
Abstract
Angiotensin-converting enzyme (ACE) inhibition can reduce stroke risk by up to 43% in humans and reduce the associated disability, and hence understanding the mechanism of improvement is important. In animals and humans, these effects may be independent of the blood pressure-lowering effects of ACE inhibition. Normotensive (Wistar–Kyoto (WKY)) and hypertensive (spontaneously hypertensive rat (SHR)) animals were treated with the ACE inhibitors ramipril or lisinopril for 7 or 42 days before 2 hours of transient middle cerebral artery occlusion (MCAo). Blood pressure, serum ACE, and blood glucose levels were measured and stroke infarct volume was recorded 24 hours after stroke. Despite greater reductions in blood pressure, infarct size was not improved by ACE inhibition in hypertensive animals. Short-term ACE inhibition produced only a modest reduction in blood pressure, but WKY rats showed marked reductions in infarct volume. Long-term ACE inhibition had additional reductions in blood pressure; however, infarct volumes in WKY rats did not improve further but worsened. WKY rats differed from SHR in having marked cortical ACE activity that was highly sensitive to ACE inhibition. The beneficial effects of ACE inhibition on infarct volume in normotensive rats do not correlate with changes in blood pressure. However, WKY rats have ACE inhibitor-sensitive cortical ACE activity that is lacking in the SHR.
Introduction
Hypertension and increasing age are the most powerful risk factors for stroke (Abbott et al, 2003; Arboix et al, 2006). Moreover, hypertension is also closely correlated with cognitive decline and dementia (Abbott et al, 2003). Globally, 9% of all deaths are attributable to stroke (Murray and Lopez, 1997), and the incidence is projected to increase by one-third over the next 10 years (Cadilhac et al, 2007). The majority (72.5%) of human strokes are ischemic, with most occurring in the major cerebral arteries, particularly the middle cerebral artery (MCA; Thrift et al, 2001).
Acute therapeutic options for stroke are limited. Although admission to a specialist stroke unit currently offers the greatest benefit to the community (Dhamija and Donnan, 2007), the mechanisms providing this benefit are ill defined. Prophylactic aspirin lessens the risk of a second stroke but offers little to the majority who are unaware of their risk of a first stroke (Dhamija and Donnan, 2007). Restoration of blood flow by thrombolysis is of benefit to only a small proportion of patients (up to 15%) who arrive at the hospital within 4.5 hours of their stroke, and who are not frankly hypertensive at initiation of treatment (Hacke et al, 2008; Vahedi et al, 2007).
There has been more success at preventing stroke by treating the risk factors for atherosclerosis. After myocardial infarction when stroke risk is high, statins, such as simvastatin (4S Group, 1994) and pravastatin (Plehn et al, 1999) that reduce cholesterol, reduce the likelihood of a subsequent stroke by up to 25% (Amarenco et al, 2006), although at the expense of a small increase in the risk of cerebral hemorrhage. It seems likely that these effects are mediated not only by cholesterol reduction but also by direct antiinflammatory and antioxidant properties (Arnaud et al, 2005). Similarly, large clinical trials (e.g., LIFE, HOPE, ONTARGET, PEACE, and PROGRESS) have shown that angiotensin-converting enzyme (ACE) inhibition to control hypertension can reduce the incidence of stroke by up to 43% (Yusuf et al, 2000) and reduce the risks of long-term disability and dependency (Fransen et al, 2003). In both animals (Stier et al, 1991) and humans (Sleight, 2000; Yusuf et al, 2000), it has been suggested that these effects may also be independent of the blood pressure-lowering effects of ACE inhibition, because normotensive patients also benefit from ACE inhibition (Sleight et al, 2001).
Despite the clear evidence of benefit from ACE inhibition in human ischemic stroke, animal data are limited and at times conflicting. In the stroke-prone spontaneously hypertensive rat (SHR), ACE inhibition results in a clear reduction in number of bleeds (Inada et al, 1995). In models of permanent focal ischemia, both moexirpil and enalapril reduced the infarct volume when introduced just 1 hour before induction of focal ischemia (Ravati et al, 1999). However, 5 days of ramipril treatment before stroke induction was reported to have no effect (Krikov et al, 2008).
