N-acetylaspartate levels, hypomyelination, arrested psychomotor development, hypotonia, and seizures (Wibom et al, 2009). We here performed an exploration of the electrical activity in postnatal (P15 to P22) mice to gain insight in the functional state and local field potential (LFP) development of the cortex and hippocampus in this early-onset brain disease.
There is little information on postnatal LFP development in mice (Wong et al, 2005). Most studies have been made in the rat. Some electrographic events and LFP frequency bands appear already during the first postnatal week (Lahtinen et al, 2002), while a transient period of hyperexcitability occurs during the second week. In parallel, a number of cellular and anatomical outbreaks take place, such as axonal reorganization (Gomez-Di Cesare et al, 1997), and peaks of expression of connexins and glutamate receptors (Rozental et al, 2000; Ritter et al, 2002). On the contrary, GABAergic inhibition remains immature. In general, cellular, synaptic, and network electrical properties progress rapidly to stabilize gradually within 1 to 2 months (Kriegstein et al, 1987; Huguenard et al, 1988). The exploration performed here in the anesthetized Aralar-deficient mice using linear multisite probes to record different types of electrical activity, as LFPs, evoked field potentials, and spontaneous firing rate of single neurons, indicates major deficit in cellular and network electrical activity compatible with progressive behavioral quiescence and status epilepticus in the period P14 to P22. While severely reduced network activity may influence negatively in the activity-dependent circuit refinement, the timeline of wild-type (WT)–KO differences indicate that deficiency in aralar initiates pathology at a very early stage.
Materials and methods
All the experiments were performed in accordance with the European Union guidelines (86/609/EU) and Spanish regulations (BOE 67/8509-12, 1988) regarding the use of laboratory animals. The experimental protocols were approved by the Animal Welfare Committee at the Cajal Institute.
Preparation and Recording
We used mice of both sex deficient for Aralar/AGC1 (Aralar−/−) and age-matched WT (Aralar+/+) littermates from heterozygous breeding pairs (SVJ129 × C57BL6). The generation of Aralar-deficient mice, obtained from Lexicon Pharmaceuticals, Inc. (The Woodlands, TX, USA), and the polymerase chain reaction analysis of genotypes have been described in detail (Jalil et al, 2005). Animals were bred in the animal house of the Centro de Biología Molecular (Universidad Autónoma de Madrid) and transported to the Cajal Institute (CSIC, Madrid) for electrophysiological study. Most of the aralar (−/−) mice die at around 23 days. At the end of the period studied P20 to P22, a progressive decline in overall exploratory behavior is observed. Tremor appeared regularly after P19 and tonic-clonic motor seizures were increasingly observed within the 2 to 3 days before animals died. The progression of decline was, however, highly variable among individuals and death time was rather unpredictable. Aralar-KO mice gained no weight within the period P15 to P22 (5±0.2 g; mean±s.e.m., n=20), while littermates did gain (8.2±0.6 at P15 and 12.1±0.3 at P22; population average: 11.1±0.5 g, n=22).
Mice were anesthetized with a mixture of ketamine (Ketolar 100 mg/kg, intraperitoneally) and xylacine (Rompun, Bayer, 5 mg/kg, intraperitoneally) and placed in a Cunningham-type mouse stereotaxic adaptor (Stoelting Co., Wood Dale, IL, USA). Supplemental doses (1/4 of the initial dosage) were administered every 45 to 60 minutes. The body temperature was maintained at 36°C by a heating element implemented on the adaptor. The stability of the animal was assessed by monitoring the heart rate. Experiments were limited to 4 hours. In general, KO mice showed a notable lability to anesthesia, especially during induction in individuals older than P19. This fact and the variable progression of Aralar-KO mice complicated the customization of groups of age (the study extended over a period of 18 months). Thus, the different types of electrical activities could not be studied on each animal. Except when indicated, at least three animals (normally 5 to 6) were employed for each quantified parameter. Elder KO animals (P20 to P23) presented a high dispersion in behavioral capability, from normally responsive to external stimuli to nearly complete quiescence.
For the reasons outlined, it was not possible to use a standard set of stereotaxic coordinates, making the exploration of hippocampal activity by evoked field potentials arduous and hardly standardizable. We thus limited their use to the best-known and most repeatable ipsilateral CA3–CA1 pathway (Schaffer collateral input). Concentric bipolar stimulating electrodes were placed in the ipsilateral CA3 field for orthodromic activation of the CA1 field. This point was approached at a 30° angle in the sagittal plane. Stimuli (0.1 ms square pulses, 0.1 to 0.8 mA) were applied to elicit the characteristic CA1-evoked potentials (Herreras, 1990). We first used glass micropipettes (1 to 4 MΩ) filled with 150 mmol/L NaCl. After stimulus site optimization, the glass pipettes were removed and linear multisite silicon probes (Neuronexus, Ann Arbor, MI, USA, A1x16-3mm50-413 and A1x16-5mm100-413) were lowered and connected to a multiple high-impedance headstage. A silver chloride wire in the neck skin served as reference for recording. The signals were amplified and acquired using low noise MultiChannel System recording hardware and software. In a few animals, at the end of the experiment, a small current (10 μA, 3 minutes) was injected through one site of the probe that served for histological verification of recording tracks.
Each animal was recorded with the linear probe in at least three consecutive placements along a single vertical track passing through the cortex, CA1, and CA3/Fascia Dentata (FD) regions in a way that electrode sites laid parallel to the main axis of principal pyramidal and granule cells. Each station aimed to span one of these subregions and covered 750 or 1,500 μm (for 50 and 100 site separation, respectively). Three epochs 3 minutes long were recorded at each station (0.1 Hz to 5 kHz bandpass) and acquired (1 kHz acquisition rate) to a computer. In a selected group of animals, the acquisition rate was increased to 20 kHz to obtain multiunit spike activity after off-line filtering (0.3 to 3 kHz). To minimize the effects of variable anesthetic plane on LFPs, all recordings were made between 10 and 35 minutes after one dose had been delivered. The energy of LFP epochs was estimated on raw signals as well as on its component frequency bands obtained by selective filtering: delta (δ), <2 Hz; theta (θ), 4 to 8 Hz; alpha (α), 10 to 12 Hz; beta (β), 13 to 30; gamma (γ), 30 to 80 Hz. As a gross measurement of the global population synaptic activity, we estimated the average energy of LFP signals (squared voltage) over identical epochs. First, we quantified the LFP power of signals recorded against a far reference electrode in three sites along the same track for each subregion, and the result was averaged. To minimize the effects of volume propagation from subregions producing large LFPs, we repeated the estimation over the same signals in differentiated cortical records (i.e., true LFP) between two consecutive sites separated vertically by 100 μm.
Quantification of Epileptiform Activity
Because of the progressive nature and varying amplitude and waveform of epileptiform activity, the classification and standardization of epileptic events was unfeasible. Thus, we only estimated the frequency of events and explored the underlying currents by current source density (CSD) analysis. The earliest visible epileptiform events (see below) were counted when they extended over at least 150 ms and contained spike activity of amplitude twofold or greater than baseline. At a later stage, epileptiform bouts were easily recognized. Three epochs 1 minute each separated by 4 minutes were quantified on selected electrodes for each animal and pooled together for statistics. To assess whether burst were local or propagated from nearby regions, we applied CSD to interictal-like spikes in the different regions and counted them as local or remote when the field burst presented or not associated current activity.
Unitary Analysis
We aimed to establish a gross relation of average LFP and global spike activity. This was quantified only in the CA1 region whose limits are reliably identified using the evoked potential profiles. Spike activity was estimated on each of the electrode sites spanning the different CA1 layers. No attempt was made to sort units or to identify principal (pyramidal) or putative interneurons. Thus multiunit activity was estimated. Yet, in the CA1 region, cells recorded in layers other than the pyramidal layer can be safely ascribed to putative interneurons (occasional spikes recorded simultaneously in this and an adjacent site were rare and therefore introduced a negligible error). Spikes were counted on filtered (100 Hz to 5 kHz) signals using a voltage threshold of 150 μV (∼2 to 4 times the baseline noise). A grand total of CA1 spike activity was estimated for each animal throughout all recording sites in a single epoch of 180 seconds.
