
Abstract
Caloric restriction (CR), resveratrol, and ischemic preconditioning (IPC) have been shown to promote protection against ischemic injury in the heart and brain, as well as in other tissues. The activity of sirtuins, which are enzymes that modulate diverse biologic processes, seems to be vital in the ability of these therapeutic modalities to prevent against cellular dysfunction and death. The protective mechanisms of the yeast Sir2 and the mammalian homolog sirtuin 1 have been extensively studied, but the involvement of other sirtuins in ischemic protection is not yet clear. We examine the roles of mammalian sirtuins in modulating protective pathways against oxidative stress, energy depletion, excitotoxicity, inflammation, DNA damage, and apoptosis. Although many of these sirtuins have not been directly implicated in ischemic protection, they may have unique roles in enhancing function and preventing against stress-mediated cellular damage and death. This review will include in-depth analyses of the roles of CR, resveratrol, and IPC in activating sirtuins and in mediating protection against ischemic damage in the heart and brain.
Caloric Restriction Activates Sirtuins and Mediates Lifespan and Ischemic Protection
Dietary restriction (or caloric restriction, CR) has been associated with increased lifespan in many animal models, including rodents, monkeys, Drosophila, and Caenorhabditis elegans (Piper and Bartke, 2008). The idea that restricting diet can increase longevity is not new. In fact, ‘longevity’ mechanisms and genes have been examined in great detail for many decades. Changes in genetic and transcriptional regulation during CR affects processes intimately involved in the aging process, such as energy metabolism, stress signaling pathways, and reactive oxygen species (ROS) production (Anderson and Weindruch, 2010). A number of proteins related to longevity have been extensively studied, such as adenosine monophosphate-activated protein kinase (AMPK), forkhead box O (FOXO) transcription factors, target of rapamycin, and SKiNhead-1 among others (Greer et al, 2007; Hansen et al, 2008). Of recent significant interest is the sirtuin family of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases, which modulate longevity in response to CR.
Sirtuins are highly conserved class III histone deacetylases with homology to the yeast silent information regulator 2 (Sir2) (Greiss and Gartner, 2009). There are seven mammalian members of the sirtuin family (SIRT1–7), which localize to different subcellular compartments and deacetylate both histone and nonhistone proteins in an NAD+-dependent manner (Figure 1). The requirement of NAD+ for sirtuin activity couples sirtuin-dependent posttranslational modifications with cellular energy levels. Thus, sirtuins regulate a diverse array of cellular functions, including metabolism, gene transcription, cell division, and cellular stress response (Finkel et al, 2009).
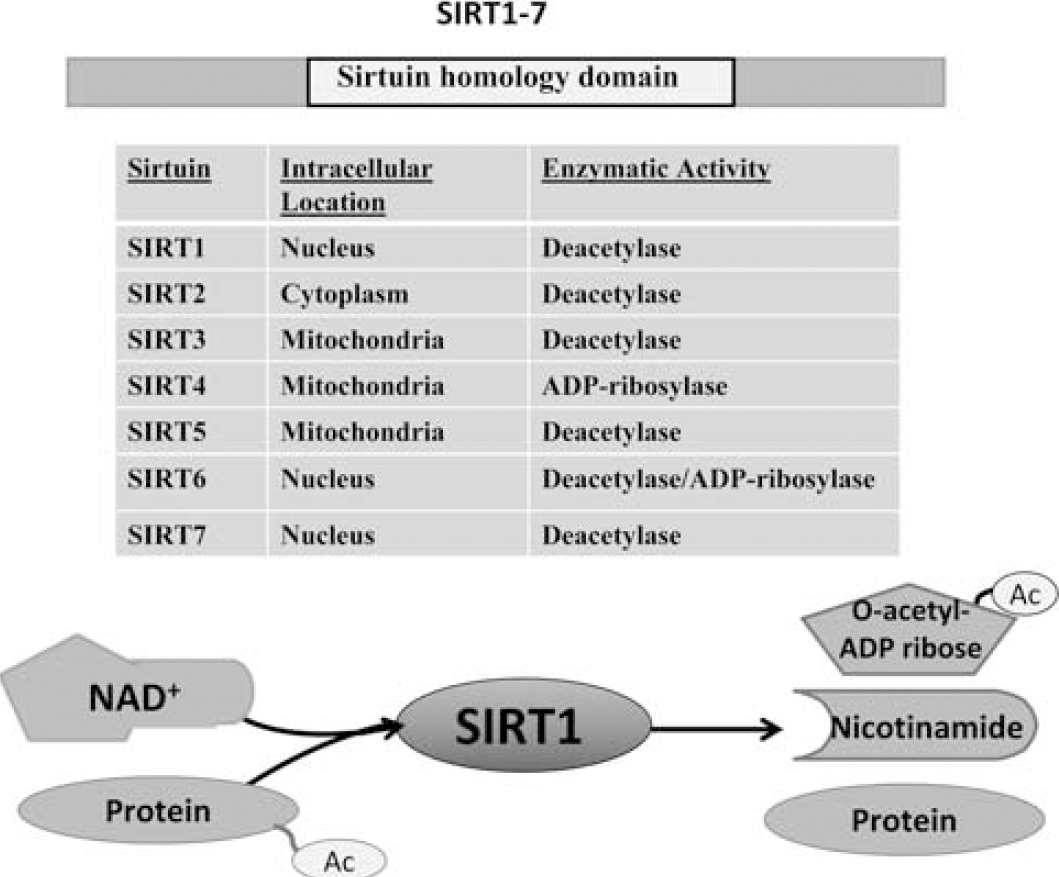
Mammalian sirtuins. The sirtuins differ in their subcellular localizations: SIRT1, SIRT6, and SIRT7 are predominantly found in the nucleus, whereas SIRT3, 4, and 5 are targeted to the mitochondria. SIRT2 is primarily found in the cytoplasm. With the exception of SIRT4, all the mammalian sirtuins have deacetylase activity. During deacetylation, the acetyl group from the target protein is transferred to the ADP-ribose moiety of NAD+ producing O-acetyl-ADP-ribose, nicotinamide, and the deacetylated protein. NAD+, nicotinamide adenine dinucleotide; SIRT1–7, sirtuin 1–7.
The evidence linking sirtuins to longevity was first discovered in yeast in which overexpression of Sir2 prolonged life, whereas Sir2 mutants displayed a shortened lifespan (Kaeberlein et al, 1999). The necessary role of Sir2 in CR-mediated longevity has also been reported in studies using Drosophila and C. elegans (Rogina and Helfand, 2004; Wang and Tissenbaum, 2006) and has been shown in mammals with sirtuin 1 (SIRT1) (Boily et al, 2008). In fact, transgenic mice overexpressing SIRT1 display a number of phenotypic markers of CR mice, such as increased metabolic activity, glucose tolerance, and reduced insulin (Bordone et al, 2007). Furthermore, evidence in rats showed that increased SIRT1 expression during CR prevents mitochondrial-mediated apoptotic signaling (Cohen et al, 2004b). These studies suggest that SIRT1 may have a significant role in CR-mediated reductions in stress signaling and enhanced mitochondrial performance (Figure 2).