We have therefore examined whether short- and long-term exposure (7 and 42 days) to the ACE inhibitors ramipril and lisinopril alters the impact of subsequent transient ischemia in aged normotensive and hypertensive rats. Follow-up experiments were then done to examine cortical and striatal ACE activity.
Materials and methods
Drug Administration
Animals were randomized to oral treatment administered each morning, with vehicle (strawberry topping) or ramipril and lisinopril, administered at 1 and 10 mg/kg, respectively. The two ACE inhibitors were selected because ramipril (1 mg/kg) is lipophilic and is thought to cross the blood–brain barrier, whereas lisinopril (10 mg/kg) is hydrophilic and is less likely to do so. The doses were selected as Cushman et al (1989) showed that they were normalized equivalent oral doses, and they have also been shown to induce renin–angiotensin system manipulation in the brain (Cushman et al, 1989; Lu et al, 2005; Sakaguchi et al, 1988). Separate cohorts received either 7 (short-term) or 42 days (long-term) of treatment until and including the day of surgery.
Blood Pressure Measurement
A noninvasive tail-cuff system (NIBP System; ADInstruments, Bella Vista, NSW, Australia) was used to measure the blood pressure throughout the experiment. All animals were acclimatized to this procedure by daily measurements for 2 weeks before the start of the experiment. Daily recordings were taken for 5 days to establish a baseline; after this, short-term animals had daily recordings whereas long-term animals were recorded three times a week.
Blood Glucose and Plasma Angiotensin-Converting Enzyme Levels
Tail vein blood (500 μl) was collected 2 hours after the final dose of ACE inhibitor, and blood glucose was analyzed on a blood spot using the ACCU-CHEK system (Roche, Dee Why, NSW, Australia). Plasma ACE activity was measured using the spectrophotometric method of Johansen et al (1987) after collection of the blood into a heparinized tube and centrifugation.
Animal Numbers and Surgical Procedures
In all, 50 adult male Wistar-Kyoto (WKY) and 62 adult male SHR rats (ARC, Canning Vale, Western Australia, Australia) aged 16 months ‘in house’ were used. A greater number of SHR animals were purchased because we were concerned that aging would have a greater effect on mortality in the hypertensive animals than in their normotensive counterparts. All methods conformed to the code of practice published by the Australian National Health Medical Research Council and were approved by the Austin Health Animal Ethics Committee.
Data from 92 animals are included in the final results (short term: WKY,
Anesthesia was induced using 5% isoflurane (50:50 air:oxygen) and then maintained with 2% isoflurane during surgery. Atropine (120 μg) was administered intraperitoneally immediately after induction of anesthesia to decrease mucosal secretions and prevent aspiration during and after surgery. Blood oxygen levels and heart rate were monitored throughout (pulse-oximeter; Nellcor, Gladesville, NSW, Australia) and temperature was maintained at 37°C using a locally manufactured rectal temperature-regulated heating pad.
Middle cerebral artery thread occlusion was performed according to the methods of Koizumi et al (1986) and Longa et al (1989) with modifications (Spratt et al, 2006). In brief, after ligation of branch arteries and creation of an external carotid artery stump, the silicone-encased suture (0.35 mm external tip diameter) was passed up the internal carotid artery approximately 18 mm from the carotid bifurcation, until resistance was felt and a decrease in cerebral blood flow was detected by laser Doppler flowmetry (Moorlabs, Devon, UK) over the right cortex (2 mm caudal and 2 mm lateral to bregma). Animals were subjected to 2-hour MCA occlusion (MCAo) without anesthesia and then the MCA-occluding suture was retracted into the external carotid stump under anesthesia to reopen the MCA. After 22 hours of reperfusion, animals were anesthetized and then transcardially perfused with saline followed by 4% paraformaldehyde. The brains were postfixed overnight, and then cryoprotected in 30% sucrose.
