
Abstract
Cortical spreading depression (CSD) and depolarization waves are associated with dramatic failure of brain ion homeostasis, efflux of excitatory amino acids from nerve cells, increased energy metabolism and changes in cerebral blood flow (CBF). There is strong clinical and experimental evidence to suggest that CSD is involved in the mechanism of migraine, stroke, subarachnoid hemorrhage and traumatic brain injury. The implications of these findings are widespread and suggest that intrinsic brain mechanisms have the potential to worsen the outcome of cerebrovascular episodes or brain trauma. The consequences of these intrinsic mechanisms are intimately linked to the composition of the brain extracellular microenvironment and to the level of brain perfusion and in consequence brain energy supply. This paper summarizes the evidence provided by novel invasive techniques, which implicates CSD as a pathophysiological mechanism for this group of acute neurological disorders. The findings have implications for monitoring and treatment of patients with acute brain disorders in the intensive care unit. Drawing on the large body of experimental findings from animal studies of CSD obtained during decades we suggest treatment strategies, which may be used to prevent or attenuate secondary neuronal damage in acutely injured human brain cortex caused by depolarization waves.
Keywords
Introduction
Cortical spreading depression (CSD) is a depolarization wave in cerebral gray matter that propagates across the brain at slow velocity, 2 to 5 mm/min (Leao, 1944). In normal brain tissue studied experimentally, CSD usually has to be induced by a deliberate perturbation of the brain such as electrical or mechanical stimulation (Leao, 1944). The CSD silences spontaneous and evoked synaptic activity for 5 to 15 minutes, and return to normal function occurs spontaneously. In hypoxic, ischemic, or hypoglycemic brain tissue, CSDs will usually occur spontaneously, and recovery occurs with a prolonged time course (Kraig and Nicholson, 1978). In this review, we use the abbreviation CSD as a generic term for all brain waves characterized by near-complete sustained depolarization of neurons (Leao, 1951). Peri-infarct depolarizations (PIDs; spontaneous) and other spreading depolarization waves accompanying brain injury may not be accompanied by depression of the electroencephalography (EEG), as the EEG is already silent in the compromised brain tissue. Thus, for these events ‘cortical spreading depolarization’ may be a more appropriate term. We shall, however, present the evidence that a clear distinction needs to be made between spreading depolarizations (1) that are accompanied by spontaneously reversible recovery of normal function and (2) PIDs—accompanied by prolonged recovery of cortical function, or no recovery at all. It is this latter variant of CSD that is most clearly a causal factor in the expansion of experimental infarcts, is demonstrable in patients with brain injury, and is suspected when present of contributing to increased long-term morbidity.
For many years, it was believed that CSD was an artifact produced in animal experiments and without significance for human neurological conditions—a viewpoint that we intend to challenge with this review. One of the reasons for the lack of interest in CSD is the difficulty in detecting the brief and relatively focal depolarization wave noninvasively in the human brain. This is explained by the high impedance of dura and skull and the evidence that it is a narrow cortical zone, only a few centimeters across, that undergoes local electrical EEG silence. This is insufficient to influence the routine scalp EEG, which records brain electrical activity from a cortical area of several square centimeters. The first evidence documenting the relevance of CSD for neurology was reports of symptoms of the migraine aura, which was explained by an excitation-depression wave propagating across the human primary visual cortex at the same rate as CSD in animals (Lashley, 1941; Leao and Morison, 1945; Milner, 1959). Later, brain scans of cerebral blood flow (CBF) in patients during migraine attacks showed what was termed a ‘spreading oligemia’, a wave of reduced blood flow that propagated across the brain at the same rate and with the same signs of vascular impairment as CSD (Olesen et al, 1981; Lauritzen, 1994; Hadjikhani et al, 2001). These studies led to the view that CSD triggers symptoms and signs in migraine patients, and that CSD is a benign phenomenon in normal human brains with preserved perfusion and energy metabolism (Figure 1).
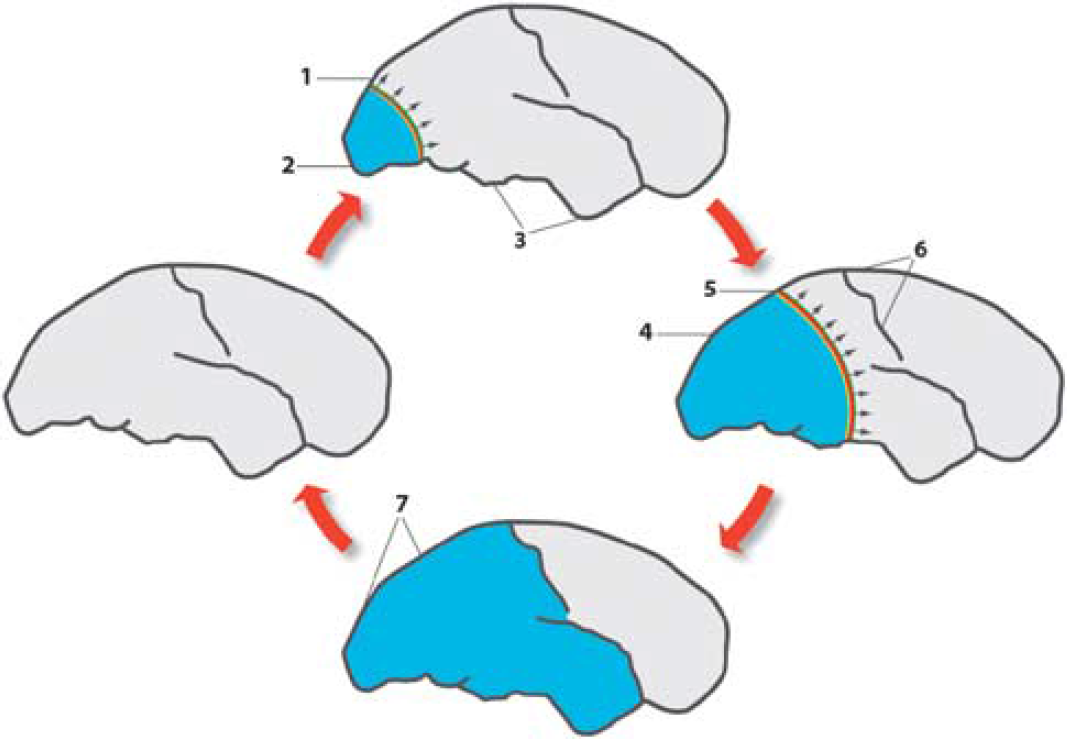
Effect of cortical spreading depression (CSD) on brain activity in normally perfused brain as exemplified in patients with migraine with aura. The figures represent lateral views of the human brain at different intervals after the beginning of the attack, spaced by ∼30 minutes. Outside of attacks, migraine brains have normal perfusion as indicated by the left cartoon. Colored bands indicate region of neuronal depolarization that causes and coincides with depression of cortical activity, a large-scale DC shift, failure of brain ion homeostasis, increased use of O2, glycogen and glucose, and rise in cerebral blood flow (CBF). Light blue area represents reduced CBF, reduced vascular reactivity and neurovascular coupling, and increased CMRO2 in the wake of CSD (Piilgaard and Lauritzen, 2009). The direction of propagation of CSD