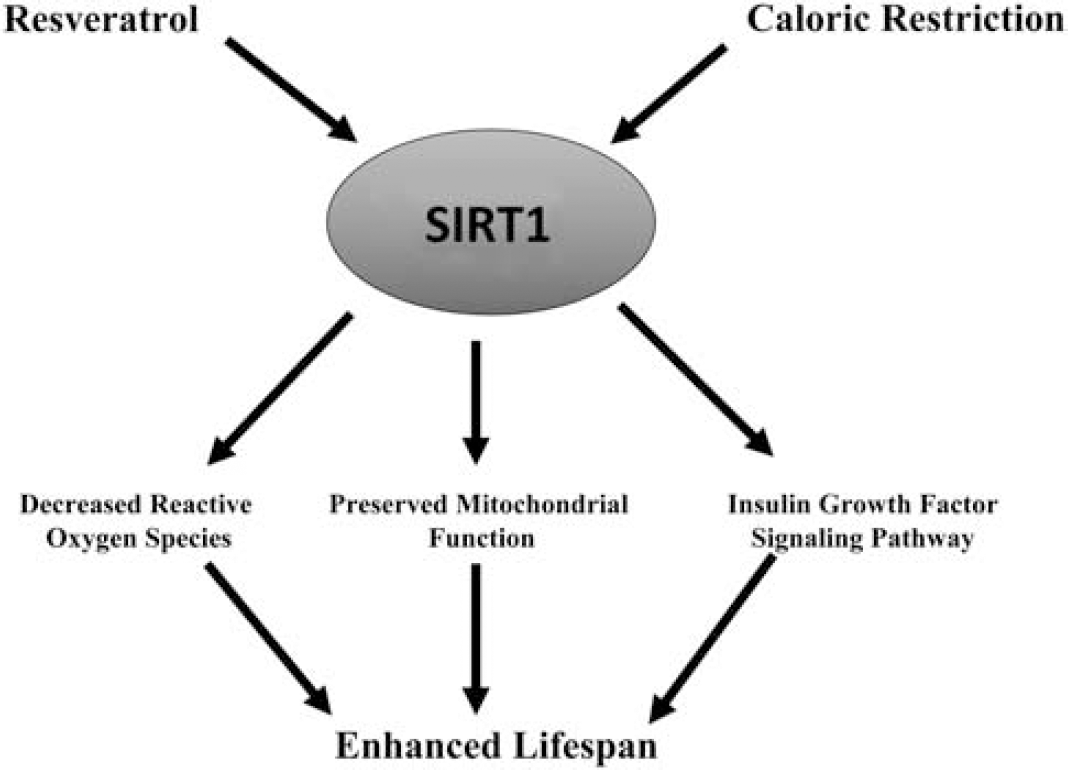
Schematic diagram of SIRT1 regulation. Resveratrol or caloric restriction enhances SIRT1 activity, resulting in decreased reactive oxygen species, preserved mitochondrial function, and enhanced insulin growth factor signaling pathway all contributing to prolonged lifespan as shown in many invertebrate and vertebrate species. SIRT1, sirtuin 1.
The benefits of CR-mediated sirtuin activation have been shown in a number of injury models, including neurodegenerative diseases and the aging heart (Schroeder et al, 2010; Marzetti et al, 2009). Recently, CR was shown to protect against ischemic injury in the brain and heart, among other tissues. For example, CR prevented learning and memory deficits despite extensive CA1 neuronal degeneration induced by forebrain ischemia (Roberge et al, 2008). Rats subjected to chronic intermittent fasting after myocardial ischemia induction were more likely to survive, showed reductions in infarct size, and had increased expression of antiapoptotic proteins (Katare et al, 2009). Furthermore, CR upregulated angiogenic factors and increased capillary density after myocardial ischemia (Katare et al, 2009) and was also shown to accelerate revascularization in mice after hindlimb ischemia (Kondo et al, 2009). Some evidence suggests SIRT1 may have an important role in the ability of CR to provide protection against ischemia. Increased SIRT1 expression after prolonged CR has been linked to improved functional recovery after global myocardial ischemia (Shinmura et al, 2008). Moreover, SIRT1 was required for CR-mediated protection against hypoxia-induced mitochondrial damage in the kidney (Kume et al, 2010), suggesting a necessary role for SIRT1 in CR-mediated protection. The specific CR-mediated SIRT1 pathways involved in ischemic protection have not been fully elucidated, although nitric oxide (NO) synthase (NOS) (Shinmura et al, 2008) and FOXO3 (Kume et al, 2010) have been implicated as possible effectors. However, as discussed later, SIRT1 has been shown to be involved in a number of signaling pathways that lead to protective mechanisms that could also be involved in CR-mediated protection (Figure 3).
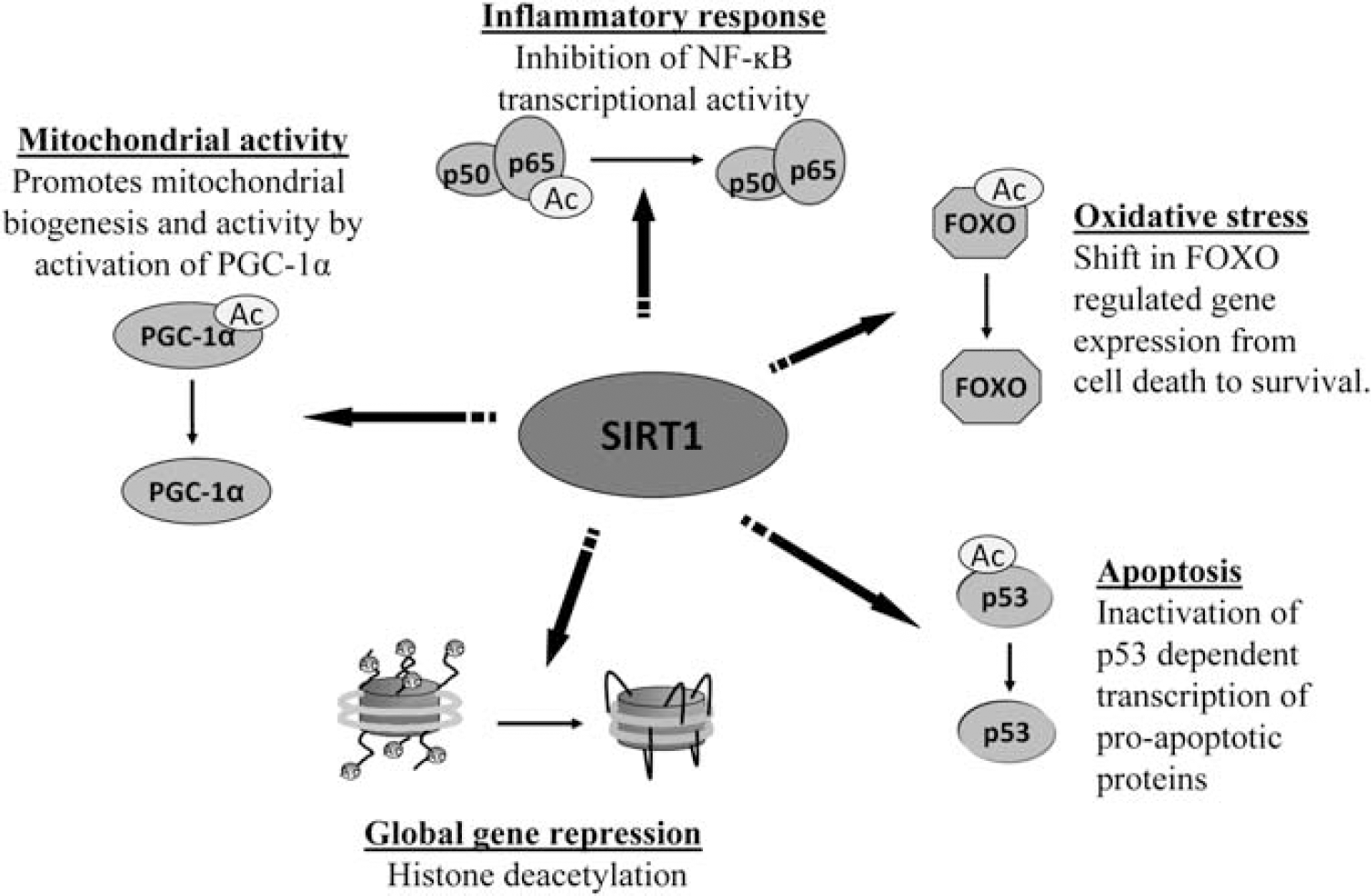
Nuclear activities of SIRT1. The regulation of gene expression by SIRT1 deacetyltransferase activity allows for the activation and inhibition of signaling pathways involved in numerous cellular functions, such as inflammation, oxidative stress, apoptosis, and mitochondrial activity. SIRT1 regulates gene expression by deacetylation of histone proteins leading to a closed chromatin configuration and by deacetylation of transcription factors, thereby activating or repressing their activity. FOXO, forkhead box O; NF-κB, nuclear factor-κB; PGC, perixosome proliferator-activated receptor γ-coactivator 1-α; SIRT1, sirtuin 1.