Stroke Lesion Volume Determination
Sections (40 μm) mounted on gelatin/chrome alum slides were stained with hematoxylin and eosin. Digital photographs were taken and infarct sizes at eight standard coronal plains were quantified by researchers masked to treatment allocation using microcomputer imaging device (MCID) image analysis software (Imaging Research, St. Catharines, Ontario, Canada). Infarct volumes were derived by calculating the average infarct area between slices and multiplying by the distance between slices. Edema corrections were calculated by dividing the ipsilateral by the contralateral hemisphere volume.
Angiotensin-Converting Enzyme Autoradiography
To determine the degree of ACE inhibitor penetration of the blood–brain barrier, a separate cohort of young (12 week old) SHR and WKY rats were dosed with ramipril, lisinopril, or vehicle (
Statistics
SPSS (SPSS Inc., Chicago, IL, USA) was used to calculate statistical differences within and between the strains after treatment by one-way analysis of variance (ANOVA; Dunnett's
Results
The use of a sugary gum as the vehicle for short-term and long-term prophylactic treatment with ramipril and lisinopril did not alter the animals' blood glucose levels, which remained within the normal range (7 to 9 mmol/L) (Petterino and Argentino-Storino, 2006). All WKY and SHR animals were of consistent weight on the day of surgery (WKY 460 ± 43 g; SHR 438 ± 11 g, mean ± s.d.). The mortality rate was 14% in WKY (5 presurgery, 1 vehicle, 4 lisinopril, and 2 surgical errors) and 9.6% in SHR (2 presurgery, both vehicle, and 4 surgical errors), which is slightly greater than previously reported from our laboratory (Spratt et al, 2006).
The baseline systolic blood pressure for WKY animals receiving vehicle was 142 to 145 mm Hg. The baseline systolic blood pressure of the SHR animals was 200 to 210 mm Hg. After short-term ACE inhibitor treatment, significant blood pressure reductions in both the SHR and WKY were observed. Blood pressure fell