Current Source Density of Evoked and Local Field Potentials
Current source density analysis of LFP profiles provides the magnitude and fine location of the net transmembrane current generated by neural elements contained within a small volume of tissue, eliminating the volume contribution of far sources inherent to LFPs. We employed here the standard unidimensional CSD approach (Herreras, 1990) by estimating the second spatial derivative over the raw LFP profiles. Although there is a notable heterogeneity of tissue resistivity in the Z axis at the level of the stratum pyramidal in mature adult brain (López-Aguado et al, 2001), we do not know the precise values of resistivity in immature brain that has much larger volume fraction (Lehmenkühler et al, 1993). Therefore, CSD values are expressed in arbitrary units. Admittedly, the unidimensional approach may produce considerable amount of spurious current when applied to raw LFPs due to mutual spatial cancellation among different LFP generators (Korovaichuk et al, 2010) or because of the reduced spatial extension of spontaneous synaptic activity that might not meet the criterion of homogeneous activation in the XZ plane. Thus, we only consider reliable the CSD results estimated for specific LFP events whose homogeneity in this plane is known to be at least twofold the interelectrode distance (i.e., 100 μm), such as the epileptic events found in Aralar−/− mice.
Measurement of Cellular Oxygen Consumption
Oxygen consumption rate was measured using a Seahorse XF24 Extracellular Flux Analyzer (Boston, MA, USA) (Qian and Van Houten, 2010). Cortical primary neuronal cultures from P16 Aralar WT and Aralar KO embryos were plated in XF24 V7 cell culture plates (Seahorse Bioscience, North Billerica, MA, USA) at 1.0 × 105 cells/well and incubated for 10 days in a 37°C, 5% CO2 incubator. The cell cultures contained >85% of neurons. Cells were equilibrated with bicarbonate-free low-buffered DMEM medium supplemented with 15 mmol/L glucose for 1 hour immediately before XF assay. Substrates were prepared in the same medium and were injected from the reagent ports automatically to the wells at the times indicated. Mitochondrial function in neurons was determined through sequential addition of 6.0 μmol/L oligomycin, 0.5 mmol/L 2,4-dinitrophenol, and 1.0 μmol/L antimycin/1.0 μmol/L rotenone. This allowed determination of basal oxygen consumption, oxygen consumption linked to ATP synthesis, non-ATP linked oxygen consumption (proton leak), uncoupled respiration, and nonmitochondrial oxygen consumption (Qian and Van Houten, 2010).
Western Blotting
Aliquots (50 μg of protein) of hippocampal tissue from 18-day-old Aralar WT and KO mice were homogenated in protease inhibitor cocktail tablet (complete Mini, EDTA-free, Roche, Indianapolis, IN, USA) and sonicated. The homogenate was used for protein determination by the Bradford protein assay and for electrophoretical separation. Samples were electrophoresed in an 8% sodium dodecyl sulfate acrylamide gel and transferred electrophoretically to nitrocellulose membranes, which were blocked in 5% (w/v) dry skimmed milk (Sveltesse, Nestle, Barcelona, Spain) in Tris-buffered saline for 2 hours, and further incubated with antibodies against glutamic-acid-decarboxilase (GAD)-65 (Chemicon polyclonal antibody, 1:1,000, Madrid, Spain) O/N and β-actin (Sigma monoclonal antibody, 1:5,000, Madrid, Spain) for 1 hour at room temperature (RT). Signal detection was performed with and enhanced chemiluminescence substrate (Western lighting-ECL; Perkin-Elmer, Waltham, MA, USA).
Histology
Animals were anesthetized by chloral hydrate (0.5 mg/g body weight; Sigma) and perfused transcardially with saline and formaldehyde (4%). The brains were removed, fixed overnight, and transferred into sucrose. Later, they were embedded in OCT (Tissue-Tek, Sakura Finetek Europe B.V., Zoeterwoude, The Netherlands), and cut (30 μmol/L coronal sections) with a cryotome. Sections were kept in cryoprotectant medium and stored (−20°C) until processing. Sections were stained with cresyl violet and examined with the light microscope.
Statistics
Data are expressed as mean±standard error of the mean (s.e.m.). Statistical analysis was performed using GraphPad Prism 5.0 software (La Jolla, CA, USA). For investigating the effect of more than one variable within each animal group, we used one-way analysis of variance (ANOVA) followed by the Bonferroni's post hoc test. For studying the effect of genotype on one variable, we used the Mann—Whitney test. The effect of genotype on more than one variable was assessed by two-way ANOVA followed by the Bonferroni's post hoc test. The significant level (α) was set at 0.05 for each statistical test.
Results
Lack of Local Field Potential Development in Aralar-Knockout Mice
Recording tracks were made through the visual (V2MM) and somatosensory cortices (S1Tr) down to the hippocampus. Under ketamine/xylacine anesthesia, the LFPs were irregular although periodic multiform activity was also present. In all animal groups, the overall LFP activity was larger in the CA3/FD region and smaller in the cortex. Figure 1A shows sample recordings of spontaneous LFP activity in young WT and KO mice at P22 along a track centered in the hilus of the FD. It can be appreciated a notable difference in the baseline amplitude that was smallest in KO mice. At this age, LFPs in WT specimens had not reached the final power of adults (compared with sample recordings in WT P60). The power of ongoing LFPs was estimated in three gross regions along the same vertical track, the neocortex, and the CA1 and CA3/FD subregions of the hippocampus, each quantified as an average of three separated locations. We also searched for possible frequency-specific bands (see sample filtered traces in Figure 1B).
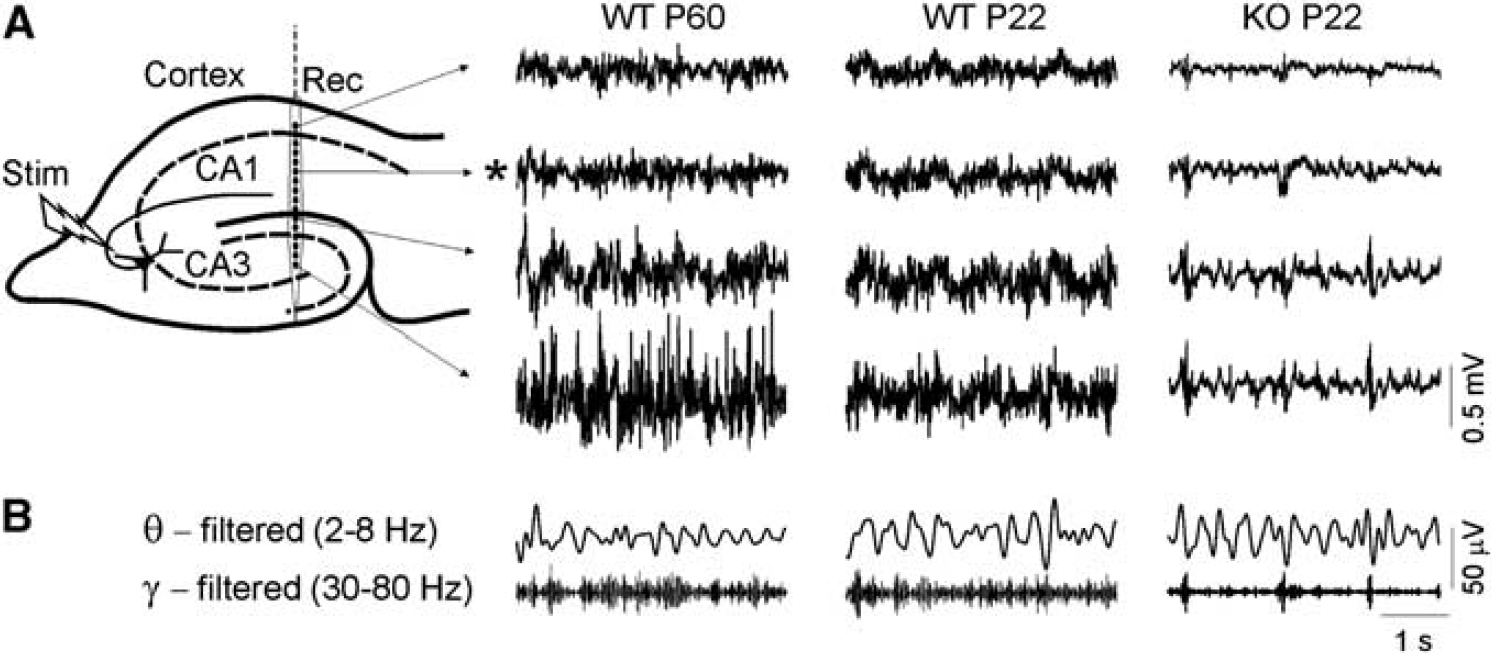
Deficient development of local field potentials (LFPs) in Aralar (−/−) knockout (KO) mice. (
Multiple comparisons yielded the following main observations. The LFP power was higher in CA3/FD and lowest in the cortex in all animal groups (Figure 2A; one-way ANOVA, P<0.0001 for all groups). Local field potential analysis (two-way ANOVA) between young (P15 to P23) and adult WT (>P60) specimens revealed that the LFP power was age and region-dependent, showing a strong age-by-region interaction (F2,27=6.38, P=0.005; Figure 2A). Bonferroni's post hoc test showed that during development, the LFP power is significantly lower in the CA3/FD region (∗∗∗P<0.001; Figure 2A).