Mechanisms of Sirtuin Activation by Caloric Restriction
The molecular mechanisms or pathways leading to sirtuin activation during CR are highly complex because of the wide range of physiologic effects linked to CR and sirtuin activity. However, as NAD+ is required as a cofactor for sirtuin enzymatic activity (Figure 1), it is clear that sirtuins can be regulated by shifts in the redox potential that modulate the cellular NAD+/NADH ratio. Increases in the NAD+/NADH ratio have been shown to stimulate the activity of sirtuins, whereas NAD+ depletion suppresses sirtuin activity (Rodgers et al, 2005; Lin et al, 2004). Evidence that shifts in the redox state could provide a potential pathway for sirtuin activation during CR was first described in yeast, which showed an increase in oxygen consumption and NADH oxidation (conversion of NADH to NAD+) during CR (Lin et al, 2002). Overexpression of NADH dehydrogenases, or other manipulations that decreased NADH levels, was shown to modulate both Sir2 activity and longevity in yeast (Lin et al, 2004). Similar results have been observed in rodents undergoing CR wherein elevated NAD+ levels correlated with increased SIRT1 expression in various tissues, including the liver and brain (Cakir et al, 2009; Rodgers et al, 2005). In liver extracts of CR mice, the absence of NAD+ decreased CR-associated SIRT1 deacetylase activity (Rodgers et al, 2005), which further suggests that increases in NAD+ availability induced by CR-mediated changes in redox potential may be a major factor in stimulating sirtuin activity.
Another possible mechanism by which CR increases sirtuin activity involves the removal of nicotinamide, a potent noncompetitive inhibitor of Sir2/SIRT1 generated by the sirtuin deacetyltransferase reaction. During deacetylation, the acetyl group from the target protein is transferred to the ADP-ribose moiety of NAD+ producing O-acetyl-ADP-ribose, nicotinamide, and the deacetylated protein (Tanner et al, 2000) (Figure 1). In yeast, nicotinamide is removed by the nicotinamidase Pnc1, which converts nicotinamide into nicotinic acid and ammonia (Ghislain et al, 2002). A study using Sir2 and Pnc1 mutant yeast strains showed that Pnc1 was both necessary and sufficient for CR-mediated lifespan extension in a Sir2-dependent manner (Anderson et al, 2003), suggesting that Pnc1 extends lifespan by activation of Sir2. The higher eukaryote homolog of Pnc1, Nampt (nicotinamide phosphoribosyltransferase), is upregulated by CR (Yang et al, 2007a) and is also a longevity mediator and activator of SIRT1 (van der Veer et al, 2007). AMPK activation by CR (Greer et al, 2007; Palacios et al, 2009) has been shown to stimulate SIRT1 through Nampt regulation (Fulco et al, 2008). Although this suggests that Nampt may also be important in CR-mediated longevity in higher eukaryotic organisms, a necessary role in SIRT1-mediated longevity/protection during CR has not yet been confirmed. Nonetheless, the evidence thus far reveals that removal of nicotinamide in addition to changes in the NAD+/NADH ratio may function to upregulate Sir2/SIRT1 during CR.
Resveratrol Protects against Ischemia by Sirtuin Activation
The population of France consumes foods high in saturated fats, yet experiences fewer cardiovascular pathologies. This is known as the ‘French paradox’ and may be partially explained by their daily consumption of red wine (Renaud and de Lorgeril, 1992). Prominent in red wine is resveratrol (3,5,4′-trihydroxystilbene), an antioxidant that has been shown to have many beneficial effects and also functions as a CR mimetic. Similar to CR, resveratrol can maintain mitochondrial integrity, reduce insulin-like growth factor-1, activate Sir2/SIRT1, and increase lifespan from yeast to mammals (Baur et al, 2006; Howitz et al, 2003) (Figure 2). Resveratrol also improves outcome after ischemic episodes in the heart and brain (Della-Morte et al, 2009; Das et al, 2006) and requires SIRT1 to mediate ischemic protection (Chen et al, 2009; Raval et al, 2006). Resveratrol-activated pathways have been shown to protect against ischemia by modulating excitotoxicity, mitochondrial functioning, blood vessel integrity, and NO signaling (Gao et al, 2006b; Gurusamy et al, 2010; Das et al, 2006; Bulhak et al, 2009).
Resveratrol-Activated Pathways Protect against Ischemic Injury
Resveratrol is a potent SIRT1 activator (Howitz et al, 2003), and both in vitro and in vivo evidence strongly suggests that resveratrol-mediated protection against ischemic injury requires SIRT1 activity. For example, resveratrol was ineffective in preventing against hypoxic-induced apoptotic signaling in cultured cardiomyocytes when SIRT1 was suppressed (Chen et al, 2009). In organotypic hippocampal slice cultures, blockade of SIRT1 inhibited the ability of resveratrol to prevent cell death after oxygen–glucose deprivation (OGD), a model of in vitro ischemia (Raval et al, 2006). Neuroprotection mediated by resveratrol in the hippocampus after asphyxial cardiac arrest (a model used to induce global cerebral ischemia) was also abolished after SIRT1 inhibition (Della-Morte et al, 2009). The protective mechanism of resveratrol against ischemic injury is not fully understood, but is likely the cumulative result of multiple pathways activated by resveratrol.
In the ischemic brain, an increase in extracellular glutamate levels results in overstimulation of glutamatergic receptors, which can increase cytosolic calcium to excitotoxic levels that deplete cellular energy. Resveratrol has been shown to suppress postsynaptic glutamatergic transmission (Gao et al, 2006b) and to increase glutamate uptake in astrocyte cultures after oxidative stress (Vieira de Almeida et al, 2008), a measure that may reduce excitotoxicity. Chronic resveratrol treatment was also shown to significantly reduce excitatory transmitter release and increase inhibitory neurotransmitter levels during ischemia–reperfusion (Li et al, 2010). Thus, by preventing excitotoxicity, resveratrol may function to protect the ischemic brain tissue against energy depletion, oxidative stress, and death signaling.
The mitochondria undergo major functional impairments after cerebral ischemia (Fiskum et al, 1999), which may be prevented by resveratrol treatment. Injection of resveratrol was shown to increase hippocampal mitochondrial oxygen consumption rates after cardiac arrest (global cerebral ischemia) in a SIRT1-dependent manner (Della-Morte et al, 2009). Resveratrol also decreased levels of uncoupling protein (UCP)-2 (Della-Morte et al, 2009), which is an inner mitochondrial membrane protein that dissipates electrochemical gradient, thereby suppressing ATP production. An additional pathway by which resveratrol may increase mitochondrial performance is by induction of autophagy, a catabolic process that maintains cellular ATP levels through the degradation of proteins and membranes. During ischemia, autophagy can also sequester the damaged mitochondria, thereby avoiding excessive generation of ROS and the release of proapoptotic proteins, such as cytochrome c and apoptosis-inducing factor (Gustafsson and Gottlieb, 2009). Recently, it was shown that resveratrol protected both H9c2 cardiac myoblast cells and the myocardium from ischemia–reperfusion injury by inducing autophagy (Gurusamy et al, 2010).
Inflammation during ischemia–reperfusion causes breakdown of the vascular endothelium, leading to increased membrane permeability and hemorrhage. Resveratrol has been reported to maintain blood vessel integrity after ischemic injury by modulating leukocyte activity and reducing expression of inflammatory enzymes. For example, matrix metalloproteinase-9 is an endopeptidase expressed during ischemia that induces inflammatory signaling and leads to degradation of blood vessels (Gidday et al, 2005). Resveratrol treatment was shown to significantly suppress matrix metalloproteinase-9 expression after OGD in cultured neurons (Cheng et al, 2009) and after focal brain ischemia (Gao et al, 2006a). A major source of matrix metalloproteinase-9 during ischemia is derived from leukocytes (Gidday et al, 2005; Justicia et al, 2003), which bind to the endothelium, infiltrate the ischemic tissue, and initiate the inflammatory response. After myocardial ischemia, resveratrol was shown to suppress molecules that mediate leukocyte adhesion, such as nuclear factor-κB (NF-κB) (Das et al, 2006), a major mediator of inflammatory signaling. It has been reported that NF-κB can be inhibited by SIRT1 deacetylation (Yeung et al, 2004), which suggests that SIRT1 may mediate defense against inflammation (Figure 3). Resveratrol has been further shown to protect coronary arterial endothelial cells against oxidative stress by activating nuclear factor-E2-related factor-2 (Ungvari et al, 2010), a transcription factor that regulates genes involved in antioxidant defense.