by 23.6% and 29.4%, respectively, for ramipril and lisinopril in the SHR cohort, and by 9.6% and 11.9%, respectively, in their WKY counterparts. Long-term ACE inhibition caused no additional reduction in blood pressure in the SHR cohorts, but did produce a significant additional reduction in the WKY rats (total blood pressure reduction 21.5% and 30.9% for ramipril and lisinopril, respectively).
In vehicle-treated rats subjected to 2 hours of MCAo, the mean corrected total infarct volume after 24 hours was the same in SHR and WKY cohorts (148.9 versus 123.7 mm3, respectively) (Figure 3). Treatment with ACE inhibitors did not significantly alter infarct volume in SHRs regardless of drug or duration of treatment (136.2 and 148.9 mm3, ramipril short term and long term, respectively). In WKY animals, short-term treatment with both ramipril and lisinopril reduced infarct volumes by > 50% (43.6 and 62.7 mm3, ramipril ANOVA, Dunnett's test
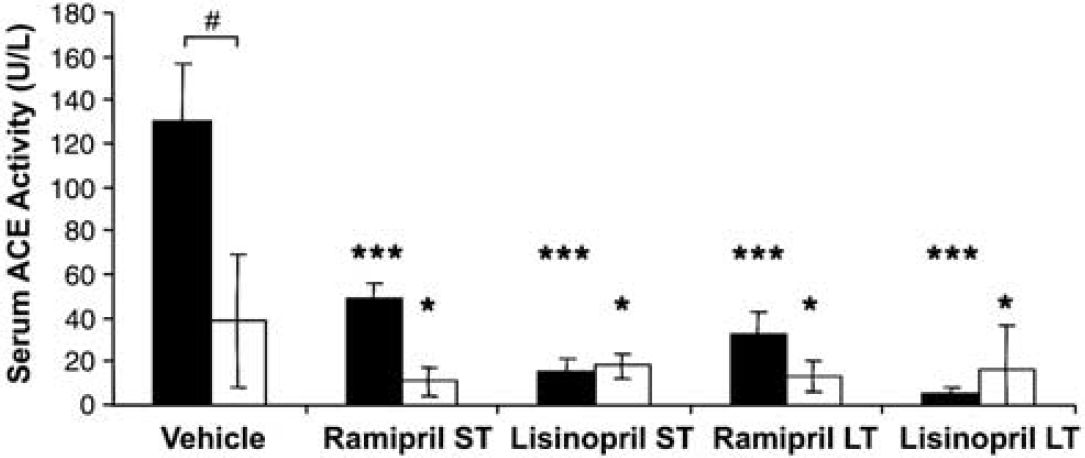
Serum angiotensin-converting enzyme (ACE) activity (U/L) in aged animals after vehicle or short-term (ST; 7 days) or long-term (LT; 28 days) treatment with ACE inhibitors ramipril or lisinopril in spontaneously hypertensive rats (SHR; □) and Wistar–Kyoto (WKY; ▪) rats (mean ± s.d.), ***
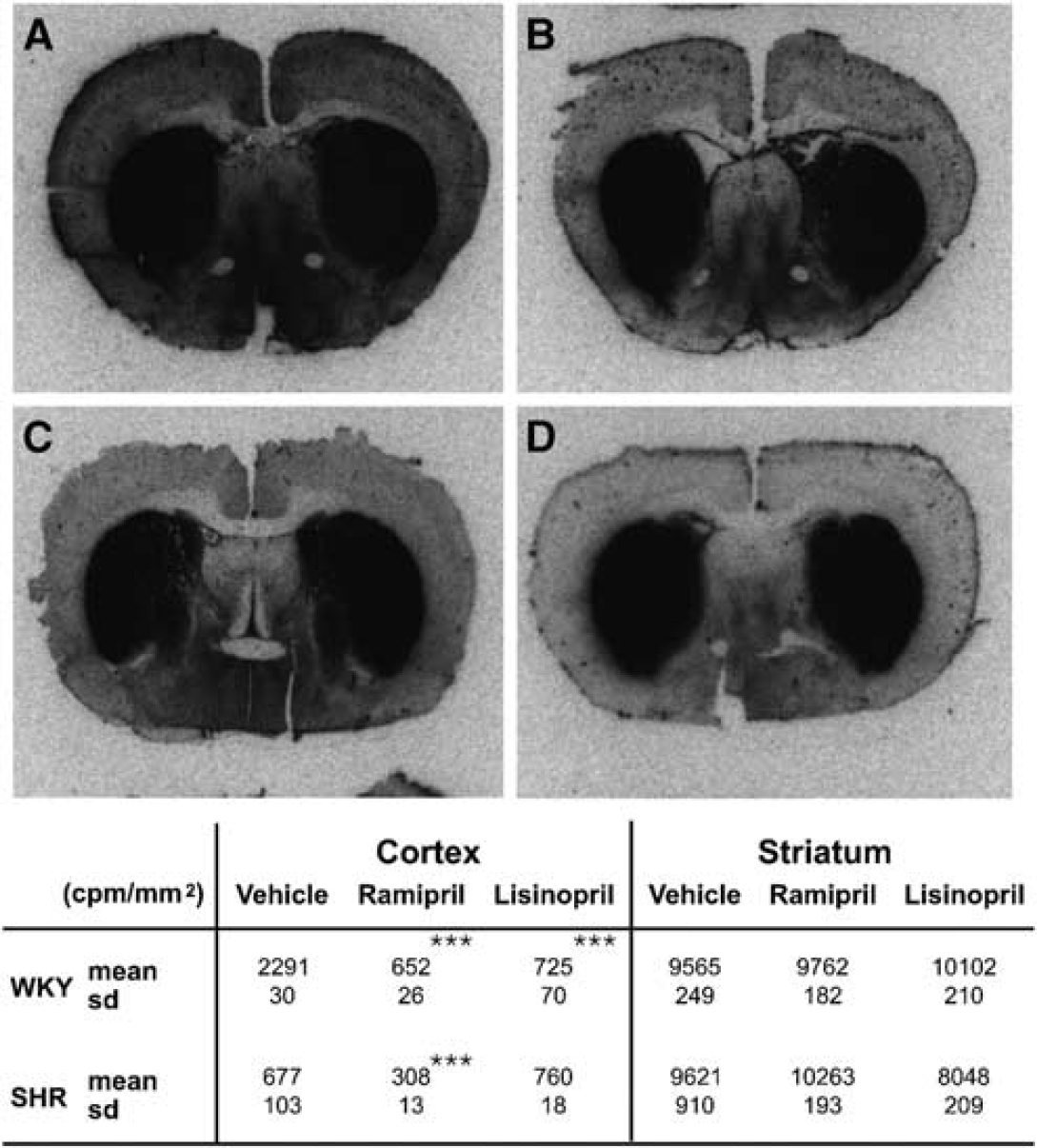
Photomicrographs of 125I-MK351A binding: (
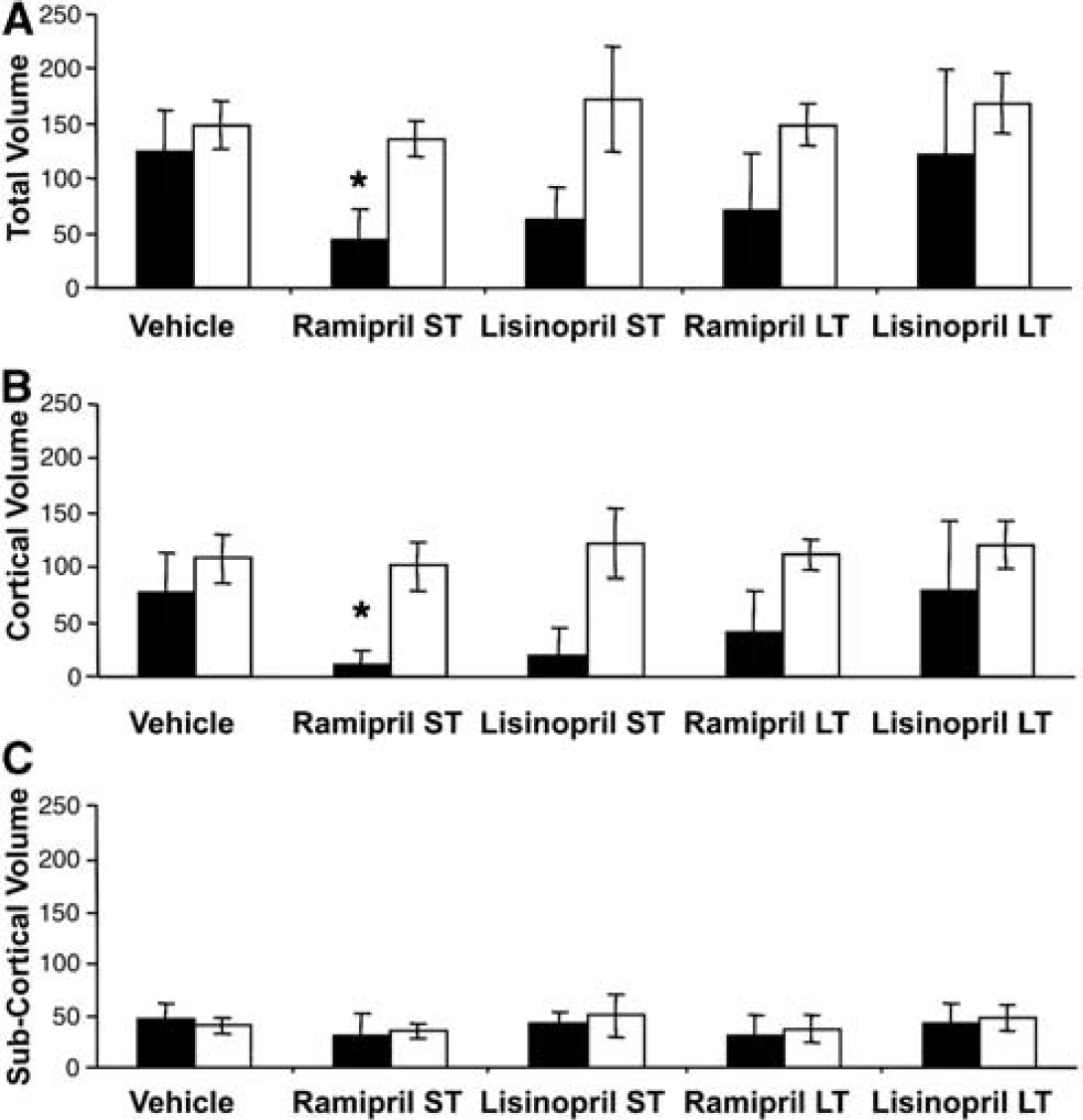
(
Baseline plasma ACE activity was consistent with previous reports in the literature (Sakaguchi et al, 1988). In WKY rats, serum ACE activity was approximately 3.5-fold greater than in the SHR cohort (vehicle 130.3 versus 38.8 U/L,
Striatal ACE activity measured by 125I-MK351A autoradiography was essentially the same in both strains, and was largely resistant to ACE inhibition. However, the two strains differed markedly in their cortical ACE activity (Figure 2). In the SHRs, cortical ACE activity was low and only slightly affected by ACE inhibition. However, WKY had > 3-fold more ACE activity, which was reduced to ∼30% of the baseline by ACE inhibition (Figure 2). It is the presence of marked cortical ACE activity and its capacity for suppression by ACE inhibition that coincide with the protective effect of ACE inhibition on infarct volume in the WKY but not SHR strain.
