
Abstract
No single animal model is able to encompass all of the variables known to affect human ischemic stroke. This review highlights the major strengths and weaknesses of the most commonly used animal models of acute ischemic stroke in the context of matching model and experimental aim. Particular emphasis is placed on the relationships between outcome and underlying vascular variability, physiologic control, and use of models of comorbidity. The aim is to provide, for novice and expert alike, an overview of the key controllable determinants of experimental stroke outcome to help ensure the most effective application of animal models to translational research.
Introduction
Human stroke comes in many forms. We can classify them by cause, location, size, and by functional impact on the patient. Thus, there is no single universally appropriate model of stroke. The purpose of this review is to describe and critique the animal models most pertinent to the most common broad subtype of human stroke caused by occlusion of the middle cerebral artery (MCA). Even this apparently simple task is complicated by the need to consider questions such as the occlusive mechanism (thromboembolism from cardiac sources versus more proximal embolic processes, such as carotid atheroma), underlying vascular anatomy (the gross anatomy of the circle of Willis, the role of communicating arteries, collateralization, and anastomotic connections within the MCA and with adjacent vascular territories), and the affect of premorbid factors, such as hypertension, diabetes, obesity, and smoking habits on all of these. Within the models themselves, we also need to consider aspects of experimental design such as animal gender, temperature control, blood gas concentrations, and anesthesia that impinge directly on stroke pathophysiology.
Different Approaches to Induction of Focal Ischemia
In this section, we describe the critical characteristics of the most commonly used models rather than their precise methodology, which is beyond the scope of this review and has been described in detail by others (Wang-Fischer, 2008).
Broadly, two surgical approaches are used to give access to the cerebral vasculature to allow generation of focal ischemia.
The first group of methods requires opening of the skull to allow direct access to the cerebral arteries. In most instances, this has involved small craniotomies that allow distal branches of vessels such as the MCA to be ligated (Crowell et al, 1981), clipped (Tamura et al, 1979, 1981), or sealed by photothrombosis (Markgraf et al, 1993) or electrocoagulation (O'Brien and Waltz, 1973). Although occlusion of the vessel is usually permanent, ligatures can be released, pneumatic cuffs deflated, and even thrombotic lesions created by electrocoagulation, or photothrombosis can recanalize to permit transient occlusion.
As there are no species-specific constraints on the use of these techniques, they have been used not only in standard laboratory animals such as rabbits and rats but also in larger domesticated animals such as cats, dogs, and pigs (Corkill et al, 1978; Imai et al, 2006; Tamura et al, 1979), as well as in both small and large primates (Del Zoppo et al, 1986; Hudgins and Garcia, 1970). The larger of these species offer the significant advantages of large gyrencephalic brains with gray/white matter proportions closer to humans (Figure 1). A disadvantage of the variants that require large craniotomies to expose more proximal portions of vessels is the unavoidable damage to structures, such as the eye, temporalis muscle, and zygomatic arch. Moreover, with the recent demonstration that hemicraniectomy can have a profound beneficial effect on survival and function after space-occupying hemispheric infarction (Hofmeijer et al, 2009), the value of methods requiring large craniectomies to expose the vessels at the base of the brain is uncertain.
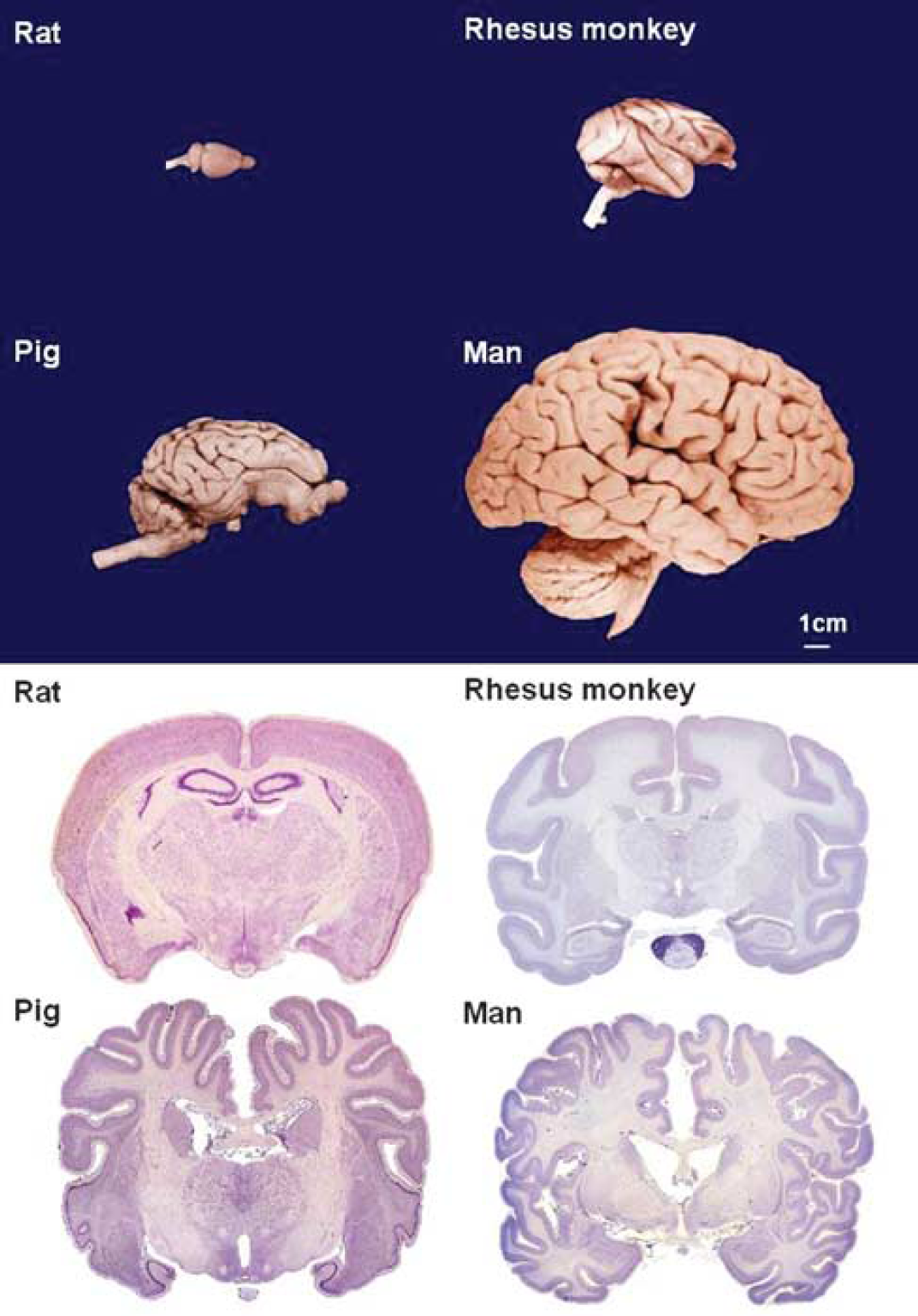
Brain size and gyral complexity.
The vasoconstrictor endothelin can also be used to reversibly occlude an artery or vascular bed (Agnati et al, 1991). Although this also requires craniotomy, the opening in the skull needs to be just large enough to introduce a fine cannula, which can be left in situ and vasoconstriction initiated long after confounding anesthesia has been withdrawn (Callaway et al, 1999). It should be noted that endothelin is about four times more potent in conscious rats than in anesthetized rats (Bogaert et al, 2000), that control over ischemic intensity and duration are limited, and that stimulation of endothelin receptors may confound the study of stroke by directly modifying the expression of key molecules, such as matrix metalloproteinases and growth factors (Koyama et al, 2003, 2007).
To avoid opening the skull, a second group of methods has used intra-arterial access to occlude cerebral arteries. The most commonly used of these is thread occlusion of the MCA. Although this method has many variants, particularly with respect to the construction of the occluding thread and closure of additional vessels to manipulate collateral blood flow, the basic technique described originally by Koizumi et al (1986) and modified by Longa et al (1989) involves introducing an occluding thread into the extracranial internal carotid artery (ICA) and advancing it until its tip occludes the origin of the MCA. Although most frequently applied to rats and mice, the method has also been used in rabbits (Kong et al, 2004), gerbils, (Baskaya et al, 1999) and marmosets (Freret et al, 2008). In baboons, the concept has been extended to the use of a balloon catheter or wire coil introduced through either the carotid (Gao et al, 2006) or femoral arteries (Hamberg et al, 2002) to occlude the MCA.