After ischemic episodes, supply of oxygen and nutrients is enhanced by angiogenesis, a process characterized by vascular endothelial cell proliferation, migration, and alignment to form new blood vessels. In the heart, resveratrol increased levels of vascular endothelial growth factor (VEGF) and its receptor FLK-1, leading to enhanced angiogenesis and reduced myocardial infarction (Fukuda et al, 2006). This protection has also been shown in the brain where administration of resveratrol elevated proangiogenic factors such as matrix metalloproteinase-2 and VEGF levels providing neuroprotection after focal ischemia in the delayed injury phase (Dong et al, 2008).
Resveratrol has been shown to prevent OGD-induced neuronal cell death by activation of peroxisome proliferator-activated receptor-α (PPAR-α) (Cheng et al, 2009), an enzyme that indirectly enhances NO, a gasotransmitter implicated in ischemic protection. For example, PPAR-α activation was shown to reduce infarct size and increase endothelial NOS expression after ischemia–reperfusion injury in type II diabetic myocardium (Bulhak et al, 2009). Similarly, resveratrol-mediated NO signaling has been shown to reduce infarct and increase endothelial NOS in the brain and heart (Tsai et al, 2007; Shen et al, 2006). Activation of resveratrol-enhanced NO signaling also increased the expression of heme oxygenase-1, an enzyme that enhances the production of carbon monoxide, a gasotransmitter with downstream consequences, such as activation of the survival signaling kinases, p38 mitogen-activated protein kinase, and Akt, leading to cardioprotection after global ischemia (Das et al, 2006). Additional evidence showed that resveratrol led to cardioprotection in apolipoprotein E knockout mice by reversing reductions in tetrahydrobiopterin (an NOS cofactor), which cause endothelial NOS uncoupling (Xia et al, 2010). Application of the sirtuin inhibitor sirtinol, prevented resveratrol-mediated enhancements of tetrahydrobiopterin (Xia et al, 2010), which suggests an important role of sirtuins in resveratrol-mediated NO signaling. Taken together, these studies show that resveratrol may both directly and indirectly modulate NO signaling, leading to protection after ischemia–reperfusion.
The diverse effects of resveratrol likely stems from its ability to alter the activity of many intracellular pathways, including signal transduction pathways, cellular reduction–oxidation reactions, gene expression, cell cycle, DNA damage response, and cell death (Raval et al, 2008). Although SIRT1 has been shown to be crucial for resveratrol-mediated protection during ischemia (Raval et al, 2006; Chen et al, 2009), the role of SIRT1 in many resveratrol pathways has not been identified. For example, resveratrol activation of AMPK and stimulation of neurite outgrowth in neuronal cultures were not altered by inhibition of SIRT1 (Dasgupta and Milbrandt, 2007). Therefore, it is possible that resveratrol may activate both SIRT1-dependent and SIRT1-independent pathways that provide protection against ischemic injury.
Mechanisms of SIRT1 Activation by Resveratrol
Resveratrol has been shown to strongly stimulate SIRT1 deacetylase activity in a dose-dependent manner by increasing its binding affinity to both the acetylated substrate and NAD+ (Howitz et al, 2003). Biochemical and structural modeling studies have shown that resveratrol binds directly to SIRT1, thereby inducing conformational change in SIRT1 protein configuration (Borra et al, 2005). However, recent research has suggested that resveratrol is not a direct activator of SIRT1. In vitro studies by Pacholec et al (2010) and Beher et al (2009) indicated that SIRT1 was activated by resveratrol when an artificial, fluorescent acetyl-peptide was used as a substrate for deacetylation. Otherwise, resveratrol was unable to activate SIRT1 when the same peptide substrate lacked the covalently linked fluorophore (Beher et al, 2009; Pacholec et al, 2010). However, these recent studies are highly controversial and do not rule out the possibility that SIRT1 is activated indirectly by resveratrol. For example, SIRT1 was shown to be phosphorylated on its carboxy-terminal at serine 659 and serine 661 by the protein kinase CK2 (Zschoernig and Mahlknecht, 2009), an enzyme that can be regulated by resveratrol (Ahmad et al, 2007). Similarly, other studies showed that sumoylation at lysine 734 (Yang et al, 2007b) or JNK (c-Jun N-terminal protein kinase) 2 phosphorylation on serine 27 (Ford et al, 2008) increased SIRT1 protein deacetylase activity. No studies have shown resveratrol-mediated posttranslational modifications of SIRT1 as of yet. However, resveratrol activates numerous pathways and could therefore modulate SIRT1 activity by regulating these signaling cascades.
Ischemic Preconditioning Activates SIRT1 and Protects against Cerebral Ischemia
Ischemic preconditioning (IPC) is an innate protective mechanism, whereby a sublethal ischemic insult protects against a subsequent lethal ischemic attack. First described in the heart by Murry et al (1986) and in brain slices by Schurr et al (1986) nearly 23 years ago, IPC has since been shown in numerous tissues and species. Ischemic preconditioning has been shown to significantly improve survival and functional recovery after severe ischemic episodes in both the heart and the brain by activating signaling pathways that maintain mitochondrial functioning, suppress ROS production, and reduce infarct (Dave et al, 2008; Saurin et al, 2002). The therapeutic potential of IPC has led to numerous investigations into the subcellular mediators and effector pathways activated by the preconditioning stimulus. The specific mechanism leading to ischemic tolerance by IPC is complex. However, our laboratory and others have identified some mediators of IPC-mediated protection.
Similar to resveratrol, IPC can counteract excitotoxicity after cerebral ischemia by modulating neurotransmitter activity. We have previously shown that IPC-mediated protection requires NO signaling (Centeno et al, 1999), a pathway that has been linked to modulation of the inhibitory neurotransmitter γ-aminobutyric acid (GABA) (Wang et al, 2006). After lethal ischemia, IPC induced the release of GABA and increased the activity of glutamate decarboxylase (Dave et al, 2005), an enzyme responsible for GABA synthesis. IPC was also shown to increase both the frequency and the amplitude of GABA receptor-mediated miniature postsynaptic currents in hippocampal brain slices (DeFazio et al, 2009). The fact that administration of a GABA agonist led to protection after lethal ischemia (Dave et al, 2005), whereas blockade of GABA receptors abolished IPC-mediated protection (DeFazio et al, 2009), confirms the protective role of GABA activity against ischemic injury in the brain. IPC has also been shown to increase inducible NOS in cardiac myocytes (Wang et al, 2002), and the effects of NO on GABA modulation have been linked to the regulation of cardiac properties (Shih and Chuang, 2007). In the hippocampus, GABA synapse activity was modulated in a protective manner by epsilon protein kinase C (εPKC) (DeFazio et al, 2009), an important signaling cascade activated by IPC.
The vital role of εPKC in IPC was shown in εPKC knockout mice, which failed to exhibit an IPC response in isolated perfused mouse hearts (Saurin et al, 2002). Similarly, pharmacological inhibition of εPKC in organotypic hippocampal slices also blocked neuroprotection by IPC (Raval et al, 2003). IPC-induced εPKC activation in vivo was shown to modulate mitochondrial properties by decreasing the production of ROS and by increasing mitochondrial membrane potential and oxygen consumption of specific respiratory chain complexes in the hippocampus (Dave et al, 2008). Furthermore, εPKC phosphorylates hippocampal mitochondrial K+ATP channels (Raval et al, 2007), which couple energy metabolism and electrical activity in the cell. In vivo studies of the heart have previously shown necessary roles of both PKC and mitochondrial K+ATP channels in IPC cardioprotection (Gaudette et al, 2000). In hippocampal slices, blocking mitochondrial K+ATP channels prevented protection by both IPC and εPKC against lethal OGD in hippocampal slices (Raval et al, 2007), suggesting that the importance of these channels can also be extended to IPC-mediated neuroprotection. Other signaling pathways implicated in IPC include the extracellular signal-regulated kinases, JNK, and p38, mitogen-activated protein kinase, NF-κB, adenosine receptors, and others (Downey et al, 2007). Our laboratory showed roles of mitogen-activated protein kinase and adenosine receptors in the εPKC signal transduction pathway (Lange-Asschenfeldt et al, 2004) and further showed εPKC-dependent phosphorylation of extracellular signal-regulated kinase1/2 (Kim et al, 2008) and NF-κB (Kim et al, 2010a) after IPC. This evidence suggests that the εPKC pathway may converge with other signaling pathways involved in IPC-mediated protection.