Discussion
At the start of the experiment, the SHRs had significantly higher systolic blood pressure (200 to 210 mm Hg) than their WKY counterparts (142 to 145 mm Hg), which is consistent with previous measures of blood pressures in these strains of rat (Fukuda et al, 2004). ACE inhibition reduced systolic blood pressure in both strains of rat, with the most pronounced effect observed in the SHRs. Both ramipril and lisinopril (1 and 10 mg/kg, respectively) were equally effective in reducing blood pressure, with long-term treatment offering no advantage over short-term treatment in the SHRs. However, in WKY rats, long-term ACE inhibition produced twice the blood pressure reduction observed after short-term treatment.
Changes in infarct volume could not be tracked using ACE inhibitor-induced changes in blood pressure. In SHRs, which showed the earliest and greatest reduction in pressure, neither ramipril nor lisinopril treatment altered the infarct volume (Figure 3). This observation is not consistent with the hypothesis that high blood pressure at the time of stroke explains larger infarcts in this strain of rat or in general (Hom et al, 2007). One might explain this observation by invoking a threshold of blood pressure reduction that must be achieved before benefit occurs. At the doses used in our experiments, it is true that ACE inhibition in the SHRs did not reduce blood pressure to the levels of the WKY rats. However, such a threshold is inconsistent with observations from the clinic that ACE inhibition has benefit across the spectrum of blood pressures (Sleight, 2000) and that blood pressure does not return to normal in many patients (Yusuf, 2002).