The great advantage of these techniques is that the thread can either be left in place for permanent occlusion or withdrawn any time to permit controlled reperfusion, and the presence of a significant ischemic penumbra early after occlusion makes them particularly suitable for studies of neuroprotection. However, despite their utility, these are not simple methods. The surgery to access and manipulate the vasculature requires skilled and experienced hands, and in practice, the results are often highly variable. Moreover, the diameter and length of the occluding proportion of the thread combine to determine which vessels off the circle of Willis are blocked and to what degree. Importantly, thread dimensions need to be adjusted for specific animal strains. For example, using a fixed-size silicone-coated 4-0 nylon monofilament thread, blood flow reduction varied markedly by strain with Long–Evans showing greater reductions in flow than Sprague–Dawley (SD) or Wistar rats (Prieto et al, 2005).
Uncoated monofilament threads and the poly-L-lysine-coated threads, that were introduced to increase the proportion of successful surgeries (Belayev et al, 1996), are each prone to high rates of subarachnoid hemorrhage (Schmid-Elsaesser et al, 1998), confounding the physiologic basis of the model and leading to high mortality (Spratt et al, 2006). The use of silicone-coated threads is recommended because these reduce the problems of subarachnoid hemorrhage (Schmid-Elsaesser et al, 1998) and variability (Aspey et al, 1998), particularly in in-bred strains, such as the spontaneously hypertensive rat (SHR) (Spratt et al, 2006). Additional coating of the silicone with poly-L-lysine may further enhance their utility (Lourbopoulos et al, 2008). Providing guidelines for selecting silicone-coated thread dimensions suitable for all circumstances is difficult because there are too many variables likely to alter arterial dimensions. For example, in mice, a 15 g increase in body weight can result in a doubling of the required thread diameter from 100 to 200 μm (Hata et al, 1998). Eleven-week-old male SD rats are 100 g heavier than equivalent Fischer 344 rats, and in both strains, the males are >60g heavier than the females (Seidel et al, 2006). A rational approach suitable for use across species and strains might be the first to establish the length of silicone coating desired by measuring the distance between the origin of the MCA and hypothalamic artery if occlusion of this vessel is to be avoided to minimize thermoregulatory disturbances (Li et al, 1999). This is easily carried out after perfusing the brain with Evans blue in gelatine (Crack et al, 2001). As the thread needs to pass through the carotid canal, starting with this measurement, the diameter is reduced to permit smooth passage into the skull without reducing the diameter beyond the point at which laser Doppler-measured MCA-territory flow starts to increase consistently.
Sprague–Dawley rats, the most widely used animals in stroke research (VO'C, personal communication), unfortunately give some of the most variable results (Spratt et al, 2006), most likely because of their highly variable MCA anatomy (Fox et al, 1993) and are thus not recommended as a starting point. Even the choice of vendor can alter outcome in the SD (Oliff et al, 1995). At present, the Wistar Kyoto (WKY) rat seems to be the best choice. It lacks the vascular variability of the SD, and does not display the extremes of inflammatory reactivity noted in the Lewis and Fischer 344 strains (Morand and Leech, 2001). Its genetic relationship to the SHR and stroke-prone SHR (spSHR) strains, which provide the most commonly used models of hypertension and spontaneous stroke, make it an ideal stepping stone for later preclinical evaluations.
In larger domestic animals (such as cats, dogs, sheep, goats, pigs, cows, and horses) that would otherwise offer significant advantages in size and cortical complexity, direct intravascular access to the MCA is prevented because blood is supplied to the cerebral hemispheres through a carotid rete (or rete mirabile), a plexus of fine freely anastomosing arteries. In dogs, this problem has recently been overcome by femoral artery catheterization and fluoroscopically controlled introduction of a platinum coil through the vertebrobasilar system to occlude the origin of the MCA (Rink et al, 2008).
Although thread occlusion and its variants effectively model induction of ischemia at the site most commonly occluded in humans, they do not model the mechanism of occlusion. Approximately 80% of human strokes are ischemic (Donnan et al, 2008), and most of the larger (nonlacunar) infarcts are caused by thromboembolism. Thus, the specific advantage of thromboembolic methods is that the mechanism of occlusion better matches that seen in a large proportion of human strokes, and that they permit the study of thrombolytic processes. However, success is highly dependent on the properties of the introduced clot and, as in humans, the timing of reperfusion can be uncertain.
Although the earliest embolic model of stroke was described in dogs (Hill et al, 1955), it was not until 1982 that an embolic model was described in rats (Kudo et al, 1982) using essentially the same surgical approach as used for intraluminal thread occlusion. The simplest embolic model injects a suspension of small clot fragments into the common carotid artery or ICA (Kudo et al, 1982). Reported mortality using this approach was low, but the foci of infarction were widely distributed and included significant numbers in the contralateral hemisphere (Kudo et al, 1982). With the aim of generating a more faithful model of human thromboembolic stroke in which the ‘obstructing emboli should be located in the proximal segment of a large feeder artery, the distal vascular bed should be open’ (Busch et al, 1997), most methods now in routine use introduce a single larger clot of carefully controlled dimensions and consistency close to the origin of the MCA. When the clot is introduced into the ICA, there is little control over where it lodges, allowing infarcts that can include MCA, anterior, and posterior cerebral artery territories. By advancing the clot-introducing catheter into the MCA itself, using laser-Doppler flowmetry to verify the placement, and then withdrawing the catheter slightly, it is possible to obtain a high proportion of animals with only MCA occlusion (MCAo) (DiNapoli et al, 2006). As one of the main reasons for using embolic models is to be able to study thrombolysis, consistency of the clot has received much attention. Clots derived from unmodified arterial blood (DiNapoli et al, 2006), arterial blood mixed with thrombin (Wang et al, 2001), and whole blood mixed with CaCl2 and thrombin and subjected to ‘osmotic shock’ (Toomey et al, 2002) have all been used. However, although both spontaneously formed and thrombin-induced clots seem to provide similar levels of occlusion, thrombin-induced clots appear more resistant to the effects of tissue plasminogen activator (tPA) (Niessen et al, 2003). Although more data are required to confirm this observation, it highlights an important choice for the experimenter. If the experimental aim is to provide a model system in which the beneficial effects of a new drug on infarct and behavioral outcome can be studied together with embolic blockade and tPA-mediated reperfusion, then the more readily thrombolyzed ‘red’ spontaneously formed clots have the advantage. With these, reperfusion occurs within a time frame that permits tissue salvage. However, if the aim is to study the mechanics of ‘clot busting’ and devise more effective ways of breaking up the embolus, the thrombin-induced and fibrin-rich ‘white’ emboli (Kirchhof et al, 2002) which probably better represent the emboli that cause human stroke (Jorgensen and Torvik, 1969; Marder et al, 2006), have the advantage. Marder et al (2006) report that the thromboemboli retrieved from the MCA or the intracranial ICA of patients with acute ischemic stroke have similar histologic components, whether derived from presumptive cardiac or arterial sources. A disadvantage of thromboembolic methods is that varying the timing of occlusion and time to reperfusion is not a certain art (as indeed is true in humans). With spontaneously formed emboli, reperfusion can take more than an hour; with thrombin-enriched emboli, this can extend to 5 hours (Niessen et al, 2003). Thread occlusion models offer much greater flexibility and certainty.
Although these models offer researchers the opportunity to study a more realistic model of stroke, even in experienced hands, poststroke mortality can be as high as 30% to 40% within 24 hours (Toomey et al, 2002) with reports of up to 85% mortality if animals are maintained up to 72 hours (Alonso de Lecinana et al, 2006). Although early reperfusion and generally smaller infarcts in most thread occlusion experiments probably contribute to this difference in mortality between models, the persistence of high mortality after embolism when early tPA therapy successfully reduced infarct volume (Toomey et al, 2002) suggests this may not be the whole story. Although there are obvious reasons for concern about the practicality of using these methods, it should be remembered that our only effective acute therapy, tPA, shows similar profiles of activity in animal and human thromboembolic stroke (Perel et al, 2007) and that new agents will almost inevitably need to be tested in the presence of tPA. Differences in the efficacy of tPA in rodents and humans are also of concern, but may lead to new avenues for therapy (Zhu et al, 2010).
An alternative, although related, approach to occlusion is direct induction of thrombus formation at the origin of the MCA or its more distal branch points. To this end, thrombin has been infused at the origin of the MCA in rats (Zhang et al, 1997) and rabbits (Jahan et al, 2008), by drawing blood into a thrombin-filled catheter and then releasing the freshly formed clot (Beech et al, 2001) or by injecting thrombin directly into the distal MCA of mice (Orset et al, 2007). In small animals in whom the skull is thin and allows passage of sufficient light, noninvasive and highly reproducible and high-throughput photothrombotic methods are also available (Watson et al, 1985). However, thrombosis can occur in any illuminated vessel containing a high enough concentration of photo-activating agent. Although proximal MCAo with these techniques is similar to other occlusion methods (Watson et al, 1985), illumination through the parietal cortex (Sugimori et al, 2004) seems unlikely to allow study of penumbral involvement if blood vessels are completely congested with aggregated platelets (Haseldonckx et al, 2000). A perceived disadvantage of photothrombotic methods is early vasogenic edema and blood–brain barrier breakdown. However, recent examinations of these phenomena have suggested similar marked blood–brain barrier disruption within an hour in both thread occlusion and photothrombotic models (Chen et al, 2009; Stoll et al, 2009).