Ischemic Preconditioning-Activated SIRT1 Protects against Cerebral Ischemia
Similar to CR, we discovered that there was an increase in NAD+/NADH levels after preconditioning in hippocampal slices (Centeno et al, 1999). Therefore, we surmised that IPC could potentially activate SIRT1, leading to neuroprotective pathways similar to that already shown by CR. The role of SIRT1 in IPC-induced neuroprotection was shown in our laboratory using both in vitro and in vivo models. Exposure of organotypic hippocampal slices to IPC increased SIRT1 enzymatic activity (Raval et al, 2006). Furthermore, SIRT1 activation was found to be neuroprotective against OGD-induced cell death because blocking SIRT1 with sirtinol abrogated IPC-induced neuroprotection (Raval et al, 2006). Similarly, IPC increased hippocampal SIRT1 enzymatic activity and neuroprotection after global ischemia induced by cardiac arrest (Della-Morte et al, 2009). Other studies have shown an important role of SIRT1 in IPC-mediated cardioprotection as well. Hypoxic preconditioning in cardiac myocytes upregulated SIRT1 expression, which was associated with induction of hypoxic preconditioning protection (Rane et al, 2009). Pharmacological inhibition of SIRT1, or suppression of the SIRT1 activator Nampt, abolished IPC-mediated protein deacetylation and cardioprotection after prolonged coronary artery occlusion (Nadtochiy et al, 2010). Although these studies in the heart and brain show an important role for SIRT1 in IPC-mediated protection, the specific SIRT1 pathways activated during IPC have not been elucidated.
Mechanisms and Signaling Pathways of SIRT1 Protection
There is a diverse array of targets activated by SIRT1, which could lead to protection during CR, IPC, or resveratrol treatment (Figure 3). During hypoxic conditions, SIRT1 modulates the activity of hypoxic-inducible factors (HIFs), transcription factors involved in mediating protective adaptations during low oxygen conditions. Sirtuin 1 has been shown to stabilize HIF-1α during hypoxic preconditioning (Rane et al, 2009) and to directly activate HIF-2α during hypoxia exposure (Dioum et al, 2009). Sirtuin 1 activation of HIF-2α increased expression of erythropoietin (Dioum et al, 2009), a regulator of red blood cell production and angiogenesis. Ischemic preconditioning elevation of erythropoietin protein expression was shown to have a key role in IPC-mediated protection (Malhotra et al, 2006). Furthermore, erythropoietin alone was sufficient to induce a preconditioned response (Meloni et al, 2006) and to protect rat CA1 neurons against ischemia (Zhang et al, 2006). SIRT1-mediated activation of erythropoietin may be important for stimulating the formation of new blood vessels and for restoring blood supply to ischemic regions. For instance, SIRT1 activation has additionally been shown to modulate endothelial functions such as endothelial cell migration and sprout formation during angiogenesis (Potente et al, 2007), a physiologic process associated with improved outcome after ischemic injury (Fukuda et al, 2006).
SIRT1 interacts with members of the FOXO family of transcription factors involved in regulating oxidative stress and cell death. FOXO signaling was shown to be important for conferring hypoxic preconditioning-mediated protection against cerebral ischemia (Zhan et al, 2010). SIRT1 activation prevented against oxidative stress by activating FOXO3a (Brunet et al, 2004), which enhanced ROS scavenging in cardiomyocytes by upregulating antioxidant agents (Tan et al, 2008). SIRT1 also directly activates perixosome proliferator-activated receptor γ-coactivator 1-α (PGC1α) (Nemoto et al, 2005), a transcriptional coactivator associated with protection against oxidative stress. SIRT1-dependent PGC1α expression was shown to reduce infarct size and improve neurologic scores after middle cerebral artery occlusion (Zhu et al, 2010a). Rodents lacking PGC1α displayed enhanced hippocampal neurodegeneration in response to oxidative stress (St-Pierre et al, 2006) and exhibited decreased ROS scavenger expression after transient global ischemia (Chen et al, 2010). This suggests that SIRT1 may protect against ischemia-induced oxidative damage by activating PGC1α. SIRT1 activation of HIF-2α enhanced the expression of mitochondrial ROS scavenger manganese superoxide dismutase (MnSOD) (Dioum et al, 2009), which further shows that SIRT1 can regulate multiple mechanisms to mediate protection against oxidative stress.
SIRT1 has also been shown to mediate survival by inhibiting apoptotic death pathways. Hypoxic ischemia significantly increases the expression of p53 (Xu et al, 2006), a tumor suppressor involved in mediating apoptotic signaling by activating proapoptotic molecules. SIRT1 activation after myocardial infarction was associated with reduced p53 acetylation, which prevents p53-mediated BAX activation and apoptosis (Vahtola et al, 2010). SIRT1 was also shown to prevent cell death in cardiomyocytes after hypoxia by inhibiting FOXO1-mediated apoptosis (Chen et al, 2009). Knockdown of SIRT1 in endothelial cells increased proapoptotic signaling by increasing mitochondrial membrane depolarization, Bcl-xL/Bcl-2-associated death promoter (BAD) and caspase activation, and release of cytochrome c (Hou et al, 2010). However, activation of SIRT1 attenuated apoptotic signaling by inhibiting these effects (Hou et al, 2010).
The neuroprotective effects of SIRT1 against ischemia may stem from modulation of mitochondrial activity. For example, in addition to its role in antioxidant defense, PGC1α has also been associated with mitochondrial protection during bioenergetic stress. PGC1α is a stimulator of mitochondrial biogenesis (Wareski et al, 2009) and an important regulator of energy homeostasis. The hearts from PGC1α knockout mice exhibited decreases in ATP production and deficiencies in the expression of genes involved in oxidative phosphorylation (Arany et al, 2005). Hypoxia was shown to stimulate increases in PGC1α and mitochondrial biogenesis in cardiomyocytes (Zhu et al, 2010b), which was associated with enhanced oxidative metabolism in the cardiac muscle (Arany et al, 2005). During oxidative stress, PGC1α was also shown to upregulate UCP2 (St-Pierre et al, 2006), a member of inner mitochondrial membrane proteins, which regulate proton electrochemical gradient. UCP2 activity has been associated with neuroprotection during IPC-induced upregulation (Liu et al, 2009b; Mattiasson et al, 2003). Uncoupling protein-2 has been shown to have a protective role in cerebral stroke by regulating ROS production and calcium elevations in the mitochondria (Mehta and Li, 2009). Conversely, SIRT1 repression of UCP2 in the hippocampus after resveratrol preconditioning was associated with protection (Della-Morte et al, 2009), which suggests that IPC and resveratrol may function in independent pathways, although they both elevate SIRT1.