is indicated with arrows. At the start of migraine attack, a CSD emerges in the occipital pole in patients with a visual aura (1) while spreading anteriorly at the lateral, mesial, and ventral sides of the brain. At the CSD wave front, the transient ionic and metabolic disequilibria trigger the neurological symptoms in eloquent cortex. (2) Following CSD, cortical CBF decreases by 20% to 30%, neurovascular coupling is disturbed, and CMRO2 is increased for >2 hours. (3) Cerebral blood flow in regions not invaded by CSD remains normal. (4) The region of reduced CBF expands as CSD moves anteriorly. (5) Somatosensory symptoms from the extremities appear when the CSD invades the primary sensory cortex at the postcentral gyrus. (6) CSD in patients with migraine usually stops on reaching the central sulcus, but in many patients it does not even propagate this far. In patients with acute brain disorders, the susceptibility of the cerebral cortex to CSD is increased and propagation patterns are more diverse. (7) Full-scale attack. The CSD has stopped and is now detectable as a persistent reduction of cortical blood flow, neurovascular dysfunction and high-energy metabolism. After the attack, the cerebral cortex returns to normal. Modified with permission after original artwork presented in Lauritzen (1987).
An entirely different scenario may apply in the human brain acutely injured for example by ischemic stroke, brain trauma, or subarachnoid or intracerebral hemorrhage (ICH) (Hansen and Lauritzen, 1984; Strong et al, 2002; Fabricius et al, 2006; Dreier et al, 2006; Dohmen et al, 2008). This has been studied recently in patients who underwent neurosurgery including craniotomy because of their acute brain condition, by using cortical electrode strips similar to the technology applied in epilepsy surgery. In this situation, perfusion and energy metabolism is compromised, and the primary brain injury may expand over time and incorporate viable tissue into the lesion over the first few days. The recent series of invasive clinical studies by the Co-Operative Study on Brain Injury Depolarizations (COSBID; see http://www.COSBID.org) group have provided evidence that CSD occurs in a large percentage of patients with acutely injured neocortex, and has shown that CSD propagates in affected and apparently normal human brain tissue and may, when it takes on the characteristics of PIDs, contribute to the expansion of brain infarcts.
This review summarizes clinical and experimental data, which highlight the vascular, metabolic, and electrophysiological identification of CSD in acutely injured human brain cortex, and summarizes the evidence for CSD as a pathological mechanism of great importance for clinical neuroscience. We postulate that CSD occurs in migraine, that CSD or CSD-like events such as PIDS feature prominently in patients with acute brain injury, and that this may contribute to secondary brain damage, impair clinical recovery, and trigger new deficits. In other words, CSD may in certain patients be the switch between life and death for nerve cells. The CSD or disturbances of the hemodynamic response to CSD are potentially amenable to therapy, which raises an important issue regarding protection of vulnerable nerve cells based on new therapeutic strategies. The key to an understanding of this notion is an understanding of the mechanisms of CSD in normal and pathological brain tissue, and an appreciation of the clinical evidence for CSD in migraine and acute brain disorders as outlined in the following sections. The acquisition of patient data reported here followed procedures that were in accordance with the ethical standards of the responsible committee on human experimentation (institutional or regional) or with the Helsinki Declaration of 1975 (and as revised in 1983).
Cortical Spreading Depression: Electrophysiological Features and Spreading Mechanism
Cortical spreading depression is characterized by depression of evoked and spontaneous EEG activity spreading at a rate of 2 to 5 mm across the cortical surface (Leao, 1944; Grafstein, 1956). The spontaneous EEG remains markedly depressed for 1/2 to 1 minute, and then returns to normal within the following 5 to 10 minutes, whereas the evoked synaptic activity usually takes longer to recover (15 to 30 minutes) (Leao, 1944). The spreading depolarization of neurons and glia is preceded by propagating field oscillations covering distances of up to 1 mm (Larrosa et al, 2006). These oscillations indicate a brief state of hyperexcitability, which may relate to the observation of seizures and CSD in the same patients with acutely injured brain cortex (Fabricius et al, 2008). The oscillations are followed by complete loss of neuronal activity, which can last for minutes, before complete recovery. Simultaneously, the local tissue potential (the DC potential) swings negative with amplitude of 15 to 30 mV for 1 minute (Leao, 1951). The negative DC-potential shift may be explained by sustained complete depolarization that is restricted to specific cell domains as there is an initial explosive opening of conductance along most of the pyramidal neuron followed by a wave-like centripetal closure towards the apical dendrites (Canals et al, 2005). Local rises in tissue resistivity may contribute as well (Makarova et al, 2008). Thus, neurons are likely to be responsible for the current signals initiating CSD, and for those involved with its propagation and termination (Kunkler et al, 2005).