Table 1 provides an overview of the advantages and disadvantages of the most commonly used models of stroke, whereas Figure 2 shows the frequency with which different model types were used in a recent analysis of neuroprotection in stroke (O'Collins et al, 2006). Of experiments using intraluminal sutures, 51.4% used heat-blunted or mechanically formed sutures, 42.2% use silicone coated sutures, and 6.4% used poly-L-lysine-coated sutures.
Overview of advantages and disadvantages of commonly used models of stroke
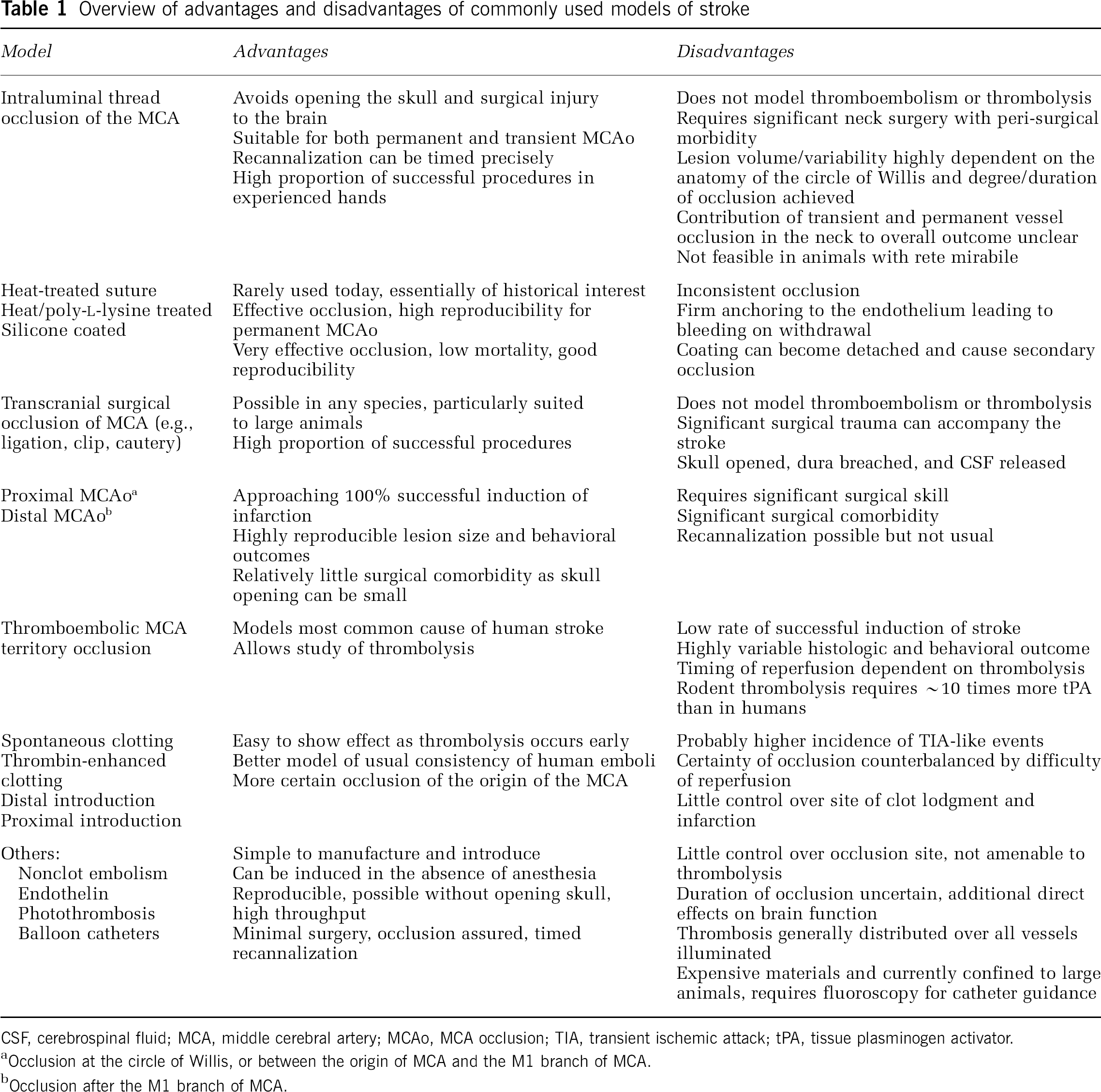
CSF, cerebrospinal fluid; MCA, middle cerebral artery; MCAo, MCA occlusion; TIA, transient ischemic attack; tPA, tissue plasminogen activator.
Occlusion at the circle of Willis, or between the origin of MCA and the M1 branch of MCA.
Occlusion after the M1 branch of MCA.
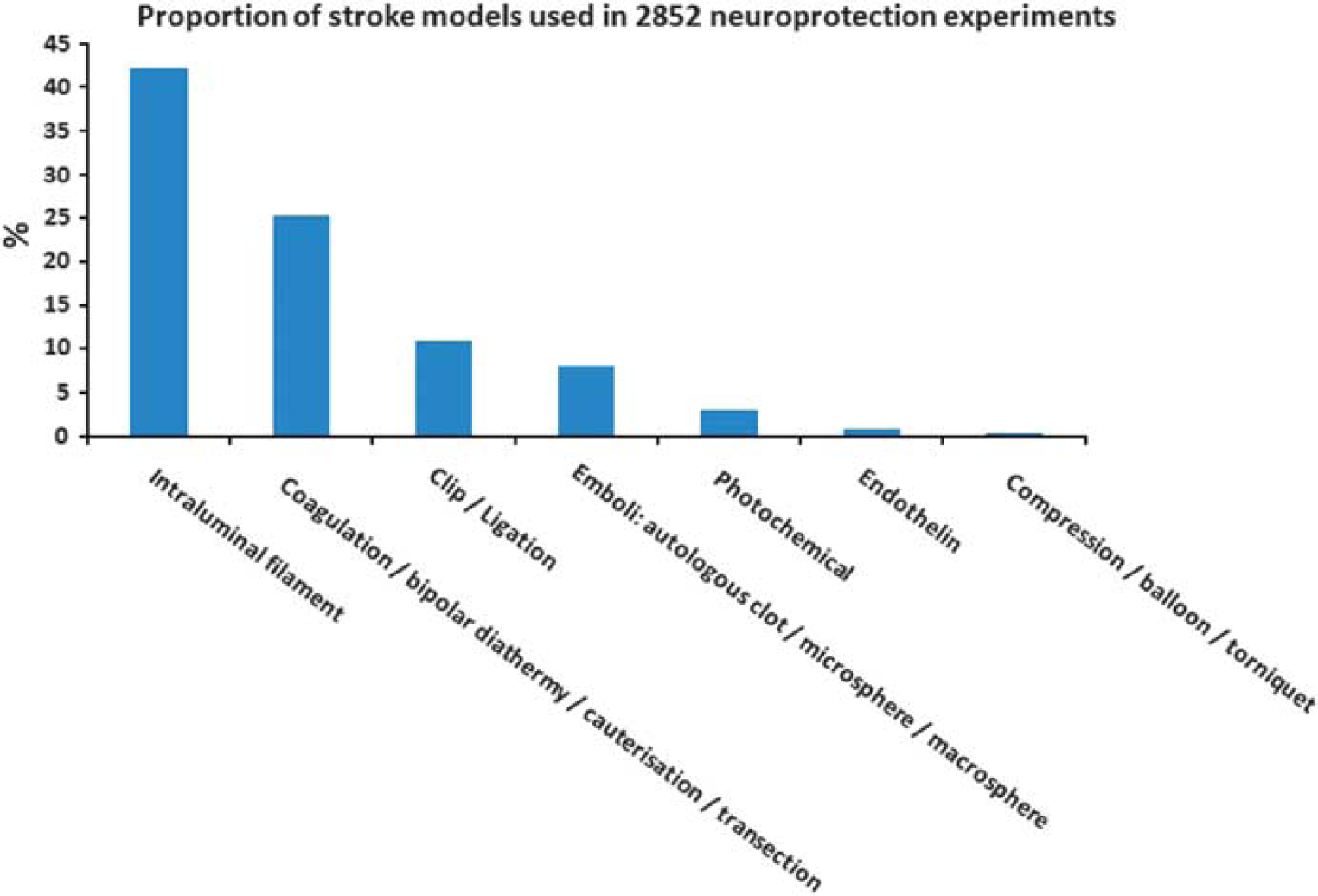
Proportion of stroke models used in 2,852 neuroprotection experiments.