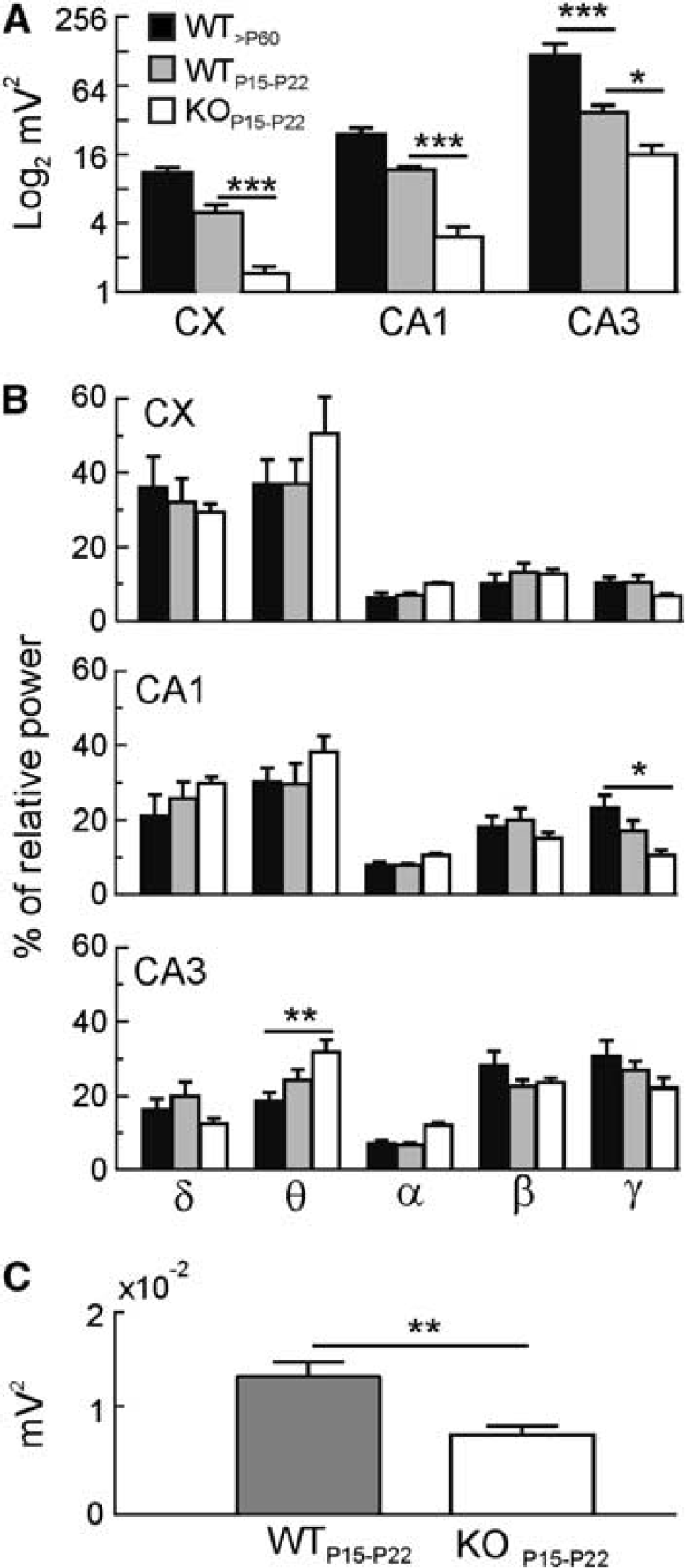
Comparison of local field potentials (LFPs) in wild-type (WT) and knockout (KO) mice. (
Knockout mice showed much weaker LFPs than WT specimens of the same age (Figure 2A, gray versus white bars). A significant genotype effect was observed (two-way ANOVA; F1,36=48.32, P<0.0001), with Aralar−/− mice showing lower LFP power in all the analyzed regions (cortex and CA1: ∗∗∗P<0.001; CA3/FD: ∗P<0.05, Bonferroni's post hoc test).
Then, we analyzed possible changes on each of the LFP frequency bands (see Materials and methods). Some peculiarities were observed (Figure 2B). In the cortex and CA1 regions, the lowest LFP frequencies (δ) augmented proportionally stronger in WT specimens, while higher frequencies (β and γ) did it in the CA3/FD region at the expense of a decrease in δ and θ content. As it concerns to KO animals, while the absolute LFP power was smaller in all frequency bands compared with WT littermates (not shown), we did not observe any significant difference in the percentage of relative power at any specific band (two-way ANOVA; Figure 2B). However, when comparing the Aralar−/− mice versus the adult WT, we observed a decrease in the γ frequency in CA1 and an increase in the θ content in the CA3/FD region (γ: ∗P<0.05; θ: ∗∗P<0.01; Figures 1B and 2B; two-way ANOVA, Bonferroni's post hoc test).
Since we found a gradation of the LFP power that increased from the cortex toward CA1 and CA3/FD regions, we explored the possibility that the estimations in cortical LFPs reflected changes in far stronger generators due to volume propagation. True local fields were thus estimated in differentiated recordings between pairs of electrodes (see Materials and methods). The analysis confirmed that cortical LFP activity was indeed weaker in KO as compared with WT littermates, albeit in a smaller degree than estimated with ground-referenced recordings (WT/KO mean power ratio was 1.74 and 3.58, respectively; compare white and gray bars in Figures 2C and 2A, CX).
The evolution of raw LFP power from P15 to P22 is shown in Figure 3. An increase was found in all regions, much more pronounced in WT animals. Two-way ANOVA analysis, followed by Bonferroni's post hoc test, showed a genotype effect in the evolution for cortex and CA1 regions (cortex: F1,10=31.9, ∗∗∗P=0.0002; CA1: F1,18=2.9, ∗∗∗P<0.0001), but not significant genotype-by-day interaction. Surprisingly, a striking reduction of power was found in P20 to P22 WT individuals in the CA3/FD region, which was not analyzed any further. A significant genotype-by-day interaction between WT and Aralar−/− mice (two-way analysis, F6,18=3.4, ∗P=0.02) was found, with a maximum difference between animals at P18 (∗∗∗P<0.0001, Bonferroni's post hoc test) followed by a steep decay in the LFPs power, much more pronounced in the WT mice.