The EEG depression coincides with and is caused by a dramatic failure of brain ion homeostasis and efflux of excitatory amino acids from nerve cells (Vyskocil et al, 1972), which has important similarities to the ischemic penumbra (see Discussion section) (Strong et al, 1983). During CSD, extracellular [K+] increases to 30 to 60 mmol/L, whereas [Ca2+] decreases from ∼1.2 to 0.1 to 0.2 mmol/L, [CI−] decreases from 120 to 50 to 70 mmol/L, and [Na+] decreases from 150 to 50 to 70 mmol/L (Kraig and Nicholson, 1978; Somjen, 2001). Simultaneously, pH declines from 7.3 to ∼6.9 and the size of the extracellular space decreases to approximately half of control values due to water movement into neurons (Mutch and Hansen, 1984). The uptake of water into dendrites leads to a reversible neuronal swelling and distortion of dendritic spines while the volume of astrocytes remains constant (Kow and Harreveld, 1972; Takano et al, 2007; Zhou et al, 2010). This is consistent with the strong involvement of neurons in CSD. Normalization of most ion concentrations and the size of the extracellular space occur spontaneously after 1/2 to 1 minute, but [Ca2+] and pHe usually take a few minutes longer to recover.
There is a massive release of amino acids, including glutamate and aspartate, during the depolarization wave (Van Harreveld and Fifkova, 1970; Fabricius et al, 1993), and voltammetric recordings have shown that the massive transmitter release follows the onset of depolarization (Moghaddam et al, 1987). This suggests that neurotransmitter release may have only a secondary role in the spreading mechanism of CSD. We do not at present have a firm idea of the mechanism of initiation of CSD, but slightly elevated levels of K+ (Grafstein, 1956) and transmitters are sufficient to trigger the spread of CSD (Kunkler et al, 2005). Furthermore, depolarization of neurons as a consequence of synaptic transmission is expected to remove the voltage-sensitive Mg2+ block of the N-methyl-D-aspartate (NMDA) receptor, and sensitize the receptor to small increases of interstitial glutamate (Lauritzen et al, 1988). Interaction of glutamate with the NMDA receptor trigger K+ and glutamate release and further neuronal depolarization that will propagate to neighboring regions and start the process all over again. This central role for K+ (Grafstein, 1956) and glutamate fits well with data showing that the susceptibility of the tissue to produce CSD is enhanced when astroglial function is hampered (Largo et al, 1997). Astroglial cells protect against CSD initiation because of their high capacity for K+ buffering and glutamate uptake (Largo et al, 1997). Repetition of CSD waves in normal tissue results in a recoverable dysfunction of electrogenesis (Herreras and Somjen, 1993), but neuronal dysfunction may become complete and permanent in tissue deprived of glial function (Largo et al, 1997).
Blood Flow and Energy Metabolism During Cortical Spreading Depression
Cerebral blood flow decreases before CSD or during the onset of depolarization, but the vasoconstriction is variable and usually brief (Fabricius et al, 1995). During return of ionic changes to normal, CBF increases by ∼100% to 200% in anesthetized animals (Piilgaard and Lauritzen, 2009). The CBF increase lasts for 1 to 2 minutes and is rapidly succeeded by persistent (1 to 2 hours) flow reduction of 20% to 30% as a result of cortical arteriolar vasoconstriction, whereas CBF in noninvaded regions remains constant. Blood pressure autoregulation of CBF is preserved in the entire brain (Lauritzen, 1984). In contrast, after CSD, the vascular reactivity is markedly impaired in response to, for example, changes in arterial carbon dioxide tension (Lauritzen, 1984), basal forebrain stimulation (Lacombe et al, 1992), vasoactive substances applied directly to the pial arterioles (Wahl et al, 1987), and corticocortical stimulation (Piilgaard and Lauritzen, 2009). The classic pattern of a brief, large hyperperfusion followed by a protracted hypoperfusion is changed in disease states. In subarachnoid hemorrhage (SAH), CSD may trigger a state of cortical spreading ischemia (Dreier et al, 1998, 2000), and the changes in cerebrovascular signature triggered by CSD differ greatly in the penumbra depending on the distance to the infarct core (Strong et al, 2007) as described in detail in the following sections. In normal rats, L-arginine pretreatment prevents the development of low CBF after CSD, and increases the rate of recovery of the cerebrovascular responsiveness after CSD (Gault et al, 1994; Fabricius et al, 1995). This finding may have clinical importance, because it may be possible to treat persistent reduction of CBF in patients having CSDs with L-arginine. In mice, CSD may cause a decrease (Ayata et al, 2004) or a minimal rise in CBF and in consequence a mismatch between O2 use and supply that may lead to tissue hypoxia (Takano et al, 2007). Similarly, young rats (examined at day 15 to 25 after birth) have been reported to display no increase in CBF in response to CSD (Chuquet et al, 2007), and the duration of the CSD is inversely correlated to blood pressure, suggesting that cerebral perfusion pressure is critical for the recovery of normal brain function after CSD (Sukhotinsky et al, 2010). In summary, CSD triggers important changes of perfusion, which may determine whether the consequence of the depolarization wave is benign (Nedergaard and Hansen, 1988) or associated with permanent damage (Dreier et al, 2009). It is therefore important to define new treatment strategies targeting systemic and local blood flow regulation to improve recovery from acute brain disorders in which CSD is involved. Preservation of normal blood flow control is important to ensure that the brain's supply of glucose and oxygen match the dramatic rise in energy expenditure that accompanies recovery of normal function after the depolarization wave.