SIRT1 May not Always Protect against Ischemic Damage
The evidence presented thus far strongly suggests that SIRT1 is an important mediator of protection against ischemic injury in the heart and brain. However, in recent years, some studies have shown that increased SIRT1 activity does not always yield beneficial effects against ischemic injury. For example, transgenic mice overexpressing SIRT1 displayed a memory deficit and were not protected against neuronal damage after focal cerebral ischemia (Kakefuda et al, 2009). These detrimental effects may be attributed to the dependence of SIRT1 on NAD+. After cerebral ischemia, there are significant reductions in NAD+, which are associated with energy depletion and cell death. Replenishing NAD+ before or after OGD in primary neuronal cultures was shown to markedly reduce ischemic injury (Wang et al, 2008). Similarly, inhibition of poly(ADP-ribose) polymerase 1, an enzyme activated by DNA damage that consumes cellular NAD+ stores, inhibited NAD+ depletion and prevented ischemic injury after middle cerebral artery occlusion (Iwashita et al, 2004). Like poly(ADP-ribose) polymerase 1, SIRT1 requires NAD+ as a cofactor for enzymatic activity. Therefore, SIRT1 could also function to deplete cellular energy. Liu et al (2009a) showed that the administration of nicotinamide, an inhibitor of sirtuin activity and NAD+ precursor, preserved cellular NAD+ and prevented cell death in neurons subjected to treatment with high levels of glutamate and N-methyl-
Roles of Mitochondrial Sirtuins in Cerebral Ischemia
After cerebral ischemia, the mitochondria undergo a number of impairments that lead to reductions in respiration rates, increased ROS generation, depolarization of mitochondrial membrane potential, and release of cytochrome c (Fiskum et al, 1999). Studies of resveratrol and IPC revealed that modifications of mitochondrial physiology are a major source of neuroprotection against ischemia-induced cellular degeneration (Dave et al, 2008; Della-Morte et al, 2009). SIRT1 modulation of nuclear targets has been shown to have a key role in protecting the mitochondria against ischemic injury by inhibiting apoptotic signaling and improving oxidative performance (Chen et al, 2009). One recent study showed that SIRT1 is present in the mitochondria where it may locally regulate mitochondrial biogenesis (Aquilano et al, 2010). The more conventionally recognized mitochondrial localized sirtuins, SIRT3–5 (Figure 1), are localized to the mitochondrial matrix, with the exception of SIRT5, which has also been found in the intermembrane space (Nakamura et al, 2008; Schlicker et al, 2008; Schwer et al, 2002) (Figure 4). SIRT4 is classified as an ADP-ribosyltransferase (Ahuja et al, 2007), whereas SIRT3 and SIRT5 exhibit deacetylase activity (Nakagawa et al, 2009; Schwer et al, 2002). Similar to SIRT1, these sirtuins have also been shown to regulate mitochondrial dynamics and are implicated in CR, aging, and metabolic stress protection.
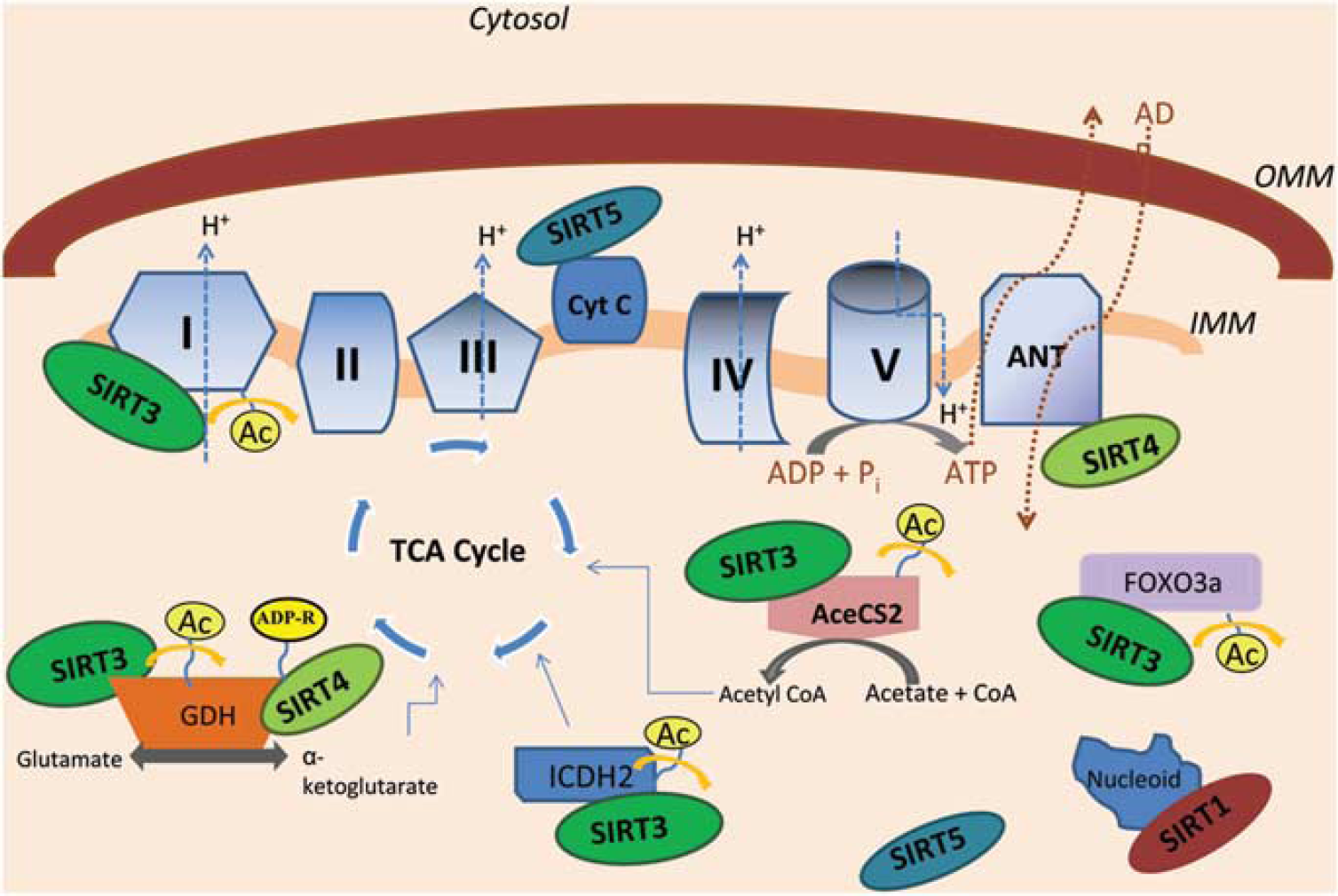
Mitochondrial sirtuins regulate substrates involved in metabolism, oxidative stress, and apoptosis. Many of the sirtuin family members localize to the mitochondria. SIRT3, SIRT4, SIRT5, and SIRT1 localize to the mitochondrial matrix, but only SIRT5 has also been identified in the intermembrane space. SIRT3 has been shown to deacetylate a protein located on complex I and FOXO3a, a transcription factor that enhances protection against ROS. SIRT3 can also activate GDH, ICDH2, and AceCS2, enzymes that produce key compounds in the TCA cycle. SIRT4 inhibits GDH activity through ADP ribosylation, and has been shown to interact with ANT, a translocase that exchanges ATP for ADP and is linked to apoptosis. SIRT5 interacts with cytochrome c, a protein linked to mitochondrial metabolism and apoptotic signaling. SIRT1 has been shown to associate with nucleoids, complexes that contain mitochondrial DNA. FOXO3a, forkhead box O 3a; IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane; ROS, reactive oxygen species; SIER1–5, SIRT1, sirtuin 1–5; TCA, tricarboxylic acid cycle.
Mitochondrial Sirtuins are Linked to Protection
As discussed previously, resveratrol and CR protect against ischemic damage by activating SIRT1 and enhancing mitochondrial functioning (Della-Morte et al, 2009; Kume et al, 2010; Raval et al, 2006). Resveratrol and CR have also been shown to modulate sirtuins located within the mitochondria. For example, heart tissues obtained from resveratrol-fed rats showed increased expression of SIRT3 and SIRT4 (Mukherjee et al, 2009). Resveratrol also enhanced SIRT3 expression in cultures of maturing adipocytes (Rayalam et al, 2008). Similarly, CR was shown to upregulate SIRT3, SIRT4, and SIRT5 in various tissues (Palacios et al, 2009; Nakagawa et al, 2009; Yang et al, 2007a). None of these mitochondrial sirtuins have been studied in relation to cerebral ischemia as of yet. However, because of their response to stress, these sirtuins may provide protective mechanisms against ischemic injury.