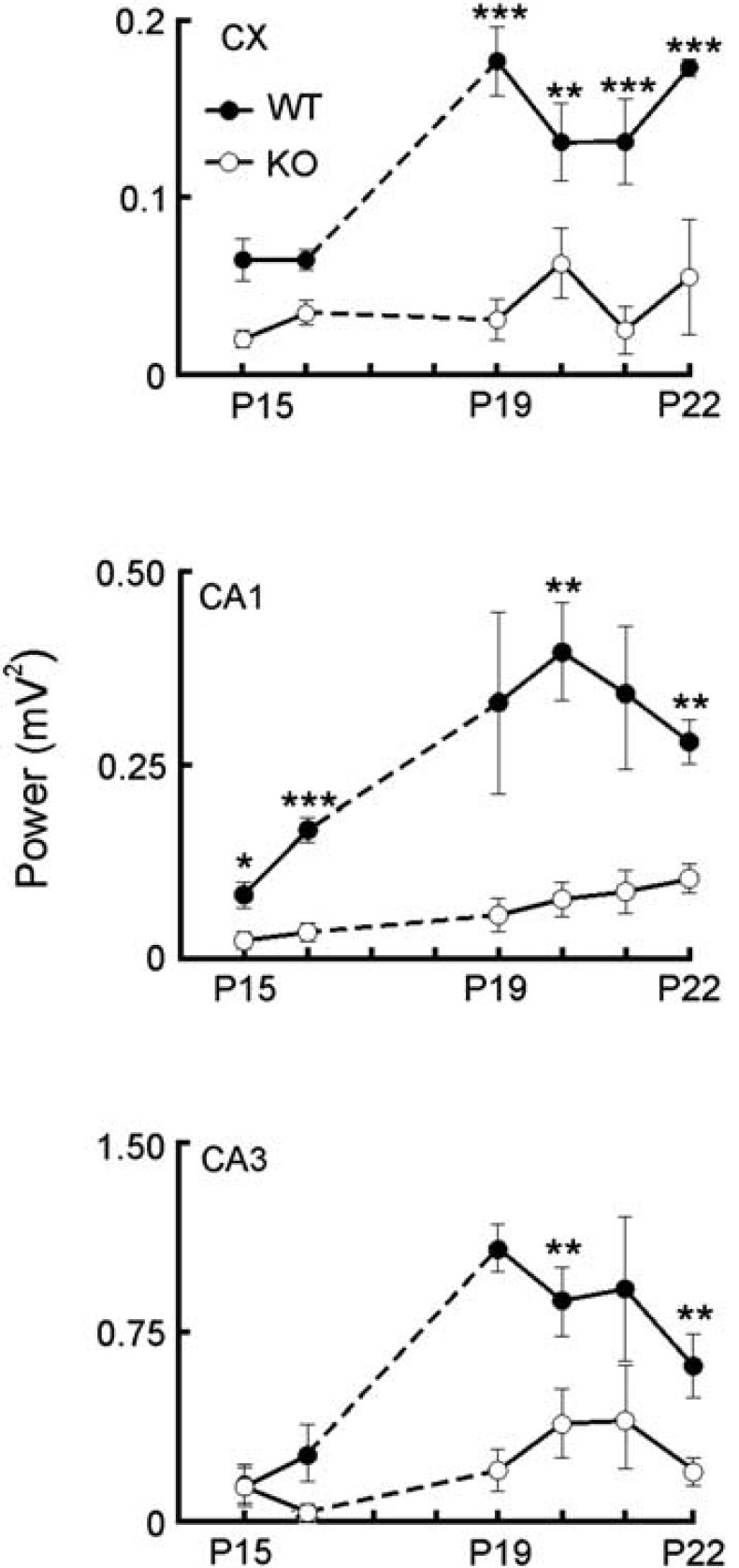
Temporal evolution of local field potential (LFP) power in young wild-type (WT) (filled circles) and knockout (KO) animals (open circles). A gradual increase of LFP power is appreciated in WT animals in all regions, while it was much smaller in KO mice. In all graphs, data represent the mean±s.e.m. of LFP power at different days. Per area (cortex—CA1–CA3), there are at least three animals per day and group. One-way analysis of variance (ANOVA) (followed by Bonferroni's post hoc analysis). ∗P<0.05; ∗∗P<0.01; ∗∗∗P<0.001.
Reduced Multiunit Activity in the Aralar-Deficient Hippocampal CA1
We next explored the mechanisms of reduced LFP power in Aralar-deficient mice. Most LFP activity in the hippocampus is likely of an inhibitory synaptic nature (Korovaichuk et al, 2010), and Aralar is conspicuously present in hippocampal interneurons in the hilus and CA1 region (Berkich et al, 2007, and results not shown), which are mainly GABAergic (Somogyi and Klausberger, 2005). Since hippocampal interneurons are often enriched in cytochrome oxidase, in a way that may correlate with their reported rates of spontaneous firing (Kageyama and Wong-Riley, 1982), we quantified the spontaneous firing rate of putative interneurons in the CA1 region. To emphasize possible age-related differences, only two age groups (P16 to P17 and P20 to P21) were employed. The spikes from all units recorded in eight sites along a recording track spanning from the alveus through the st. lacunosum-moleculare were pooled together. Most spike activity was found in dendritic layers (Figure 4). Multiunit activity in WT animals was similar in the two age groups (152±13 and 150±28 spikes/min; n.s., P>0.1), while it was much lower in Aralar-KO animals whose CA1 seemed nearly silent, especially at P20 to P21 days (27.7±16 and 4.5±3.2 spikes/min, respectively; Figure 4).
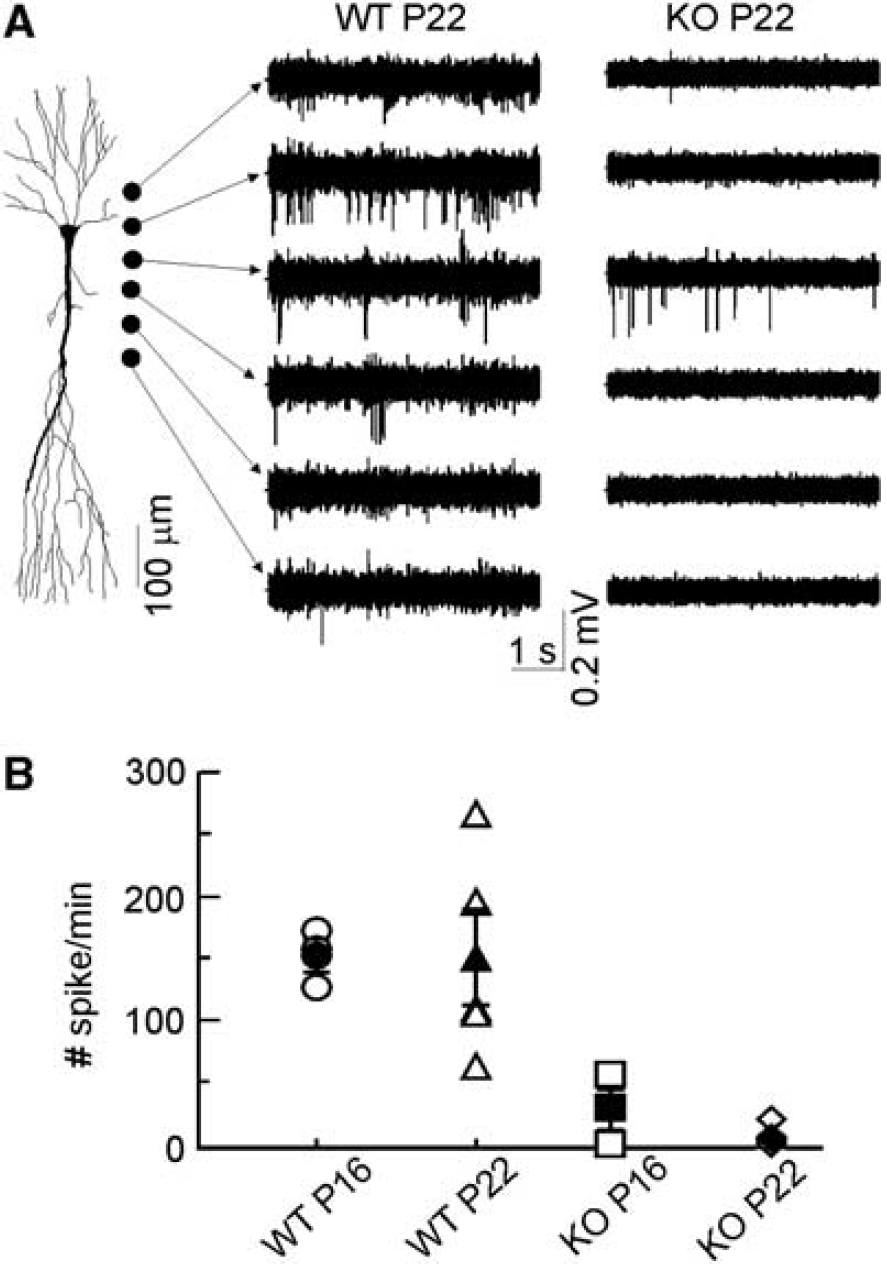
Unitary activity in the CA1 hippocampal subfield. Unitary activity was counted throughout CA1 hippocampal layers. (
Altered Electrogenesis in the CA1
The synaptic and spike electrogenesis of individual neurons was studied in the CA1 pyramidal population by CSD of the CA3-evoked field potential profiles. Adult animals showed consistent CSD spatiotemporal profiles upon supramaximal stimuli (Figure 5A). The synaptic (CA3-evoked) sink of current (sk1 in Figure 5) lasted 15 to 20 milliseconds and was centered in the mid stratum radiatum (sample fEPSP marked by two arrows in Figure 5A), surrounded by passive sources in strata pyramidale and lacunosum-moleculare (sc1 and sc2 in Figure 5, respectively). This sink was interrupted by a stronger and shorter sink (sk2) corresponding to dendro-somatic action currents, that is, those originating the PS (population spike; single arrow in Figure 5A). These originated in the apical shafts ∼100 μm below the cell body layer and propagated forwardly to the soma/axon region (curved arrow in Figure 5A). Immediately following