The data from the WKY strain are also inconsistent with blood pressure at the time of stroke being a determinant of infarct size. After short-term ACE inhibition, which produced a modest reduction in blood pressure, infarct volumes were markedly reduced (Figure 3). However, after long-term ACE inhibition, which produced significantly greater reduction in blood pressure, no additional benefit was observed. Indeed the data suggest that despite a profound effect on blood pressure, long-term ACE inhibition was less rather than more effective (Figure 3). All these changes occurred because of alteration in cortical infarction; however, striatal infarction did not change (Figure 3). Despite the lack of a relationship with blood pressure, there does seem to be a relationship with ACE activity. Plasma ACE activity in the WKY rats was consistent with previously reported data (Zhang et al, 1996), but 3.5-fold greater than measured in the plasma of SHRs (Figure 1). Although there are reports of early developmental changes in ACE activity (Costerousse et al, 1994), activity is thought to remain stable for most part of the life. In WKY rats, plasma ACE activity is reported to be stable from 4 weeks of age (Costerousse et al, 1994), whereas in SHRs it is reported to be stable for at least the first 20 weeks of life (Campbell et al, 1995a), which includes the period when this strain develops hypertension (Burrell et al, 1995). Moreover, in the clinic there is no suggestion that ACE inhibitor dose needs to be adjusted with age. Within the brain, the data obtained by Mendelsohn et al (1980) from approximately 4-month-old SHR and WKY rats are nearly identical to our data obtained at 3 months. Therefore, it seems likely that our measurements of ACE activity in cohorts of young adult rats are representative of activity in the aged cohorts in which infarct volumes were measured. Using young adult animals in this follow-on experiment was a pragmatic decision based on cost and the absence of data, suggesting that ACE activity would change significantly with age.