Vascular Anatomy and Concordance with Human Disease
Stroke incidence and subtype proportion vary considerably between communities (Feigin et al, 2006), but overall, occlusion of (a branch of) the MCA is the most commonly identified type of human ischemic stroke (Olsen et al, 1985) and thus the most common target for animal models.
Although the human brain sits at one end of the spectrum of mammalian brain complexity, it still adheres to the basic mammalian pattern of neural and vascular organization. Thus, strokes induced in laboratory animals look very similar to those in humans. Blockage of the origin of the MCA in most mammals studied results in infarcts, which incorporate the gray matter of the motor and somatosensory cortex, the underlying white matter tracts, and the basal ganglia (caudate-putamen and thalamus), which have blood supplied by the small perforating arteries that branch from the MCA or adjacent segments of the circle of Willis.
Despite the broad similarities, there are important differences between species and strains of animals that can affect the experimental outcome. In humans, a pronounced anterior communicating artery usually completes the circle of Willis providing some capacity to redistribute blood between hemispheres. Although relatively uncommon (Kapoor et al, 2008), when the anterior communicating artery is absent or of reduced bore, ischemic stroke outcome may be worse (Jaramillo et al, 2006). Similarly, patients (∼30%) who have absent or hypoplastic posterior communicating arteries appear to be at a greater risk of stroke (Chuang et al, 2008). Cross-sectional studies in humans also suggest that an incomplete circle of Willis distinguishes patients with symptomatic and asymptomatic ICA stenosis (Waaijer et al, 2007).
Similar anatomic variation is seen in most species that have been used to model stroke. A recent examination of the incidence of anatomic variation in the circle of Willis in humans, cows, sheep, goats, and pigs illustrates that although variation is greatest in humans (probably because a deeper genetic pool was sampled), it is also present in most other species (Ashwini et al, 2008). Rats and mice, the animals most commonly used to model stroke, exhibit similar variation. In rats, differences in the posterior communicating artery bore influences of the outcome of white matter injury induced by chronic cerebral hypoperfusion, with Wistar rats with small-diameter vessels more susceptible to ischemic damage than SD rats (Kim et al, 2008). In mice, which are becoming increasingly important because of the availability of transgenic animals, it has been reported that only 10% of C57Black/6 mice have a complete circle of Willis (McColl et al, 2004). In CD1 mice, patency of the posterior communicating artery is not only highly variable but also correlated with the extent of ischemic injury (Zhen and Dore, 2007). Similarly, the increasing sensitivity of BDF (F(1) hybrids of C57BL/6 and DBA/2 normal strains) <CFW (SwissWebster) <BALB/C mice to MCAo seems to be dependent on a decreasing frequency of patent posterior communicating arteries (Barone et al, 1993). It seems likely that possession of a patent posterior communicating artery permits maintenance of residual cortical perfusion above the ischemic threshold in territories that would otherwise die after MCAo (Kitagawa et al, 1998). Restricting collateral blood supply by occlusion of additional vessels can have a profound effect on the regions infarcted and on experimental variability (Chen et al, 2008). Indeed the gerbil has been promoted as a model species for stroke studies, specifically because their lack of posterior communicating arteries and an absence of an anterior communicating artery in ∼20% of the gerbil population leads to more consistent infarct volumes (Oostveen et al, 1992).
Vascular variability and plasticity in more distal parts of the cerebral circulation also has the potential to alter human stroke outcome and animal modeling. Sprague–Dawley rats (∼60% of all neuroprotection data come from this strain (VO'C, personal communication) display a much greater range of infarct sizes in thread occlusion models than strains, such as the SHR and WKY (Figure 3), probably because of the highly variable origin and branching pattern of the MCA in this strain (Fox et al, 1993). Similarly, although changes in vascular contractility and blood–brain barrier permeability undoubtedly contribute to the propensity to stroke in SHR and spSHR strains (Knox et al, 1980), the observation that anastomotic vessels linking the anterior cerebral artery andMCA territories are narrower in spSHR than in normotensive rats is probably a major determinant of blood flow to the threatened territory and of the amount of tissue that can be protected from infarction by collateral circulation (Coyle and Heistad, 1991).
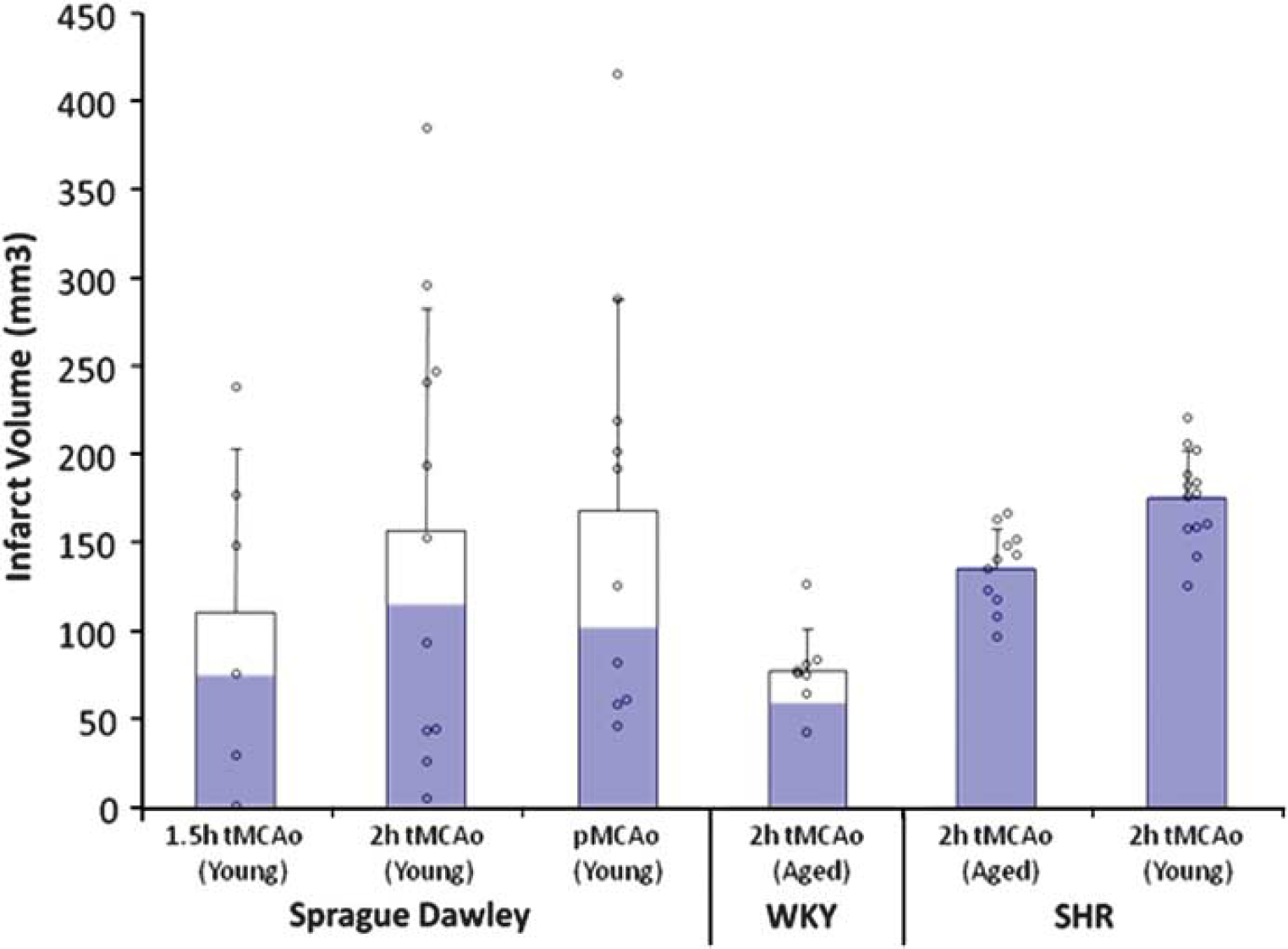
Not all rats were created equal: infarct volume variability after thread occlusion in Sprague–Dawley (SD), Wistar Kyoto (WKY), and spontaneously hypertensive rat (SHR) under a range of experimental circumstances. Blue bars represent the proportion of animals with cortical infarction. Mean and s.d. plus individual data points.
Although it is possible to exploit these species/strain differences to reduce experimental variability in infarct volume, the relevance to human stroke subtypes must always be considered if the aim is the evaluation of a drug's potential to treat human stroke.
Experimentally Controllable Physiologic Variables that Affect Outcome
In addition to differences in model construction and the underlying vascular anatomy, controllable variables such as regulation of blood flow, temperature, and blood gas concentration all have the potential to affect experimental outcome.