the spike sink, a distinct slow passive sink developed in the st. oriens (sk3) that indicated the presence of somatic and perisomatic active sources corresponding to recurrent inhibition mediated by basket cells (these overlapped to the passive sources from excitatory synapses in the st. radiatum, i.e., sc1). This spatiotemporal profile is essentially the same as in the adult rat (Herreras, 1990). Younger animals showed less consistent CSD maps, in part due to technical difficulties (see Materials and methods) but also to rapidly changing electrogenesis at this period of development. However, a few qualitative differences were consistent enough to be pointed out. Strikingly, the synaptic sink was significantly stronger in Aralar than WT littermates (−2.2±0.1 versus −0.9±0.3 mV/mm2 at P16 to P17 and −2±0.3 versus −0.9±0.1 at P20 to P22, respectively). A nonsignificant increase occurred with age in WT animals, while a decrease was appreciated in the KO.
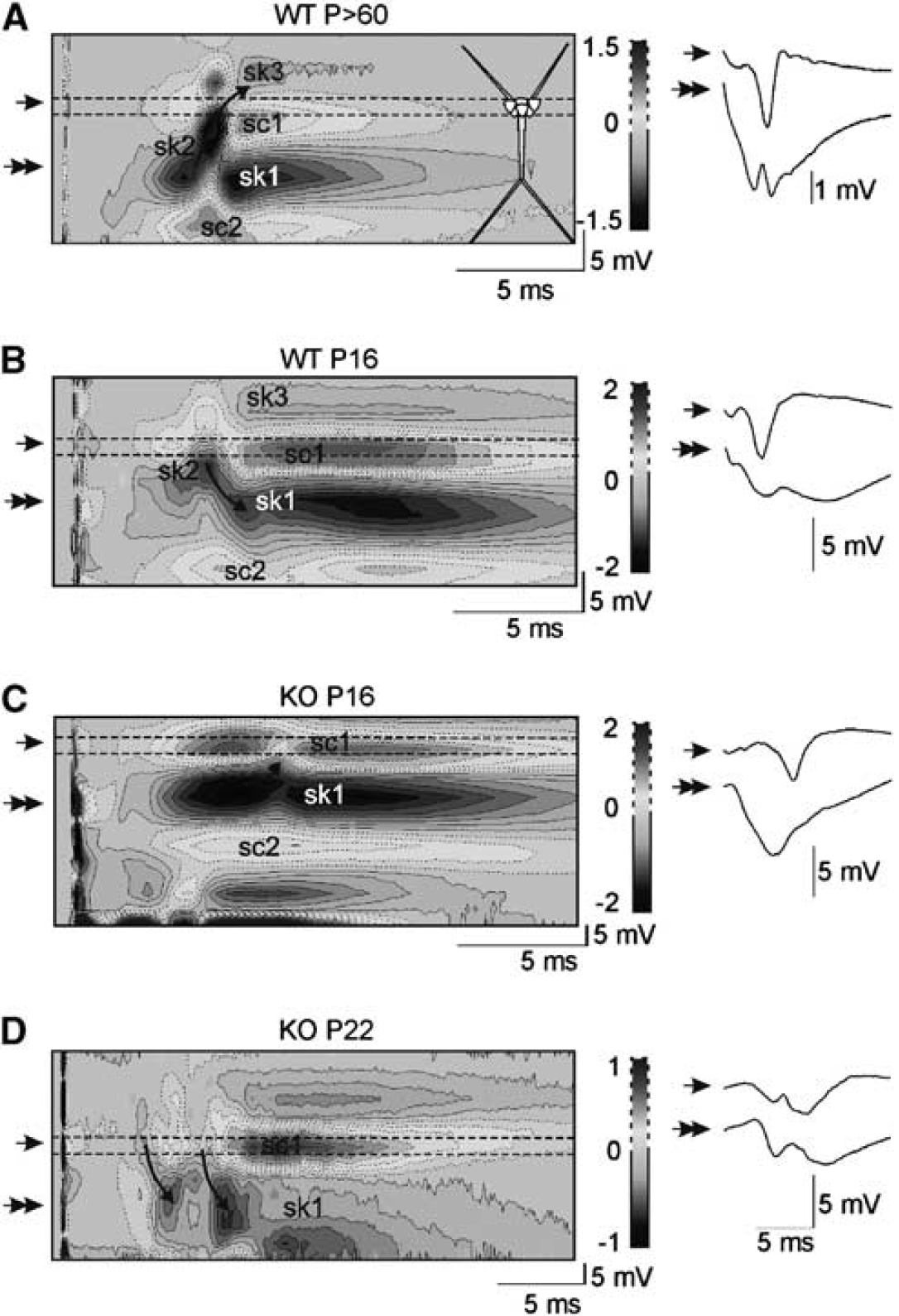
The spike electrogenesis in the CA1 pyramidal population becomes aberrant in knockout (KO) mice. Current source density (CSD) analysis of CA3-evoked field potential maps was employed to unveil the origin and direction of action potential currents. Sample evoked field potentials recorded at the cell body layer and the st. radiatum (single and double arrow, respectively) are to the right of CSD color-coded contour maps. Sinks and sources of current are depicted in blue and yellow/red tones, respectively. (
Wild-type animals showed a well-shaped somatic PS at P16 (4.6±0.6 mV; range: 3.4 to 5.4; n=4), while it was highly variable in KO animals (5.3±2.3 mV; range: 0 to 11; n=6). At P20 to P22, the PS had increased larger in WT (8.2±1.9 mV) than in Aralar-KO mice (6.4±0.7 mV). Population spike amplitude, however, was less reliable (see below) than the spatiotemporal CSD maps that provide the origin and direction of propagation of action currents along the somatodendritic axis. The spike currents in P16 to P17 of WT specimens were initiated in the soma layer and back-propagated into the apical dendrites for ∼100 μm (Figure 5B). On the contrary, younger KO animals showed a small current sink (curved arrow in Figure 5C) fanning out from the upper border of the main Schaffer collateral synaptic sink that indicated mild dendritic spike currents propagating forwardly and inconsistently to the soma (i.e., they failed to invade the soma/axon). At P20 to P22, the origin of spike currents in WT animals shifted to adult-like locations within the apical dendrites and propagated both somatically and distally, indicating stronger dendritic spike electrogenesis. Meanwhile, KO animals were highly variable. Some (n=3) strikingly presented a similar CSD map as WT specimens, while others (n=4) displayed strongly altered somatodendritic spike sinks of current (see an example in Figure 5), with one or two dendritically initiated small sinks propagating variably and mostly distally.