The absolute fall in ACE activity on ACE inhibition was greatest in the WKY rats, the proportional fall was similar in both strains of rat, and there was no change after long-term ACE inhibition that might explain the apparent lessening of impact on infarct volume (Figures 2 and 3). This dependence on duration of therapy suggests that reports of acute efficacy (1 hour before induction of stroke) after moexirpil and enalapril treatment (Ravati et al, 1999) but absence of effect after 5 days of ramipril treatment (Krikov et al, 2008) may not be mutually exclusive. However, the low dose of ramipril (0.01 to 0.1 mg/kg) used in the latter experiment may provide adequate explanation for the lack of effect.
In the brain, autoradiography revealed that ACE activity is highest in the striatum. The level of activity was the same in both WKY and SHR strains and was not affected by ACE inhibition (Figure 2). ACE activity in the cerebral cortex of WKY rats was, as in the plasma, more than three times greater than that observed in SHRs. Importantly, although ACE inhibition had only a modest effect on cortical ACE activity in the SHRs, it caused profound suppression of ACE activity in the WKY rats, reducing activity to the levels observed in the SHRs (Figure 2).
The effect of ACE inhibitor treatment on components of the renin–angiotensin system, such as angiotensin II (AngII), AngI, and renin, are complex. Studies in humans have shown that inhibition of circulating AngII by ACE inhibitors is dependent on reactive increases in plasma renin levels (Gadsboll et al, 1990). The central actions of AngII are also thought to be exaggerated in SHR compared with WKY rats (Nelson, 1988). Administration of ACE inhibitors to young SHR is reported to result in significant increases in plasma renin levels and increased brain AngI and Ang(1–7) but not brain AngII (Campbell et al, 1995b; Kohara et al, 1993). The greater reduction in cortical ACE activity observed in the WKY rats by ACE inhibitor treatment probably reduces AngII generation and thus improves tissue survival by reducing AngII vasoconstriction and potentially also by reducing thrombosis through decrease in levels of plasminogen activator inhibitor-1 (PAI-1; Skurk et al, 2001).
These observations strongly suggest a direct effect in the brain. One mechanism of action might be mediated by cortical angiotensin receptors, as brief ACE inhibition in young SHR decreases central responses to AngII and AngI, suggesting alterations in receptor expression and postreceptor mechanisms (Mizuno and Fukuchi, 1981; Wu and Berecek, 1993). This is further supported by the observations that AngII AT1 receptor antagonists such as candesartan protect against stroke induction (Inada et al, 1997) and cerebral ischemia (Ito et al, 2002), and that angiotensin type-2 receptor mRNA level increases after global brain ischemia (Shibata et al, 1998).
Although this is not the first study to report that ACE inhibitors improve outcome after stroke in animals, it is the first to use aged hypertensive and normotensive animals. Age and hypertension are among the most important risk factors for ischemic stroke (Abbott et al, 2003; Arboix et al, 2006). Our data confirm that ACE inhibition provides benefit that is independent of blood pressure and suggest that this benefit accrues because of direct effects in the brain that may be mediated by inhibition of cortical ACE. The apparent waning of effect with duration of therapy suggests that a strategy combining blood pressure reduction with agents that do not penetrate the blood–brain barrier and acute central nervous system (CNS) ACE inhibition might maximize the benefit to patients.
Footnotes
The authors declare no conflict of interest.