Blood Flow
One of the most important variables is the reduction of blood flow achieved by thread or embolus occlusion. In most species studied, including humans, the evidence suggests that unless blood flow is reduced to below a flow of ∼0.12 ml/g per min for a significant period, infarction is not inevitable. Above this threshold, electrical activity and normal function may be suppressed, but there is sufficient metabolic reserve to preserve cellular integrity (Astrup et al, 1981; Sakoh et al, 2000). It is not uncommon for animals to have acute, but transient, functional deficits upon waking from anesthesia, which do not progress to frank infarction (Sicard et al, 2006).
In laboratories without access to high-resolution computed tomography or magnetic resonance imaging, laser-Doppler flowmetry is widely used to judge whether blood flow reduction has been sufficient to induce infarction and reveal when spontaneous reperfusion is a cause of failed experiments (DiNapoli et al, 2006; Schmid-Elsaesser et al, 1998), emphasizing the need for frequent and long-term monitoring. Moreover, it has been reported that excessive and sustained reduction of cortical blood flow (93.6% ± 5%) after thread occlusion suggests subarachnoid hemorrhage (Woitzik and Schilling, 2002). When occlusion is successful, both thread and embolic methodologies produce similar reductions (70% to 85%) in blood flow detected over the parietal cortex (Chen et al, 2008; DiNapoli et al, 2006; Schmid-Elsaesser et al, 1998; Woitzik and Schilling, 2002). Endothelin-1-induced occlusion of the MCA produces a similar reduction in blood flow (Bogaert et al, 2000), but local induction of thrombosis by thrombin seems to be less effective, producing deficits of only 40% to 50% of baseline (Orset et al, 2007). In most laboratories, laser-Doppler flowmetry probes are placed over the parietal cortex of rodents because the lack of musculature and a relatively flat skull make probe attachment easy. However, these sites sample a varying mixture of MCA and anterior cerebral artery territory flow. Harada et al (2005) have reported that sampling MCA flow specifically by placing a flat probe between the temporalis muscle and the lateral aspect of the skull allows more successful induction of stroke with smaller variation. Although the numbers of animals studied were small (12 per cohort), the suggestion warrants further attention.
Temperature
Temperature is an important determinant of mammalian cell function and survival. Our biochemistry has evolved to function most effectively within narrow temperature ranges, and we have evolved specific mechanisms to help maintain an optimal body temperature and to limit damage to our proteins if we overheat.
In small mammals, a precipitous decrease in body temperature is common during anesthesia because their high-surface-area-to-mass ratio makes thermoregulation difficult, a phenomenon compounded by the use of unwarmed gases during inhalational anesthesia (Haskins and Patz, 1980). With agents such as sodium pentobarbital, the core temperature can decrease by 3.5°C to 4.5°C within an hour and brain temperature can be 0.3°C to 0.4°C lower (Kiyatkin and Brown, 2005). As cooling can be profoundly neuroprotective (van der Worp et al, 2007), preclinical evaluation of neuroprotectants should at some stage incorporate an evaluation of the impact of body or brain temperature. Although some recommend measuring both brain and body temperatures (Busto et al, 1989), the observation that for isoflurane, the differential between the two measurements is constant (R2 = 0.9996) (Zhu et al, 2009) suggests that less-invasive measures of core temperature may suffice. It would be foolish to simply constrain animal temperature to the normal range during experiments as the induction of hypothermia, or other changes that lead to it, might be the mechanism of action of a new drug. A number of candidate pharmacological neuroprotectants such as the AMPA (α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate) receptor antagonist 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione) (NBQX) (Nurse and Corbett, 1996) and the sedative clomethiazole (Visser et al, 2005) probably involve hypothermic effects in their mechanism of action, whereas others such as Mg2+ (Campbell et al, 2008) and tacrolimus (FK506) (Nito et al, 2004) are reported to have their effects enhanced by even mild hypothermia. Even the prototypical antiexcitotoxic dizocilpine (MK-801) alters body temperature and can have its effects masked by concomitant hyperthermia (Memezawa et al, 1995).
Hyperthermia is a common and significant complication in stroke. In humans, a core temperature of >37.5°C on the first day after stroke onset has been reported in up to a third of patients and is a strong predictor of poor outcome. Conversely, low body temperature on admission is associated with good short-term outcome (den Hertog et al, 2007). In rats, increasing the body temperature to 40°C 24 h after stroke has been reported to cause up to a three-fold increase in infarct volume (Kim et al, 1996). Furthermore, there are fluctuations in endogenous temperature of up to 1.3°C which have been found to correlate with differences in stroke size found when surgery is conducted at different times of the day (Vinall et al, 2000). In experimental stroke, hyperthermia is a particular problem when the thermoregulatory centers of the hypothalamus are damaged by infarction. The body temperature can increase quickly (within 15 minutes of the onset of ischemia), reach 39°C to 40°C within an hour, and can be sustained for >24 hours (Reglodi et al, 2000). Because of the mechanics of occlusion, this problem is usually only seen when thread occlusion blocks the multiple vessels which contribute to hypothalamic perfusion (Li et al, 1999).
Anesthesia
Anesthesia is required at some stage in all models of stroke, which require surgery for access to the vasculature or the brain. For the most part, anesthetics act through two principal mechanisms: an increase in inhibition through GABA A receptors (such as barbiturates, benzodiazepines, propofol, isoflurane, etomidate, enflurane, and halothane) or a decreased excitation through NMDA (N-methyl-D-aspartic acid) receptors (such as nitrous oxide, ketamine, and xenon) (Traystman, 2010). However, anesthesia itself seems to have both neuroprotective and preconditioning effects mediated by the inhibition of spontaneous depolarization (Patel et al, 1998), activity as antioxidants (Wilson and Gelb, 2002), antagonism of NMDA receptors (Harada et al, 1999), GABA potentiation (Harris et al, 1994), and alteration of cerebral blood flow redistribution (Warner et al, 1989). Early clinical observations that patients under general anesthesia were more tolerant of ischemia than were unanesthetized patients (Wells et al, 1963) support this view.
Data obtained from animals are however difficult to interpret. The impractibility of unanesthetized surgery makes experimental control difficult, and the precise mechanism is difficult to ascertain when effects on the cerebrovasculature, brain metabolism, brain electrophysiology, temperature, and blood pressure can all interact (Traystman, 2010). Observations that some agents induce neuronal apoptosis which can potentially make these agents neurotoxic (Ikonomidou et al, 1999) confuse the picture further. Importantly, anesthesia can interact with neuroprotectants to increase apparent efficacy (Macleod et al, 2005a, b ). Whether this is caused by enhanced induction of hypothermia, suppression of metabolism, modulation of blood flow, or specific neurochemical interactions is not always clear.
A practical approach is to avoid using anesthetics with marked intrinsic neuroprotective properties (Anderson and Sundt, 1983; Macleod et al, 2009) such as barbiturates and ketamine, which also make the depth and duration of anesthesia difficult to control. Instead, inhalational anesthetics such as isoflurane are recommended because of the ease with which the depth of anesthesia can be controlled and animals recovered, even though they also have neuroprotective properties (Warner et al, 1993). If mechanical ventilation and if possible continuous pCO2 monitoring for dose adjustment are available, this approach is recommended (Zausinger et al, 2002). Although spontaneous breathing of inhalational anesthetics gives less experimental control and larger infarcts (Zausinger et al, 2002), it provides a practical solution for smaller laboratories. Whatever the route of administration, overuse is to be avoided, and with inhalational agents, the staff must be protected from inadvertent exposure. As human stroke patients are not routinely anesthetized, developing methods that avoid anesthesia during stroke induction, as is possible with thromboembolic (Zivin et al, 1985) and endothelin (Callaway et al, 1999) models, should perhaps receive more attention. To date, there does not seem to have been a formal comparison of different drugs with or without anesthesia at the time of stroke induction in the thromboembolic or endothelin models.
Blood Gases, Blood Pressure, and pH
In an ideal world, changes in partial pressure of O2 and CO2, pH, as well as blood pressure and anesthetic concentration would be monitored constantly during stroke modeling and adjusted minute by minute to ensure that blood and oxygen supply to the tissue beds only changed because of the stroke, and not by some inadvertent vasoconstriction or dilation of collateral blood vessels caused by our experimental machinery. We know that blood gas concentrations influence experimental stroke outcome (Browning et al, 1997; Zausinger et al, 2002), that increasing blood pressure slightly improves blood flow and oxygen metabolism (Shin et al, 2008), and that pH also influences outcome (Anderson and Meyer, 2002). However, we currently lack the data and sophistication required to allow us to effectively control all of these parameters. Monitoring them is a starting point and many believe this should be mandatory. However, to be of value, the frequency of monitoring needs to be high enough to detect a number of relatively short periods of decompensation during occlusion. A similar scenario is seen when laser Doppler measurements of cortical flow velocity reveal transient periods of premature reperfusion, which keep an animal above the threshold for infarction (Schmid-Elsaesser et al, 1998).