Progressive Development of Status Epilepticus
In parallel to the above-described changes in LFPs, multiunit activity, and CSD maps in KO animals, we found a progressive development of spontaneous epileptic activity in all three regions examined, cortex, CA1, and CA3. The earliest anomalous activity appeared as bouts of fast γ-like activity lasting ∼100 to 150 milliseconds, and had amplitude slightly larger than ongoing LFPs at P16 to P17 (see Figure 6). Such activity evolved in latter days to become a full-blown interictal-like pattern. Notably, interictal spikes were multiform and their amplitude and complexity increased with age (Figure 6). The frequency of events was significantly higher in CA3/FD and lowest in the cortex (cortex: 8±3; CA1: 34±13; CA3/FD: 73±15 events/min, ∗∗P<0.01, one-way ANOVA followed by Bonferroni's post hoc test; n=8, P18 to P22; Figure 6C, 1). However, within each region, the rate of events did not significantly change from P18 to P22 (Figure 6C, 2, two-way ANOVA).
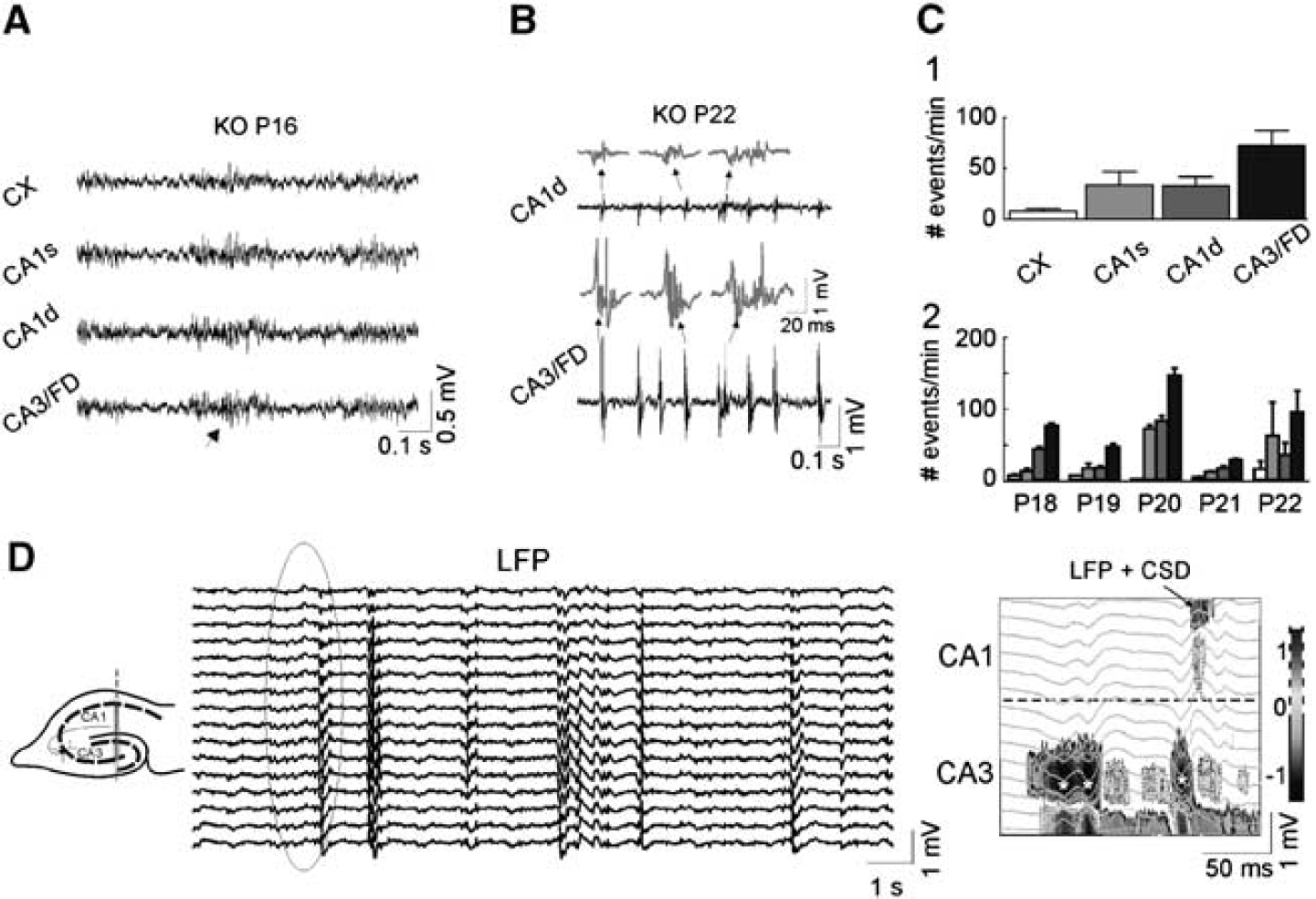
Development of epileptic activity in knockout (KO) animals. (
We here also suspected a strong volume contribution of epileptic spikes in the CA3/FD region toward distant regions (note the much reduced amplitude in pairs of simultaneous recordings in Figure 6). We therefore performed a detailed exploration using CSD analysis in multiple LFP segments to better localize the source region for epileptic activity. It was found that in all cases, the field epileptic complex begun in the CA3/hilar region. Strikingly, in two out of five animals, these field bursts did not propagate into the CA1, as noted by the absence of currents at the expected times, that is, the local field activity in the CA1 was not generated locally but volume propagated (see example in Figure 6). In epileptic CA3 field spikes examined in five animals, only 16.3%±5.1% propagated to the CA1. The CSD of epileptic bouts in the overlying cortex never showed any local current; hence the small epileptic field bursts were also remote, that is, from the hippocampus.
Bioenergetic Characterization of Intact Aralar-Knockout Cultured Neurons
Basal and coupled oxygen consumption rate in neuronal cultures from Aralar-KO mice were only slightly diminished with respect to those from WT mice (Figure 7). However, the maximal respiration obtained in the presence of the uncoupler dinitrophenol is very much reduced in Aralar-deficient neurons. This maximal respiratory activity is limited by the supply of substrates to mitochondria (Brand and Nicholls, 2011), and thus reflects the limitation in glucose-derived pyruvate supply to mitochondria in the absence of a functional malate-aspartate shuttle. This reduced maximal respiratory activity will probably limit ATP production upon neuronal activation and spiking activity of fast firing neurons in vivo.