Restoration of the ionic gradients after CSD is energy demanding, and the rise in metabolism during CSD is among the largest observed in the brain (Shinohara et al, 1979). A brief rise in tissue partial pressure of oxygen (PtiO2) concomitant with the initial rise in CBF at the time of tissue depolarization (Piilgaard and Lauritzen, 2009) is consistent with a slight decrease in cerebral metabolic rate of oxygen (CMRO2) at onset of depolarization wave (Lukyanova and Bures, 1967). The rise in PtiO2 indicates that it takes a few seconds before oxidative phosphorylation reaches such high speed that CMRO2 exceeds O2 supply. In animals, PtiO2 may decrease to anoxic levels lasting for tens of seconds (Takano et al, 2007; Piilgaard and Lauritzen, 2009). This rise in CMRO2 corresponds to the phase where extracellular [K+] drops from 60 to 3 mmol/L and cells repolarize because of a pronounced increase in Na+/K+-ATPase activity (LaManna and Rosenthal, 1975), which doubles glucose use (Shinohara et al, 1979) and lactate release (Cruz et al, 1999). The huge rise in cytosolic Ca2+ accompanying CSD depolarizes neuronal mitochondria and triggers the rise in oxygen use via the Ca2+ uniporter in the inner mitochondrial membrane (Zhou et al, 2010). The CSD also activates glycolytic pathways, as evidenced by the accompanying prolonged rise in brain lactate and reduction in brain glycogen, while the energy charge (i.e., the brain concentration of energy-rich phosphate compounds) remains preserved in normal brains (Lauritzen et al, 1990; Parkin et al, 2005). The activation of glycolytic pathways is consistent with intracellular alkalinization of astrocytes during CSD (Chesler and Kraig, 1987), which stimulates glycogen phosphorylase (Hof et al, 1988) and lactate production (Cruz et al, 1999). Whether energy failure prevails and delays recovery of cortical function depends on limitations in the blood-borne O2 and glucose supply, which may vary between species because of variations in the acute CBF response (Ayata et al, 2004).
Migraine
Migraine is an important disorder encompassing both evidence of transient focal pathology and pain (Lauritzen, 2001). The migraine aura may be any neurological disturbance that appears shortly before or during the development of a migraine headache, and migraine auras may have different features, suggesting that different brain regions are involved (Lauritzen, 1994). Migraine auras are most often visual, apparently starting in area 17, which is the brain region with the highest neuronal density, and the relatively lowest density of astrocytes that may protect the cortex from CSD (Largo et al, 1997). Both the migraine aura and CSD propagate along the cortical surface (Hadjikhani et al, 2001) (Figure 1). For the human aspects, it is important that CSD tends to become extinct when propagating into a sulcus (Bowyer et al, 1999). The rate of spread of the migraine aura corresponds to a propagating velocity of the CSD of 2 to 6 mm/min, that is the rate of spread of CSD is variable, especially in gyrencephalic animals. Patients with migraine are reliable reporters of the characteristic spread of each symptom and the sequence of different symptoms, which suggests that CSD is the mechanism underlying migraine (Russell and Olesen, 1996). Interestingly, patients with acute brain disorders described in the following sections who had documented episodes of CSD while being conscious did not report a march of focal symptoms similar to a migraine aura, illustrating the complexity of the relation between CSD and the accompanying neurological symptoms. The idea of CSD as the underlying mechanism of migraine has been supported by neuroimaging studies recording changes in CBF or the BOLD signal in migraine patients (Lauritzen, 1994; Hadjikhani et al, 2001).
The discovery of causative genes for migraine has proven to be crucial for animal and clinical research into migraine (Barrett et al, 2008). The genetic forms of migraine (familial hemiplegic migraine) are rare diseases, which involve defects in sodium or calcium channels, or in the Na+-K-ATPase (Barrett et al, 2008). The well-established genetic basis of some familial hemiplegic migraine phenotypes has provided a unique opportunity for studies of the cellular migraine mechanisms (Tottene et al, 2009). The cortex of migraineurs is believed to be hyperexcitable due to impaired habituation, which may explain the increased vulnerability to CSD in these patients (Schoenen et al, 2003). Likewise, the basic pathology of CSD is an increase in excitability of cortical neurons, which is brought about by the cooperative action of persistent Na+ currents plus NMDA receptor-controlled currents. CSD is ignited when the sum of persistent inward currents exceeds persistent outward currents so that total membrane current turns inward (Somjen, 2001). Mice models carrying mutations of genetic migraines, for example the pore-forming subunit of neuronal Ca(V)2.1 Ca2+ channels display an exquisite sensitivity to CSD, with a vastly reduced triggering threshold, an increased propagation velocity, and frequently multiple CSD events after a single stimulus (van den Maagdenberg et al, 2010). Patients with familial hemiplegic migraine may display a particularly low threshold for CSD, a tendency to respond with multiple CSD events to a mild head trauma or ICH, and to deteriorate much more severely following such incidents than in the general population, but this issue will need to be addressed in further studies.