Of the mitochondrial sirtuins, SIRT3 has been the most extensively investigated. SIRT3−/− tissues displayed markedly elevated levels of mitochondrial protein lysine acetylation, (Lombard et al, 2007), which suggests that SIRT3 is a major mitochondrial deacetylase with multiple protein targets. Previously, acetylation of lysine residues was mainly viewed as an epigenetic regulator of gene expression (Loidl, 1994). However, recently, lysine acetylation has been recognized as an important posttranslational modification in the regulation of proteins located outside the nucleus. For example, a large proportion of mitochondrial proteins were found to contain acetyl-lysine residues, many of which regulate metabolism and longevity (Kim et al, 2006). During CR, the mitochondria undergo significant alterations in mitochondrial protein lysine-acetylation (Schwer et al, 2009), which suggests that this may be an important mitochondrial adaptation for conferring mitochondrial protection. As a major mitochondrial deacetylase, SIRT3 may function as a key regulator of protective mitochondrial functions. Both SIRT4 and SIRT5 activities have also been linked to resveratrol, CR, and metabolic stress response, which suggests that these sirtuins may also have roles in modulating the mitochondria during cellular stress.
Mitochondrial Sirtuins Protect against Oxidative Stress
Oxidative stress and the production of ROS are a major source of injury after ischemia–reperfusion (Siesjo et al, 1989). Under normal physiologic conditions, hydrogen peroxides and superoxide radicals produced by the mitochondria are detoxified by ROS scavengers, such as superoxide dismutase, glutathione peroxidase, and catalase. However, reoxygenation during reperfusion causes a significant increase in levels of mitochondrial ROS, which is associated with p53 activation and apoptotic signaling, genotoxic stress, and exacerbation of infarct (Chan, 2001). SIRT3 expression significantly increases in response to oxidative stress (Sundaresan et al, 2008) and may protect against ROS-mediated damage.
SIRT3 may boost antioxidant defense by activating isocitrate dehydrogenase 2 (Schlicker et al, 2008), which produces NADPH (NAD phosphate), a substrate required for antioxidant regeneration by glutathione reductase in the mitochondria. The SIRT3 overexpression has also been shown to increase levels of the ROS scavengers, namely MnSOD and catalase (Jacobs et al, 2008; Sundaresan et al, 2009). The enzymatic activity of both scavengers is significantly reduced in cells lacking SIRT3 expression, which suggests that SIRT3 may be an important regulator of MnSOD and catalase activity (Sundaresan et al, 2009). Mouse embryonic fibroblasts isolated from SIRT3 knockout mice display elevated mitochondrial superoxide levels, which increase in response to exogenous stressors, such as radiation and respiratory chain inhibitors (Kim et al, 2010a). Cells expressing enzymatically inactive SIRT3 exhibited signs of an overall oxidized intracellular environment in addition to increased intracellular superoxide levels (Jacobs et al, 2008). At the basal level, SIRT3 knockout cardiomyocytes produced twice the amount of ROS in comparison with wild types, which increased even further in response to exposure to pharmacological stress (Sundaresan et al, 2009). These results suggest that SIRT3 activity is integral for preventing ROS production in response to oxidative stress.
SIRT1 has been shown to protect against oxidative stress by activating the FOXO3a transcription factor (Brunet et al, 2004), a role that may also be shared with SIRT3. Recent evidence showed that overexpression of SIRT3 increased the DNA-binding activity of FOXO3a and induced FOXO3a-mediated gene expression (Jacobs et al, 2008). Furthermore, FOXO3a was found to bind directly to SIRT3 in the mitochondria (Jacobs et al, 2008) (Figure 4), which suggests direct interactions between SIRT3 and FOXO3a. This is corroborated by experiments in cardiomyocytes, which showed that SIRT3 directly binds and deacetylates FOXO3a in response to oxidative stress, thereby preventing accumulation of free radicals in the mitochondria (Sundaresan et al, 2009).
SIRT3 overexpression was also associated with increased expression of the transcriptional coactivator PGC1α (Palacios et al, 2009), a major mediator of oxidative stress protection during ischemia (Chen et al, 2010). SIRT3-deficient mice expressed lower levels of PGC1α in the skeletal muscle (Palacios et al, 2009) and blocked PGC1α upregulation of ROS scavengers in myotubes (Kong et al, 2010). SIRT1 directly binds and activates PGC1α expression in the nucleus (Nemoto et al, 2005), and a recent study has revealed that SIRT1 and PGC1α are also localized in the mitochondrial matrix in which they associate with nucleoids (aggregates of mitochondrial DNA) (Aquilano et al, 2010) (Figure 4). This could possibly suggest a new role for SIRT1 and PGC1α in the regulation of mitochondrial genes in addition to their already well-defined roles in the nucleus. It remains unknown whether SIRT3 can directly interact with PGC1α, but like SIRT1, SIRT3 may also protect against oxidative stress by regulating the transcriptional activity of FOXO3 and PGC1α.
Roles of Mitochondrial Sirtuins in Mitochondrial Metabolism
Impairments in mitochondrial respiratory machinery after cerebral ischemia result in energy metabolism failure by inhibiting the ability of the brain to maintain electrochemical gradients, which leads to cell death (Fiskum et al, 1999). The location of SIRT3, SIRT4, and SIRT5 in the mitochondria and their dependence on cellular NAD+ availability suggest that these sirtuins are involved in modulating energy and metabolism in response to stress. Acetyl CoA synthetase 2 and glutamate dehydrogenase have been identified as specific mitochondrial targets activated by SIRT3 (Hallows et al, 2006; Schlicker et al, 2008). Acetyl CoA synthetase 2 uses acetate and coenzyme A to produce acetyl CoA, an enzyme in the citric acid cycle important for mitochondrial respiration. The expression of acetyl CoA synthetase 2 was shown to significantly increase during conditions that mimic CR (Fujino et al, 2001), suggesting a significance of SIRT3 activation during metabolic stress. Glutamate dehydrogenase is involved in the production of α-ketoglutarate, an important intermediate in the citric acid cycle. SIRT4 inhibited glutamate dehydrogenase activity by ADP ribosylation (Ahuja et al, 2007), which may oppose activation of glutamate dehydrogenase by SIRT3 deacetylation. SIRT4 has additionally been shown to coimmunoprecipitate with ATP/ADP translocases 2 and 3 (Ahuja et al, 2007), which are carrier proteins that exchange mitochondrial ATP for cytosolic ADP. SIRT5 binds and deacetylates cytochorome c (Schlicker et al, 2008), a molecule involved in electron transfer important for respiration (Figure 4). These targets could signify important roles of SIRT3–5 in regulating metabolic dynamics. However, although SIRT3 seems to have a significant role in regulating energy production, the significance of SIRT4 and SIRT5 activity in metabolism is not yet known.
Similar to SIRT1, SIRT3 may also improve mitochondrial function and oxidative performance through activation of PGC1α (Palacios et al, 2009), a stimulator of mitochondrial biogenesis and a regulator of oxidative performance (Arany et al, 2005). In myotubes, SIRT3 overexpression increased mitochondrial biogenesis and was essential for PGC1α induction of biogenesis (Kong et al, 2010). Furthermore, SIRT3 was required for PGC1α induction of respiratory chain components, such as ATP5c and cytochrome c (Kong et al, 2010). A study by Ahn et al (2008) showed that knockout of SIRT3 resulted in significant reductions in basal ATP levels in the heart, kidney, and liver. Cells lacking SIRT3 also exhibited decreases in complex I state 3 respiration, which could be restored by expressing SIRT3 in the mutant tissue. Furthermore, SIRT3 interacted with a specific component of subunit 9 on complex I, which suggests a functional role for SIRT3 in the modulation of electron transport chain activity (Ahn et al, 2008). More recent studies have shown that SIRT3 deacetylates the succinate dehydrogenase flavoprotein subunit and increases complex II activity (Cimen et al, 2010). SIRT3 was also shown to directly bind and possibly deacetylate ATP5a (Law et al, 2009), a subunit of complex V. This evidence further supports a role for SIRT3 in regulating energy metabolism, which could provide beneficial effects during ischemic episodes.