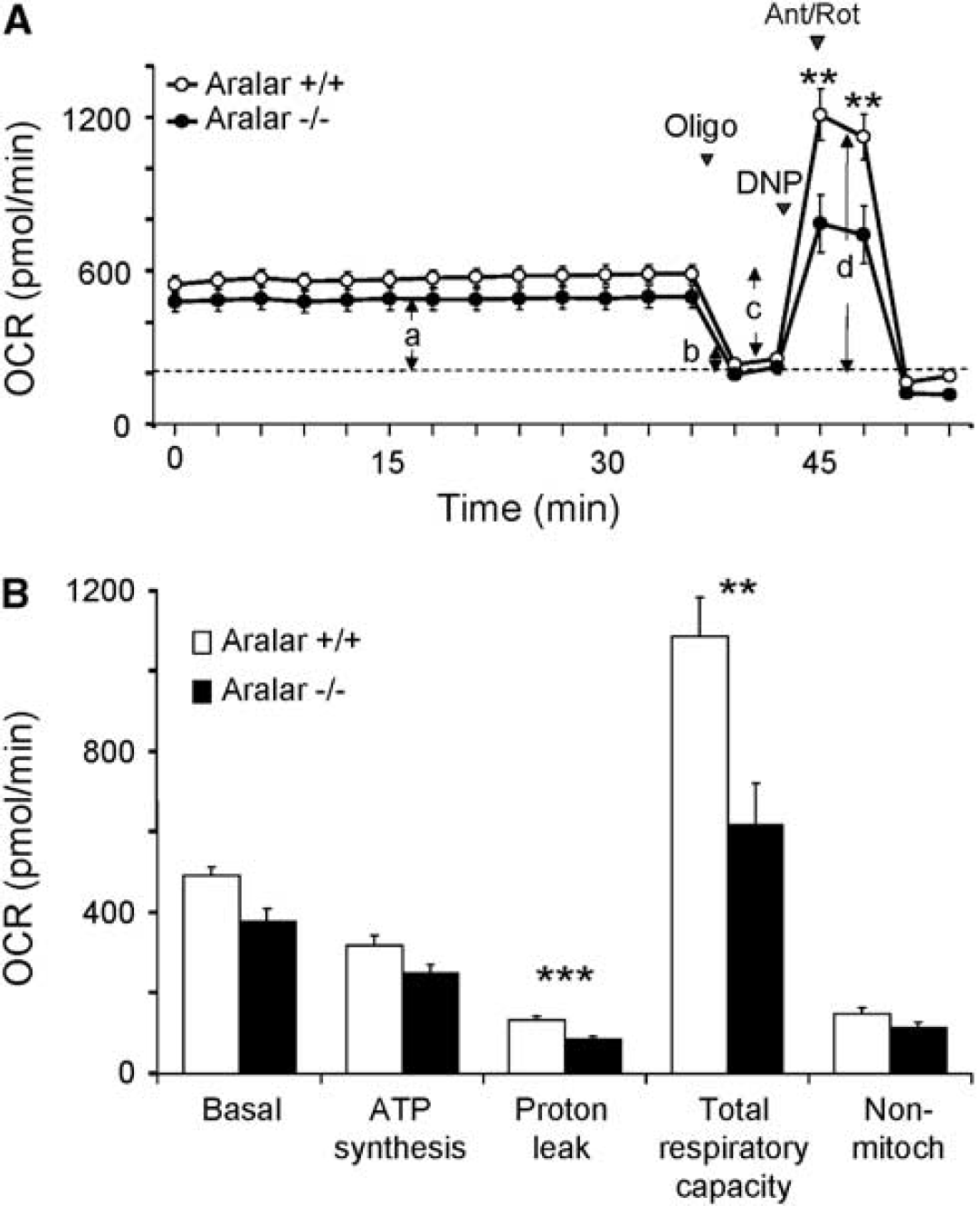
Bionergetic profile in primary neuronal cultures from wild-type (WT) and Aralar-knockout (KO) embryos. (
Expression of Glutamic-Acid-Decarboxilase
The gross anatomy was examined in cresyl violet-stained sections (Figure 8A). We did not find noticeable changes in the main architectural features of the cortex and hippocampus in Aralar WT versus KO mice, in agreement with former studies (Jalil et al, 2005; Ramos et al, 2011). In light of the above results showing the progressive instauration of an epileptic phenotype, we performed a study to determine GAD in the hippocampus (Figure 8B), the enzyme involved in the synthesis of the inhibitory neurotransmitter GABA (γ-aminobutyric acid). The levels of GAD decreased notably in KO animals (ratio of GAD-65/β-actin: 1.5±0.3 WT versus 0.8±0.2, n=5; P<0.05, Student's t-test) that may result in deficient operation of the inhibitory GABAergic system.
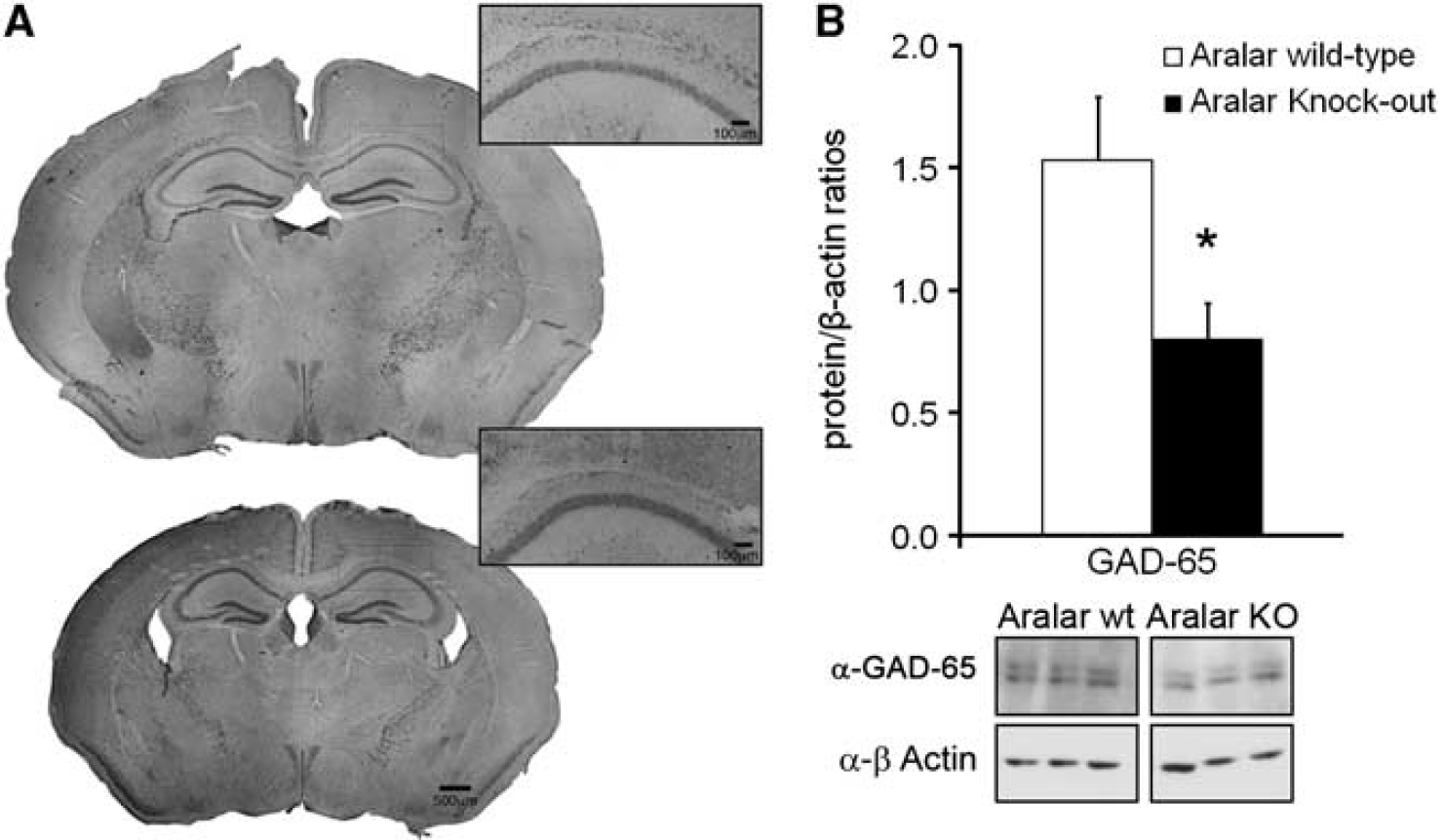
Structural characterization and glutamic-acid-decarboxilase (GAD)-65 quantification in hippocampus from aralar wild-type (WT) and knockout (KO) mice. (
Discussion
The brain uses complementary pathways for energy and metabolite production that may be more or less active throughout the animal's life. Here, we show that genetically modified Aralar-deficient mice lacking mitochondrial NADH malate-aspartate functional shuttle that affects directly to neuroglial metabolic coupling in the brain, develop abnormal electrogenesis in critical postnatal periods that lead to status epilepticus and death. Thus, Aralar-KO mice show a poor development of the LFP in cortex and hippocampus in parallel to progressive silencing of spike activity and altered somatodendritic spike electrogenesis. We infer that the reduced synaptic and abnormal spike electrogenesis lie beneath the altered and proto-epileptic phenotype.