Malignant Hemispheric Stroke
Transient elevations of extracellular potassium resembling CSD were demonstrated as early as 1977 in a baboon focal ischemia model (Branston et al, 1977), and not much later, CSD-like depolarizations were described in stroke models in cats (Strong et al, 1983) and rats (Nedergaard and Astrup, 1986). These waves of depolarization appeared spontaneously in the surrounding of freshly developing ischemic infarcts in tissue that was functionally and metabolically compromised, but not yet irreversibly damaged—the so-called ischemic ‘penumbra’ (Astrup et al, 1981) and were termed peri-infarct depolarization (PID) (Mies et al, 1993; Hossmann, 1996). PIDs have quite similar features compared to artificially elicited CSDs, including cortical DC shifts of around −20 mV, massive disturbances of membrane ion homeostasis, and propagation velocities of typically 3 to 5 mm/min. Expansion of infarct size has been shown to correlate with the number of PIDs (Mies et al, 1993) or with their cumulative duration (Dijkhuizen et al, 1999). In a study combining focal ischemia with CSDs evoked in normal brain outside the penumbra, Busch et al (1996) were able to show directly that acceleration of infarct growth is related to the number of induced events spreading into the penumbra and there believed to take on the characteristics and pathogenicity of PIDs. The basis for this relationship may lie in the imbalance between metabolic workload and delivery of oxygen and glucose: metabolic workload is abnormally high because of restoration of cell membrane ion gradients after depolarization events, leading to depletion of the tissue glucose pool (Nedergaard and Astrup, 1986; Hopwood et al, 2005; Hashemi et al, 2009; Feuerstein et al, 2010) and insufficient oxygen delivery to the tissue (Back et al, 1994). On the other hand, oxygen and glucose supply is reduced due to microvascular constriction in response to the depolarization (Dreier et al, 1998; Strong et al, 2007; Luckl et al, 2009). Early formation of vasogenic edema within the first 3 hours is another factor contributing to progressively disturbed tissue perfusion in the penumbra due to mechanical compression of adjacent microvessels and veins in hemispheric experimental stroke in rats (Walberer et al, 2008; Gerriets et al, 2009). CSD alters blood–brain barrier permeability even in healthy tissue by activating brain matrix metalloproteinases, in particular matrix metalloproteinases-9 (Gursoy-Ozdemir et al, 2004). In the penumbra, therefore, blood–brain barrier disruption and edema formation are also linked to the occurrence of PIDs and their destructive role. Delayed repolarization subsequent to the negative shift of the cortical DC potential may develop from the combination of these deteriorative processes (Back et al, 1994). The harmful role of PIDs may be augmented by the fact that they often appear in clusters as shown in middle cerebral artery occlusion in cats, for example (Ohta et al, 2001), up to 16 hours after arterial occlusion (Saito et al, 1997). The higher the rate of recurrence of PIDs, the earlier did the cortical DC potential convert into terminal depolarization as an indicator of functional deterioration and infarct growth (Saito et al, 1997).
For several decades, it has been speculated whether spreading depolarizations arise in human ischemic stroke. A first attempt to discover the phenomenon by repetitive imaging of the apparent diffusion coefficient failed (Back et al, 2000), presumably because the successful demonstration of propagating PIDs by repetitive magnetic resonance (MR) diffusion mapping in experimental stroke in rats (Gyngell et al, 1994; Hasegawa et al, 1995) could not be easily reproduced in humans. The time period covered by apparent diffusion coefficient imaging amounted for example to only 15 minutes per patient, providing little opportunity to capture a depolarization wave. Another, more promising approach emerged after a first report of CSD in the periphery of traumatic contusions in human patients (Strong et al, 2002), based on invasive recording of the subdural electrocorticogram (ECoG). In stroke patients, a chance to use such an invasive technique arose from the introduction of decompressive surgery (hemicraniectomy) as a treatment option for large, hemispheric stroke (Hofmeijer et al, 2006; Juttler et