Protection against Genotoxic Stress and Cell Death
Genotoxic stress after ischemic injury damages DNA and induces apoptotic signaling (Chen et al, 1997). Increasing NAD+ levels before or after OGD in neurons protects against DNA strand breaks and apurinic/apyrimidinic abasic sites, while also enhancing the activity of enzymes that repair damaged DNA (Wang et al, 2008). After cerebral ischemia, there is a significant reduction in NAD+ in the brain (Iwashita et al, 2004), which may have a major role in cell death caused by genotoxic stress (Yang et al, 2007a). Little is known about the mechanism by which NAD+ is synthesized in the mitochondria. However, it was recently shown that Nampt, an enzyme involved in the synthesis of NAD+, can maintain mitochondrial pools of NAD+ during genotoxic stress (Yang et al, 2007a). Nampt was shown to increase in response to hypoxia and protected against genotoxic-induced cell death through the activity of SIRT3 and SIRT4 (Yang et al, 2007a). Knockdown of either SIRT3 or SIRT4 with small-interfering RNA inhibited the ability of Nampt to prevent cell death mediated by a genotoxic alkylating agent (Yang et al, 2007a). The fact that mitochondrial levels of NAD+ were not depleted in response to genotoxic stress (Yang et al, 2007a) suggests that protection against DNA damage can be mediated in the mitochondria by SIRT3 and SIRT4 even after depletion of NAD+ from other cellular compartments. In cardiomyocytes, SIRT3 overexpression prevented DNA alkylation and also reduced expression of poly(ADP-ribose) polymerase, a marker of DNA damage (Sundaresan et al, 2008). Mouse embryonic fibroblasts isolated from SIRT3 knockout mice exhibited an increase in aneuploidy in response to infrared radiation in comparison with wild-type mice (Kim et al, 2010b), suggesting that SIRT3 can also maintain chromosomal stability. SIRT3 seems to be important for preserving the integrity of mitochondrial DNA as livers obtained from SIRT3 knockouts displayed increased mitochondrial DNA damage with age (Kim et al, 2010b). Prevention of genotoxic stress by mitochondrial sirtuins could enhance survival after ischemia by reducing apoptotic signaling. Further evidence suggests that mitochondrial sirtuins may also directly modulate factors intimately involved in apoptotic signaling.
Translocation of BAX to the mitochondria after ischemic brain injury initiates apoptotic signaling and cell death (Cao et al, 2001). SIRT3 has been shown to prevent apoptosis through direct interaction with Ku70 (Sundaresan et al, 2008), a nuclear DNA-repair factor that binds to BAX, thereby inhibiting its translocation from the cytoplasm to the mitochondria (Sawada et al, 2003). During stress conditions, Ku70 becomes acetylated, thus preventing its ability to bind BAX, which allows BAX to enter the mitochondria and initiate apoptosis (Cohen et al, 2004a). SIRT3 was shown to prevent acetylation of Ku70 in cardiomyocytes subjected to lethal oxidative or genotoxic stressors (Sundaresan et al, 2008). In addition, SIRT3 was found to be localized in the cytoplasm and nucleus in addition to the mitochondria, which could partially explain the ability of SIRT3 to directly deacetylate Ku70 in vitro and in vivo (Sundaresan et al, 2008). Although SIRT3 is generally defined as a mitochondrial sirtuin, this study and others have suggested that SIRT3 may have a role in the nucleus (Nakamura et al, 2008; Scher et al, 2007; Sundaresan et al, 2008). Therefore, SIRT3 may protect the mitochondria by directly modulating both nuclear and mitochondrial proteins.
After cerebral ischemia, BAX binds to adenine nucleotide translocator (ANT), forming a heterodimer complex implicated in triggering apoptotic signaling (Cao et al, 2001). The ANT is a translocator localized in the inner mitochondrial membrane that has been shown to coimmunoprecipitate with SIRT4 (Ahuja et al, 2007). It was identified as a major component of the mitochondrial permeability transition pore (Vieira et al, 2000), which has a major role in apoptosis. Inhibition of ANT prevented the activation of caspases after cerebral ischemia (Cao et al, 2001). The interaction of BAX with ANT was shown to trigger cell death by increasing membrane permeability, resulting in the release of proapoptotic molecules from the mitochondria, such as cytochrome c (Bhola et al, 2009). SIRT5 has been shown to bind and deacetylate cytochrome c (Schlicker et al, 2008), an important signaling molecule in the apoptosis cascade. The physiologic relevance of SIRT4 interaction with ANT and SIRT5 deacetylation of cytochrome c has not been explored, but may involve regulation of apoptosis. However, as previously discussed, SIRT4 and SIRT5 may also regulate some aspect of metabolism by binding to these molecules. Although SIRT3, SIRT4, and SIRT5 are implicated in certain paradigms of apoptotic signaling (Figure 4), their roles in apoptosis after cerebral ischemia have not been defined.
SIRT6 and SIRT7 Protect against DNA Damage and Cell Death
SIRT6 is predominantly localized in the nucleus and is highly expressed in the heart and brain (Liszt et al, 2005). It has been shown to display deacetylase activity on histones (Mostoslavsky et al, 2006), and more recent reports have shown that SIRT6 associates with telomeric chromatin and deacetylates lysine 9 of histone H3 (McCord et al, 2009; Michishita et al, 2008; Tennen et al, 2010). Similar to the other sirtuins already discussed, SIRT6 seems to have important roles in longevity, metabolic stress response, and DNA repair. Mice overexpressing SIRT6 are protected against impairments in fat and glucose metabolism caused by diet-induced obesity (Kanfi et al, 2010), whereas SIRT6-deficient mice have shortened lifespan and are highly susceptible to DNA damage (Michishita et al, 2008; Mostoslavsky et al, 2006). SIRT6 was shown to form a complex with DNA-dependent protein kinase (McCord et al, 2009), an enzyme involved in the repair of DNA double-strand breaks. Binding of SIRT6 stabilized DNA-dependent protein kinase at chromatin and promoted the activity of DNA-dependent protein kinase (McCord et al, 2009). Furthermore, SIRT6 was required for the base excision repair pathway under normal physiologic conditions (Mostoslavsky et al, 2006). On the basis of this evidence, SIRT6 could be involved in protection by regulating the cellular response to metabolic and genotoxic stress during cerebral ischemia.
SIRT7 is found in the nucleus where it is highly concentrated in nucleolar regions (Michishita et al, 2005). It has a role in activating RNA polymerase I (Ford et al, 2006) and has been shown to directly interact with the upstream binding factor (Grob et al, 2009), a transcription factor that regulates rDNA transcription. SIRT7 was shown to be highly expressed in metabolically active tissues, but displayed low basal levels in the heart and brain (Ford et al, 2006). However, in cardiomyocytes, SIRT7 directly deacetylated p53 in vitro, and depletion of SIRT7 led to hyperacetylation of p53 resulting in significant increases in basal apoptosis in vivo (Vakhrusheva et al, 2008). Furthermore, the fact that SIRT7-deficient cardiomyocytes were more susceptible to oxidative and genotoxic stress (Vakhrusheva et al, 2008) suggests that SIRT7 may also have a role in protection during ischemic episodes.
Concluding Remarks
Sirtuin activity seems to have a vital role in the therapeutic potential of CR, resveratrol, and IPC against ischemic injury. Research has shown that sirtuin activity is both necessary and sufficient for protection against lethal cerebral ischemia. Sirtuin 1 had been studied extensively in many experimental models, whereas the involvement of other sirtuins in neuroprotection is less defined. However, the sirtuins discussed are stress-responsive enzymes that can be linked to modulating protective pathways against oxidative stress, energy depletion, DNA damage, and apoptosis. Thus, although SIRT3–7 have not been directly implicated in ischemic protection or neurodegenerative disorders, these sirtuins exhibit significant roles in enhancing cellular function against stress-mediated damage, some of which have been associated with ischemia–reperfusion injury. The functional roles of sirtuins are still being investigated, and major research lies ahead in determining the importance of these enzymes in modulating ischemic protection.
Footnotes
The authors declared no conflict of interest.