Postnatal Electrophysiological Development Is Arrested in Aralar-Knockout Mice
It is known that an important refinement of brain circuits and function takes place after birth. Our electrophysiological monitoring began immediately after weaning, when LFPs have already appeared (Gramsbergen, 1976), and extended through the third postnatal week, since Aralar mice do not survive longer. In this period, LFP growing is drastically slowed down in Aralar-KO mice. An exhaustive interpretation of LFP power is unfeasible as some relevant parameters are unknown, particularly the variable cancellation of mixing sources (Korovaichuk et al, 2010). One factor likely having a role during this period is tissue impedance since the extracellular volume fraction reduces from about 45% at birth to 15% to 20% during the first month (Lehmenkühler et al, 1993). The end of gliogenesis (Riol et al, 1992) plus the continuing arborization of neuron dendrites explain the impedance increase by reduction of the volume fraction. While the gross anatomy of the Aralar-KO mice appears normal (Jalil et al, 2005), the delayed development or degeneration of neuronal processes observed in these mice (Ramos et al, 2011) and/or altered gliogenesis may be involved. Additional factors appear to be at play. On one side, it is known that Aralar-KO mice are strongly hypomyelinated in central but not peripheral nervous system, most likely because of a shortage of N-acetylaspartate, a neuronal precursor of myelin lipids that requires aspartate produced in neurons as a precursor (Jalil et al, 2005). Myelination peaks during the third postnatal week in the rat, which may explain the motor and exploratory deficits in Aralar-KO mice 1 to 2 days before death by generalized failure of action potential conduction and sensory-motor integration. Similar phenotypes have been reported in other mutants for myelin components (i.e., the shiverer and jimpy mutants; Honke et al, 2002; Saher et al, 2005).
We find that hippocampal unitary activity is strongly depressed, even arrested in elder Aralar mice. Although the depression may be overestimated by anesthesia, it explains the reduction in hippocampal LFP power. Thus, most unit activity belongs to putative interneurons recorded outside the pyramidal layer (Ranck, 1973), which in the hippocampus, and possibly the cortex too, are responsible for most of the LFP variance (Korovaichuk et al, 2010). The resting Na+–K+ ATPase current density in fast spiking interneurons, was found to be three- to sevenfold larger than that of cortical pyramidal neurons, indicating a high energy demand in interneurons (Anderson et al, 2010). This is in concordance with the higher number of mitochondria and stronger cytochrome c staining of interneurons compared with principal cells (Gulyás et al, 2006). About a dozen different types of interneurons are recognized in the CA1 subfield (Somogyi and Klausberger, 2005) whose activity tailor finely the synaptic integration from afferent nuclei in the projection pyramidal cells. One of LFP frequency bands dominated by interneuron activity is γ oscillation (Bartos et al, 2007), which is known to be reduced in several pathophysiological disorders, including epilepsy and ischemia (Barth and Mody, 2011). This frequency band showed a marked reduction in Aralar-KO mice, a result compatible with the reported exquisite dependence of γ oscillations in LFPs on the mitochondrial oxidative capacity (Kann et al, 2011).
The very low LFP activity in CA1 in Aralar-KO animals may reflect a limitation in the production of GABA by the interneurons, as indicated by the decreased GAD immunostaining. On the other hand, neurons from Aralar-KO mice have a clear metabolic impairment in glucose utilization due to the lack of a functional malate-aspartate shuttle, which results in an increase in lactate production (Pardo et al, 2011). We have now observed that maximal respiration is very much reduced in Aralar-KO neurons, and this will result in a reduced ATP production at the expense of glucose. Therefore, the much reduced LFP activity in CA1 is likely associated with a lack of input from the fast firing interneurons due to failure of these cells to sustain maximal respiration and ATP production in Aralar-KO animals.
On the other hand, although we have not estimated unitary activity of outgoing pyramidal cells, we rarely found spike activity in the cell body layer of Aralar-KO mice. Besides, CSD analysis indicated that KO mice had abnormal spike electrogenesis in these cells in half of the animals tested, consisting on failure of action potential generation in the soma while dendritic action currents are still present (though aberrant too). This may suggest an altered development/localization of Na+ or other channel types during these critical developmental periods, which may have a role in the final performance of the aralar-KO mouse and perhaps in human AGC1 deficiency (Wibom et al, 2009).
We found epileptogenic activity in Aralar-KO mice originating in CA3/hilar region. It may arise from a deficient or delayed maturation of the GABAergic system, which in the rat switches from depolarizing to hyperpolarizing in the second postnatal week (Tyzio et al, 2008). In the rat, an early rhythmic bursting activity has been reported in the CA1 that ends by P15 to P16 (Garaschuk et al, 1998). An increased propensity to seizure activity has been reported for this period (Mohns et al, 2007). The slowed development in Aralar-KO mice may extend the period of natural bursting activity, which may then become pathological (see Le Van Quyen et al, 2006).
Alternatively, the epileptic activity may have a different origin. Expectedly, a decreased inhibitory background (silenced interneurons) would render pyramidal cells hyperexcitable. This would not be reflected in evoked or unitary activity in the CA1 region because of the accompanying strong alterations in the intrinsic electrogenesis of pyramidal cells themselves. It was however clearly visible in the CA3 region that undergoes a progressive instauration of status epilepticus. Strikingly, while hypersynchronous activity arises from the CA3, it arrives in an inconsistent manner to the CA1. The frequent failure of CA1 postsynaptic activation by Schaffer collaterals during CA3 bursting activity is unexpected. One possibility is that Schaffer collaterals may drive action potentials inconsistently due to deficient myelination (Jalil et al, 2005). In support of this idea, evoked excitatory currents weakened in elder Aralar-KO mice. However, the strongly reduced CA3–CA1–cortex flow of activity does not rule out completely a hippocampal origin for seizure activity in the awaken animal.
We cannot ascertain whether seizures cause the animals death. We never recorded an electrographic seizure under anesthesia. Also, the epileptic activity in the cortex turned out to be volume propagated from the hippocampus. Our speculation is that anesthesia not only depressed ongoing activity, but also raised the threshold for seizure activity. Since the interictal pattern worsened with age in our recordings in anesthetized animals, we may speculate that motor seizures were at some point lethal in awaken animals. Occasionally, we often witnessed a typical motor seizure with tonic-clonic components from which the animal did not recover.
Functional and Clinical Implications
We cannot fix the time point at which electrophysiological abnormalities begin in the Aralar-KO mice since the LFP and evoked field potential differences with control animals were already observed at the earliest postnatal day studied (P15). Postnatal development is a continuum process in which structural (dendritic and axonal arborization and pruning), cellular (receptor and ion channel expression, and insertion in the membranes), and dynamic (activity-dependent synapse stabilization, wiring refinement, activation of silent synapses) factors interact finely (Monyer et al, 1994; Bolshakov and Siegelbaum, 1995; Represa and Ben-Ari, 2005; Aizenman and Cline, 2007). Some of these processes evolve in a continuous manner while others have a critical peak in time. Poorly developed early activity in Aralar-KO neurons may thus constitute a negative signal leading to abnormal subcellular progress of the electrogenic mechanisms in neurons, severe decline of neuron spiking, and functional disconnection of circuitry. Astrocytes processes wrap tightly around synaptic neurons and there is increasing evidence for signaling between astrocytes and neurons by way of the release of transmitter molecules such as glutamate (Perea and Araque, 2007). Whether the progressive decline in the synthesis of glutamate and glutamine in astrocytes from Aralar-KO (Pardo et al, 2011) is also involved in the abnormal progression of electrogenic mechanisms in aralar-KO neurons is an open question.
Footnotes
Acknowledgements
The authors thank Belén Larrosa for her participation in pilot experiments, and Isabel Manso and Barbara Sesé for technical support. IL-F held a fellowship from the Comunidad de Madrid, Spain.
The authors declare no conflict of interest.