Effects of Comorbidities on Outcome
Perhaps the most powerful strategy in animal modeling is to identify and analyze the key subcomponents of a problem and thus reduce the complexity of human disease to manageable proportions. Conversely, to evaluate the therapeutic potential of a new therapy, we may need, at some point, to model those complexities. In the development of stroke drugs, we have often failed to consider the impact of risk factors such as hypertension and diabetes that are present in a large proportion of patients with ischemic stroke (Fisher et al, 2009a).
Co-morbidities in the Clinic
Hypertension can account for 30% to 40% of the risk of stroke (Lawes et al, 2004). A 10mmHg increase in arterial blood pressure increases stroke risk by 20% to 30% (Alberts and Atkinson, 2004) and a blood pressure above 120/80mmHg doubles the lifetime risk of stroke (Kelly et al, 2007). Similarly, hyperglycemia and diabetes are common in stroke patients. Approximately one-fourth of stroke patients have a history of diabetes (Kaarisalo et al, 2005), whereas hyperglycemia is detected in up to 40% of stroke patients on admission (Williams et al, 2002). Both type 1 and type 2 diabetes are associated with an increased risk of stroke (Jeerakathil et al, 2007; Sundquist and Li, 2006) with newly treated type 2 diabetes doubling the short-term risk of stroke in one study (Jeerakathil et al, 2007). In the United States, it has been estimated that 37% to 42% of all ischemic strokes may be attributable to the effects of diabetes alone or in combination with hypertension (Kissela et al, 2005). Moreover, although hypertension and type 2 diabetes increase stroke risk independently, their combination appears to increase the risk drastically (Hu et al, 2005). As for hypertension, evidence is beginning to emerge which suggests that better diabetic control reduces stroke risk (Boden-Albala et al, 2008).
Although perhaps not strictly comorbidities, age and gender have a profound influence on stroke biology. Between 19 and 77 years of age, each additional year of age increases stroke risk by 9% in women and 10% in men (Asplund et al, 2009). Although the risk is greater in men, because they live longer, women are more likely to experience a stroke and to have a more disabling stroke (Reeves et al, 2008).
Clearly, it is important to know whether candidate stroke drugs retain efficacy in the face of these comorbidities and how they influence the pathophysiology of stroke. However, of 3,142 animal experiments on neuroprotection abstracted from the literature (O'Collins et al, 2006), only 11% involved testing in hypertensive and only 1% in diabetic animals.
Hypertension
At least 20 models of hypertension have been reported. These range from surgical ligation of arteries supplying a kidney, through pharmacological or genetic manipulation of vascular reactivity, to selective breeding of hypertensive rabbits and rats (Lerman et al, 2005). Despite the range of available methodologies, only a few have been used in stroke modeling.
The Dahl salt-sensitive rat develops hypertension dependent on the salt content of their diet (Meneely and Ball, 1958). On a high-salt diet (8.7% NaCl2), marked hypertension (∼200mmHg systolic) develops in ∼4 weeks and blood–brain barrier disruption, stroke, and death can quickly follow (Payne and Smeda, 2002). At lower salt concentrations, the same end is reached but over a longer time frame, with animals fed a 1% NaCl2 diet starting to die at ∼5 months of age (Rapp and Dene, 1985). Arterial lesions characterized by histiocytic, eosinophilic, and neutrophilic infiltration and frank coagulation are prominent in the mesentery, pancreas, intestine, testis, heart, and kidney but absent from the brain and lung (Rapp and Dene, 1985). When thread occlusion for 120 minutes was used to occlude the MCA of Dahl salt-sensitive rats after 5 weeks of high-salt diet (8%), 80% died or experienced intracranial hemorrhage within 24 hours. Reducing occlusion time to 90 minutes still left 40% of the animals with hemorrhage (Bright et al, 2007). These losses are substantially higher than reported with similar occlusion times in SHRs (21 or 3% using poly-Llysine- or silicone-coated filaments, respectively) (Spratt et al, 2006).
The SHR and related spSHR, which were both selectively bred from the WKY strain, are the most widely used hypertensive animals in stroke research. Although normotensive at birth, SHRs start to develop hypertension in the first 2 to 4 months of life and usually reach a stable systolic blood pressure plateau of ∼200mmHg by 6 months. The phenotype of the SHR and spSHR is complex. In addition to hypertension, these animals have smaller brains (Tajima et al, 1993), enlarged ventricles (Bendel and Eilam, 1992; Tajima et al, 1993), hypertrophy (due to increased smooth muscle cell number) of the large cerebral arteries (Mangiarua and Lee, 1992), increased circulating monocyte number, increased endothelial macrophage infiltration, and inflammatory marker expression (Liu et al, 1996).
The spSHR was derived by inbreeding of the offspring of SHRs that died of stroke until >80% of the population developed stroke characterized by multifocal microvascular and spongy-cystic parenchymal lesions (Fredriksson et al, 1988). Similar to the Dahl rat, spSHRs are prone to salt-sensitive renal injury which precipitates hypertension (>240mmHg) and rapid onset of hemorrhagic stroke (Lee et al, 2007). Approximately 70% of strokes occur in the gray matter of the cortex (Yamori et al, 1976). Magnetic resonance imaging studies confirming vasogenic edema and blood–brain barrier breakdown without metabolic impairment have confirmed the absence of ischemia as a precipitating event (Guerrini et al, 2002). The cortical focus for the lesions is believed to reflect abnormal vascular structure and vascular reactivity (Baumbach et al, 1989; Coyle, 1987). After MCAo, nitric oxide production is also impaired and correlates with increased infarct size (Kidd et al, 2000).
After induction of focal ischemia, blood flow reductions are more pronounced and brain injury as measured by infarct volume and behavior after focal stroke in SHR and spSHR is significantly larger and more reproducible than in normotensive rat strains (Barone et al, 1992; Spratt et al, 2006). Unlike the gerbil (Baskaya et al, 1999), this reproducibility is not due to the absence of the posterior communicating arteries (Figure 4) (Ogata et al, 1976) and the constraints on collateral blood flow this imposes. However, larger than normal infarcts in the spSHR are independent of blood pressure, age, or sex and appear to result from inadequate cortical collateral blood flow (Gratton et al, 1998). Moreover, because SHRs have smaller body and brain weights than the WKY strain throughout life and successful thread occlusion is highly dependent on the relationship between thread and vessel size, using the WKY as a normotensive control for the SHR or spSHR presents a significant challenge in its own right.
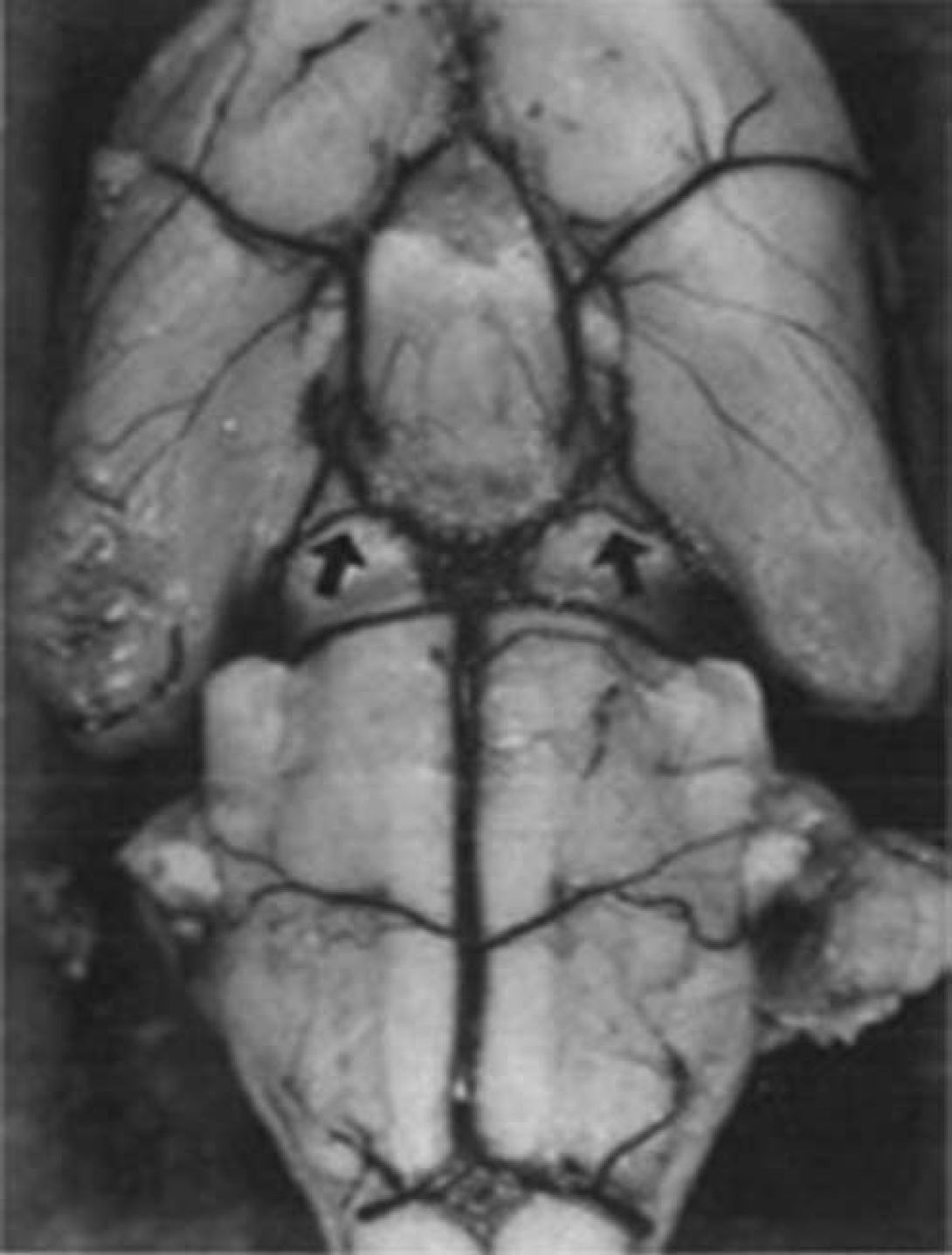
Circle of Willis and posterior communicating arteries of the spontaneously hypertensive rat (Ogata et al, 1976).