al, 2007; Vahedi et al, 2007a, b ). This space-occupying so-called ‘malignant’ hemispheric stroke occurs in around 10% of patients with supratentorial infarcts. The mass effect, due to blood–brain barrier disruption and progressive edema formation, leads to a rise in intracranial pressure with transtentorial herniation and death in up to 80% of victims (Hacke et al, 1996). Acute ischemia of at least two thirds of the middle cerebral artery territory and optionally additional infarction of the territories of the posterior or anterior arteries identified by computed tomography or MR imaging serve as criteria to decide for hemicraniectomy (Huttner et al, 2008).
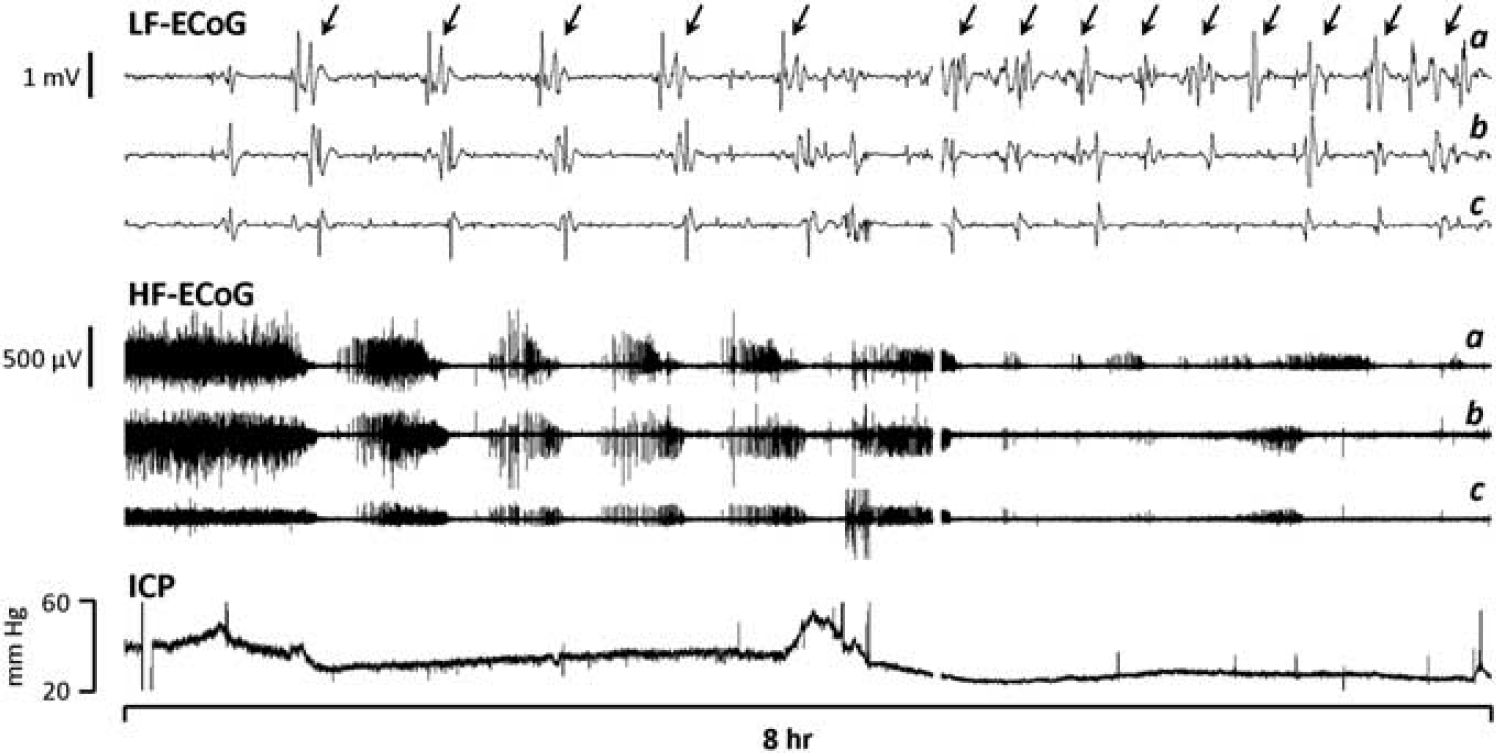
Four centers of the COSBID group conducted a first study in 16 ‘malignant’ stroke patients (Dohmen et al, 2008). In this study, polyphasic slow potential changes of the ECoG (corresponding to the negative slow voltage variation described by Leao (1947) and by Hartings et al (2006) and simultaneous transient ECoG suppressions were recorded as signatures of CSD; slow potential changes in the absence of ECoG background activity were recorded as signatures of PID (Fabricius et al, 2006). This differentiation between CSD and PID follows the original definition of the penumbra as a condition with impaired electrical function but preserved membrane integrity (Astrup et al, 1981). The CSD/PID events were detected over the course of 7 days after stroke in all but two of the ‘malignant’ stroke patients. More than 90% of the total of depolarization events occurred in temporal clusters. The very high incidence of CSD/PID in these patients was underlined by the fact that in the two patients without recording of depolarization waves, retrospective analysis confirmed that the electrode strip had been located above already infarcted and not, as intended, on peri-infarct tissue. In individual cases, transition from CSD to PID and progressive loss of remaining PID events on channels near the infarct core served as an indirect indicator of the energy status of the tissue and documented tissue deterioration, as did prolongation of the duration of ECoG suppression until recovery. In clusters of CSD/PID in particular, the duration of ECoG suppression tended to increase over time, indicating progressive worsening of the hemodynamic or metabolic status (Figure 2). Serial high-frequency imaging of perfusion (laser speckle) of the exposed cortex surrounding a small distal middle cerebral occlusion infarct in rats has now shown that a single PID can cycle in regular repetition around the core infarct, raising for the first time the possibility that PIDs might behave similarly in the ischemic human brain (Figure 3). This behavior would explain the periodicity of events as seen with electrode strips in patients, and suggests that such cycling can serve to amplify the adverse effects of PIDs in the penumbra described above. Lesion progression has recently been demonstrated by sequential MR imaging in a malignant stroke patient exactly in the zone experiencing some 100 PIDs over a period of 5 days (Nakamura et al, 2010). Similar deterioration has recently been observed in SAH patients: measurement of tissue oxygen partial pressure revealed progressive stepwise decline with subsequent CSDs within clusters (Bosche et al, 2010).
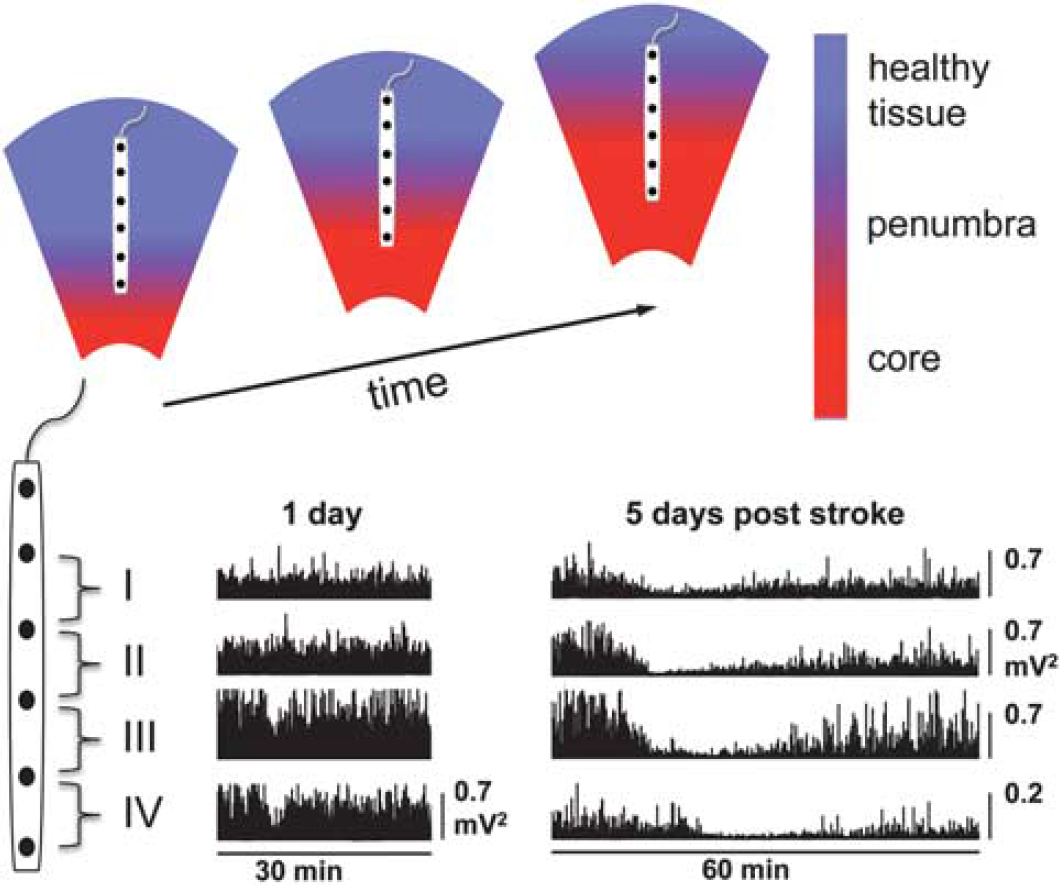
Infarct growth associated with prolongation of the duration of electrocorticogram (ECoG) suppression with repetitive peri-infarct depolarization. The schematic diagram (upper panel) shows the temporal and spatial expansion of infarcted core tissue and the ischemic penumbra along the electrode strip into the healthy periphery of the infarct. This structural deterioration progresses over several days and corresponds to the progressive prolongation of the depolarization along the electrode strip during successive peri-infarct depolarizations. The two examples of ECoG recordings (lower panel: power of the ECoG, high-pass filtered at 0.5 Hz) taken in a patient suffering from malignant ischemic stroke 1 and 5 days after ictus indicate this worsening of ECoG recovery after depolarization. Note that on day 1 after stroke, brief episodes of ECoG suppression are present only on channels IV and III, whereas on day 5, ECoG suppression is prolonged and present on channels I to IV.