Of 493 drugs tested in 45,512 animals with focal cerebral ischemia (VO'C, personal communication), 409 were tested only in normotensive animals, just 56 were tested in both normotensive and hypertensive animals, whereas 28 appear to have been tested only in hypertensive animals. The majority of this testing was performed in the SHR. For six drugs reviewed systematically, only 10% of publications included the modeling of efficacy in animals with high blood pressure or diabetes. Where efficacy was reported in the context of comorbidity, it was generally substantially lower. Disufenton sodium (NXY-059) was less effective in SHRs than in healthy animals (17.6% versus 47.8%; P < 0.001) (Macleod et al, 2008). Tissue plasminogen activator had no beneficial effect on either infarct volume or neurobehavioral score but did increase the observed odds of hemorrhage in SHRs (ESS, personal communication). Nicotinamide was less effective in animals with diabetes or hypertension (21.8% versus 30%; P < 0.01) (Macleod et al, 2004) as was FK506 (17% versus 33.3%; P < 10−10) (Macleod et al, 2005b). In contrast, hypothermia was slightly more effective in SHR than in normotensive SD and Wister rats (van der Worp et al, 2007). Melatonin was not tested in hypertensive animals (Macleod et al, 2005a).
Of the many other models of hypertension, renovascular models (induced by various combinations of renal artery clipping and kidney removal) are the next most commonly used. By keeping both kidneys in place and clipping one renal artery (two-kidney one-clip, 2K1C) only mild and relatively unstable hypertension is achieved, yet this model provided one of the earliest reports that hypertension exacerbates ischemic injury (Fujishima et al, 1978). When a kidney is removed and the other renal artery clipped (one-kidney one-clip, 1K1C) animals often die of acute renal failure accompanied by diffuse edematous lesions in the brain (Nag, 1984). To avoid these problems, Zeng et al described clipping both renal arteries without kidney removal (two-kidney two-clip) to model stroke in hypertension. All SD rats developed stable hypertension without acute renal failure or diffuse cerebral lesions. Within 40 weeks, 62% had developed spontaneous stroke, significantly more than in 2K1C or 1K1C models. The strokes, which were a mixture of small infarcts with clear evidence of thrombotic occlusion and hemorrhagic lesions caused by bleeding from the arteriolar wall of fibrinoid necrosis or ruptured microaneurysms, correlated with the presence of vascular pathology in small arteries or arterioles (Zeng et al, 1998). Using serial magnetic resonance imaging to assess the consequences of inducing hypertension by partial occlusion of both renal arteries, showed a tight relationship between the degree of hypertension and development of cerebral lesions. Below a mean systolic pressure of 210mmHg, rats never had brain lesions, but when pressure exceeded 276mmHg, rats consistently developed brain lesions (Del Bigio et al, 1999).
Similar to the spSHR and renovascular models, rats made hypertensive by treatment with deoxycorticosterone (a mineralocorticoid receptor agonist) and salt can also have spontaneous strokes when blood pressure is high (Sukamoto et al, 1980), but they are more readily protected by the antihypertensive captopril against the effects of acute MCAo than spSHR (Coyle, 1984). Experiments in young Wistar rats suggest that these effects may be mediated in part by remodeling of the cerebrovasculature as treatment with deoxycorticosterone acetate alone (without salt) for 6 weeks stiffens and narrows the MCA, induces mild hypertension, and renders the rats more sensitive to MCAo (Dorrance et al, 2006).
Little is known about the impact of most other methods of inducing hypertension on stroke. For example, the New Zealand, Milan hypertensive, and Lyon hypertensive rats do not die due to strokes or cardiovascular disease like the spSHR and do not appear to have been used in the study of MCAo (Bianchi et al, 1984; Phelan, 1968; Vincent et al, 1984). To date, no stroke studies seem to have been undertaken in hypertensive transgenic rats expressing an extra copy of the renin gene (Wagner et al, 1997). Ischemic stroke has been induced in Cynomolgus monkeys made hypertensive by surgical coarctation of the aorta in animals fed an atherogenic diet (Prusty et al, 1988), but this would not seem to be a practical model for widespread use.
Diabetes and Hyperglycemia
As for hypertension, there are numerous models of diabetes. Although surgical removal of the pancreas has been used since the 1880s, diabetes is most commonly induced by selectively poisoning pancreatic β-cells. This can be achieved using the uric acid derivative alloxan, but streptozotocin which is isolated from the soil bacterium Streptomyces achromogenes is more widely used and mimics most of the major hallmarks of clinical type 1 diabetes, including hyperglycemia, elevated HbA1c concentration, weight loss, polydipsia, and polyurea. Other models including the Non-Obese Diabetic mouse, the BioBreeding rat, and the Zucker Diabetic Fatty rat have been generated by selective inbreeding (Rees and Alcolado, 2005). Defects in the db gene on mouse chromosome 4 and the fa gene on rat chromosome 5 both lead to leptin receptor defects (Chen et al, 1996; Takaya et al, 1996). The db/db mouse develops severe diabetes by 6 weeks of age, characterized by hyperglycemia, hyperinsulinemia, and obesity (Vannucci et al, 2001). Rats homozygous for an amino-acid substitution in the fa gene become obese (reaching ∼500 g at 6 months of age), hyperlipidemic, and develop insulin-resistant hyperglycemia (400 to 500 mg/dL) when 7 to 10 weeks old. The Goto-Kakizaki rat offers a model of spontaneous type 2 diabetes generated by inbreeding glucose-intolerant Wistar rats (Ergul et al, 2007).
Both diabetes as a metabolic condition and hyperglycemia independently have been used in conjunction with animal models of stroke. Intraperitoneal dextrose to increase blood glucose to > 15 mmol/L accelerates and extends infarct development after transient (thread occlusion) and permanent (distal MCA cautery) MCAo (Liu et al, 2007). Others have found that hyperglycemia enlarged infarcts but only in the cortex (Martin et al, 2006). In cats, hyperglycemia led to a three- to four-fold increase in infarct size after permanent MCAo and increased death due to edema upon reperfusion (de Courten-Myers et al, 1989). Similar observations have been made in dogs (Palmon et al, 1995), rabbits (Kraft et al, 1990), and rats (Dietrich et al, 1993). Damage to the blood–brain barrier may be relevant to reports that hyperglycemia exacerbates injury after tPA therapy (Ribo et al, 2007). Damage to the insular cortex and hyperglycemia as a result of endogenous stress responses further complicates our understanding of the impact of hyperglycemia (Allport et al, 2004).
Although early attention focused on acidosis as the mechanism of hyperglycemia-enhanced neuronal injury, other targets have also gained favor. After thread occlusion of the MCA in rats, hyperglycemia was reported to lead to a progressive reduction in cerebral blood flow and enhanced blood–brain barrier permeability (Kawai et al, 1998). After chemical poisoning of pancreatic β-cells with alloxan or streptozotocin, the results are similar with increased edematous change (Kamada et al, 2007), exacerbation of transient and permanent ischemic lesions with increased speed of lesion development, and continued growth upon reperfusion (Huang et al, 1996; Kittaka et al, 1996). Studies of the mechanism of damage also implicate altered inflammatory responses with exaggerated leukocyte-endothelial cell adhesion (Panes et al, 1996), and increased interlukin-1 and intercellular adhesion molecule-1 expression (Ding et al, 2005). Blood–brain barrier dysfunction has also been attributed to increased oxidative stress and matrix metalloproteinase-9 activation (Kamada et al, 2007). Interestingly, it has been reported that although acute hyperglycemia has no effect on endogenous tPA expression, a similar but persistent elevation of blood glucose (∼ 15mmol/L) in streptozotocin-treated rats led to a complete depletion of tPA protein and more than six-fold loss of tPA mRNA expression (Kittaka et al, 1996).
In genetically determined models of diabetes, such as the BioBreeding Rat and db/db mouse, the effects of hyperglycemia on ischemic injury are gender specific. In the BioBreeding rat, cortical injury is the same in diabetic and control animals, but males had larger and females smaller subcortical infarction (Toung et al, 2000). Similar observations have been made after unilateral common carotid artery ligation combined with systemic hypoxia in the db/db mouse wherein even though female diabetic mice were more hyperglycemic and acidotic than the males, they were more resistant to damage (Vannucci et al, 2001).
The recently described Goto-Kakizaki rat, generated by inbreeding of glucose-intolerant Wistar rats, develops mild hyperglycemia at 6 weeks of age, but is also unusual in producing significantly smaller infarcts after extended (3-hour) thread occlusion of the MCA (but high rates of subcortical hemorrhagic transformation) than nondiabetic controls, which is interpreted as the result of diabetes-induced vascular remodeling (Ergul et al, 2007). However, the shape of the infarcts is reminiscent of the hypothalamic lesions that can be produced when only hypothalamic-perforating arteries but not the MCA are occluded (He et al, 1999), and suggests that reduced effectiveness of thread occlusion in the more tortuous vessels of the Goto-Kakizaki rat (Ergul et al, 2007) might provide an alternative explanation.
Using insulin to reduce blood glucose is reported to induce marked neuroprotection (Hamilton et al, 1995). Similar protective effects on infarct volume were reported when insulin was used together with tPA in normoglycemic animals to treat thromboembolic strokes. Others have reported that tight glycemic control does not improve infarct size in male BioBreeding rats (Toung et al, 2000). Moreover, despite reduced infarct volumes, others have reported that mortality was as high after insulin treatment alone (47%) as it was when combined with tPA (38%) (Meden et al, 2002).
Age and Gender
It is surprising that we perform almost all of our testing in male animals and have little representation of half the human population. The main reason for this choice seems to be simply that lack of an estrus cycle (in male rats) might reduce the overall experimental variability. However, even in male rats, the influence of testosterone seems to be age dependent, with castration conferring protection in the young and supplementation conferring protection in the middle aged (Cheng et al, 2009) and the effects of estrogens, which are far from clear (Strom et al, 2009), may be dependent on interactions with specific elements of models of hypertension (Carswell et al, 2000, 2005) and diabetes (Vannucci et al, 2001), which may possibly be linked by differences in vascular reactivity (Miller et al, 2007). Moreover, as most strokes in women occur after menopause (average age of menopause and average age of incident stroke in women have been reported as 49 and 80 years, respectively) (Lisabeth et al, 2009), ignoring half of our species because of a possibly spurious advantage in the laboratory seems very unwise.
Our understanding of the influence of animal age in the laboratory is also limited. Although age-dependent increases in infarct size are most often reported (Davis et al, 1995; Driscoll et al, 2008; Hachinski et al, 1992), others report reductions in behavioral deficits in aged rats (Shapira et al, 2002). Whether this reflects our lack of understanding of stroke biology or of the models we use is unclear. Differential responses to candidate therapeutics with aging (Won et al, 2006) suggest that we would be wise to learn more. Why has aging been studied so little when the majority of stroke patients are old? The answer would seem to be just the cost of maintaining animals for longer periods. It is certainly feasible to induce consistent strokes in aged animals even when they are also diabetic and hypertensive (Rewell et al, 2010).
Conclusions
The utility of animal models of stroke is governed by many factors, and it is clear that no single model can encompass all of the variables known to affect human stroke. Which model we choose is determined by a series of compromises and questions we have to ask about the aims of our experiments. Do we wish to occlude single or multiple vessels, do we need control of the timing of reperfusion, or do we need to investigate how a drug interacts with the natural process? Are we trying to determine whether a drug has an effect or trying to define the limits of its efficacy. If the later, we need to consider the common comorbidities of age, atherosclerosis, hypertension, and diabetes. Is limiting experimental variability more important than demonstrating robust effects across a range of genetic backgrounds? Does variability in vascular anatomy matter to your experiment? Brain temperature can dramatically alter infarct size but should we control it or monitor it, do we lose valuable insight by performing all experiments at a fixed temperature that might not be relevant in the clinic? Owing to space limitations, this article could not discuss all the possible variables. For example, blood gas concentrations are often measured and used as part of the physiologic work-up to show equivalence of experimental cohorts or to exclude animals with hypoxia from further analysis. However, how do the subtleties of blood gas concentration encountered during different forms of ventilation and their interaction with anesthesia impact on outcome? Hypertension, hyperglycemia, and diabetes have been considered where specific stroke modeling has been performed, but the broader implications of metabolic syndrome and atherosclerosis remain largely unexplored in animal stroke modeling. There is still much to learn and every experimenter faces many choices. Figure 5 provides a guide to matching model characteristics to experimental aim and an outline of the experiments likely to be needed to move from identifying a candidate drug through to clinical trial. These figures provide only a framework for the questions that will be encountered and decisions that will need to be made as we learn more about stroke and move closer to introducing new and more effective therapies. The deliberations of the Stroke Therapy Academic Industry Roundtable (Fisher et al, 2009b; STAIR, 1999) and those of Macleod et al (2009) are recommended for their insight into the problems of stroke translational medicine and avoidance of bias at the bench.
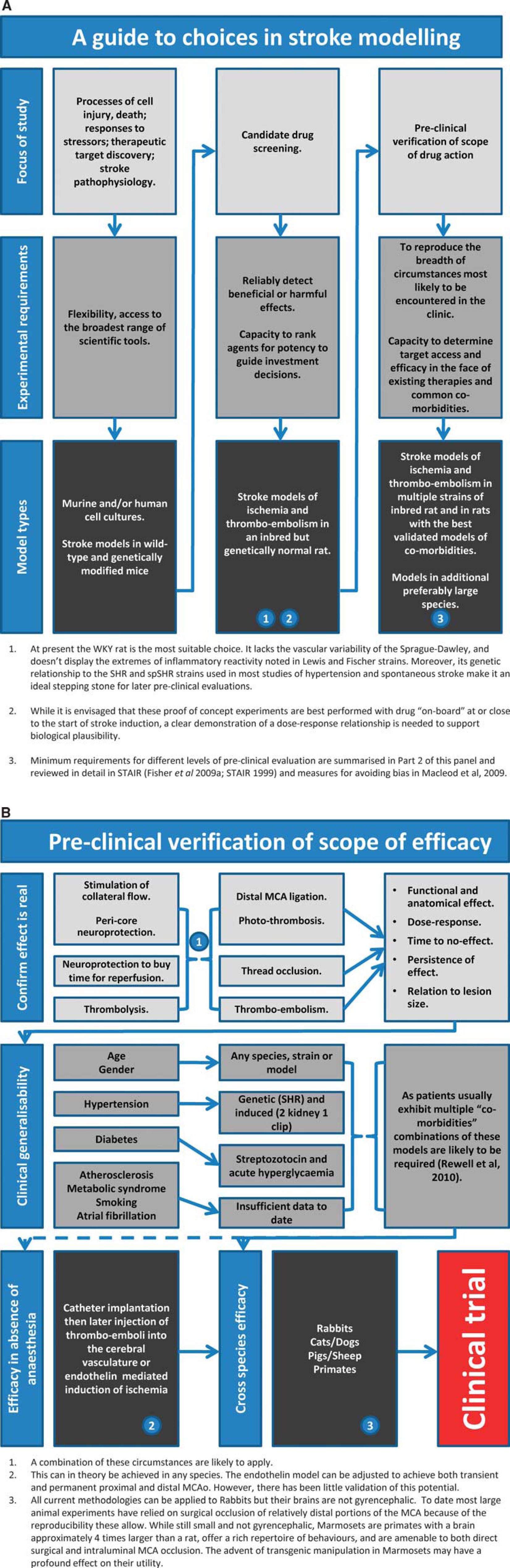
Guidelines for stroke modeling. (
In conclusion, ‘The lack of translation between the animal work and clinical benefits does not lie in the animal models, but in how we use the models and how we apply this knowledge to design of clinical trials' (Willing, 2009).
Footnotes
The authors declare no conflict of interest.