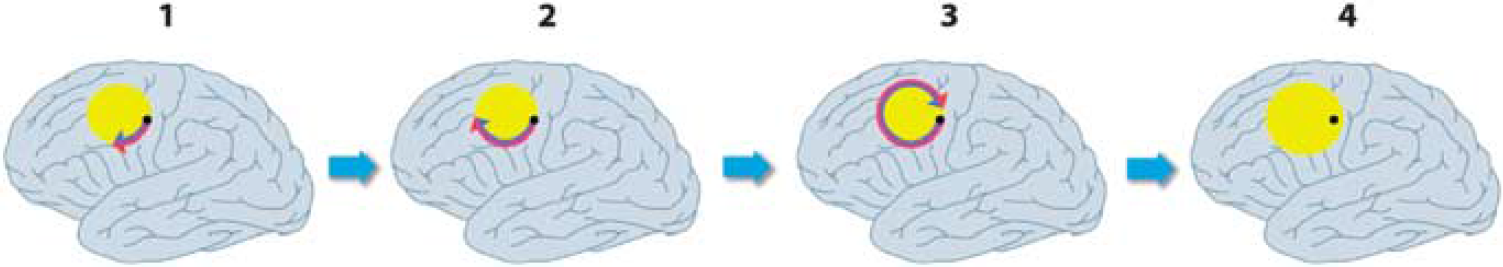
Schematic model of cycling of a peri-infarct depolarization around an ischemic core, and its effect on perfusion, and expansion of the core. Panel 1: A depolarization starts at the edge of the core (proposed to be a stochastic event most probably influenced by factors such as partial neuronal depolarization, limited availability of oxygen and glucose, and brain temperature). Panels 2 and 3: The cortical spreading depression (CSD) propagates most prominently around the edge of the core, where tissue is most susceptible to depolarization (Dahlem and Hadjikhani, 2009). Here, spread is clockwise only as anticlockwise spread is prevented by slow recovery from a previous event. The residual effect of the depolarization on perfusion is profound vasoconstriction nearest the core (blue component of arrow), sufficient to preclude repolarization and hence expanding the core. However, vasoconstriction further from the core (red section of arrow) is less severe or sustained, probably allowing repolarization from this event, but leaving tissue at this location vulnerable to a future depolarization. The net effect is shown in panel 4, with the core now expanded to include the original penumbral point of depolarization (Nakamura et al, 2010).
In conclusion, the demonstration and high incidence of CSD/PID in ‘malignant’ human stroke resembles results in numerous experimental models of focal ischemia. These effects may be generated by several pathophysiological mechanisms linked to CSD/PID such as metabolic decline, inverse coupling of regional perfusion to depolarization, and formation of brain edema. In hemispheric stroke not treated by hemicraniectomy (or even despite decompression), such detrimental effects will most probably accelerate the transition into malignant status.
Subarachnoid hemorrhage
Aneurysmal SAH is a life-threatening condition and patient outcome is determined mainly by brain damage induced by the initial hemorrhage and by the occurrence of delayed cerebral ischemia (DCI). Delayed cerebral ischemia shows peak occurrence around day 7 and develops in about 25% of patients surviving the initial hemorrhage (Macdonald et al, 2008). The impact of DCI research comes from the notion that DCI is a disease model for ischemic stroke (Dreier et al, 2009). This idea is based on the evidence that DCI develops while the patient is on the intensive care unit where proof-of-concept studies on neuroprotectants and neuromonitoring can be performed. The risk for DCI correlates with the amount of blood in the initial computed tomography scan (Brouwers et al, 1993), and its onset coincides with the period of peak subarachnoid hemolysis (Pluta et al, 1998). Therefore, erythrocyte products in the subarachnoid space presumably cause DCI. However, the pathophysiology of DCI is complex.
It has been increasingly questioned whether vasospasm of major cerebral arteries alone determines DCI. Initially, dominance of cortical infarcts over infarcts corresponding to territories of supply of major cerebral arteries in the pathoanatomical literature gave rise to this debate (Dreier et al, 2002). This controversy gained momentum with the finding that the endothelin receptor antagonist clazosentan showed a 65% relative risk reduction of angiographic vasospasm while the patient outcome did not improve significantly (Macdonald, 2008). Chronic vasospasm of distal arteries/arterioles, microthrombosis, spreading depolarization and spreading ischemia are under intense study as complementary factors in the pathogenesis of DCI (Ohkuma et al, 2000; Stein et al, 2006; Dreier et al, 2009).
Cat experiments with intracortical potassium- and calcium-sensitive microelectrodes provided first evidence that spreading depolarizations occur during SAH (Hubschmann and Kornhauser, 1980, 1982). This was later confirmed in rats using diffusion MR imaging (Beaulieu et al, 2000). These studies were restricted to the initial hours after the hemorrhage. The phenomenon of spreading ischemia was discovered in a rat model replicating the delayed conditions after subarachnoid hemolysis after SAH (Dreier et al, 1998). Spreading ischemia results from local microvascular dysfunction, which causes an inverse hemodynamic response to the cortical depolarization. With the inverse response, severe microvascular spasm instead of vasodilatation is coupled to the depolarization (Dreier et al, 1998; Petzold et al, 2005; Shin et al, 2006). Consequently, spreading ischemia delays the cortical repolarization. Thus, a prolonged negative cortical DC shift is the defining electrophysiological feature for spreading ischemia (Figure 4). Severe spreading ischemia was sufficient to induce widespread cortical necrosis in rats (Dreier et al, 2000). Based on the experimental evidence, it was speculated that spreading ischemia is a phenomenon involved in DCI (Dreier et al, 1998).
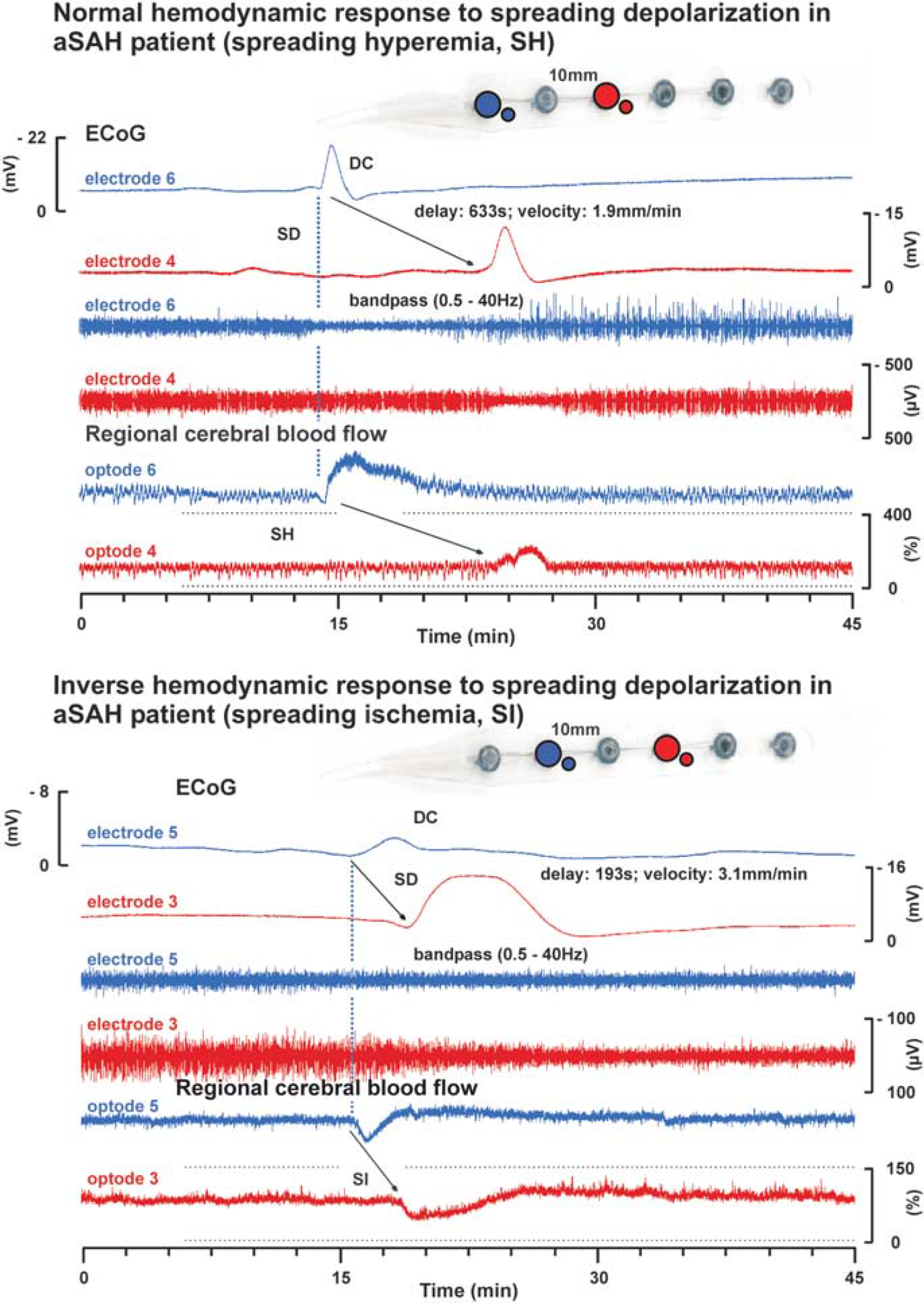
Normal and inverse response to cortical spreading depression (CSD) in patients with aneurysmal subarachnoid hemorrhage (aSAH). The upper half of the figure shows the normal hemodynamic response to spreading depolarization (CSD) in the human brain in a patient with aSAH. The subdural opto-electrode strip is shown in the upper right corner. The six traces represent simultaneous recordings of a single spreading depolarization that propagates from opto-electrode 6 (blue) to 4 (red). The calibration bar of trace 4 also applies to trace 3 and that of trace 6 also applies to trace 5. The four upper traces identify the spreading depolarization electrophysiologically. Traces 1 and 2: direct current (DC) electrocorticogram (ECoG) with negative shift of spreading depolarization. Traces 3 and 4: band-pass filtered ECoG (0.5 to 45 Hz) with spreading depression of activity. The spreading depolarization propagates at a rate of about 1.9 mm/min assuming an ideal linear spread along the recording strip. Traces 5 and 6: Normal spreading hyperemia in response to spreading depolarization recorded by laser-Doppler flowmetry as reported previously (Dreier et al, 2009). The lower half of the figure demonstrates the inverse hemodynamic response to spreading depolarization in another patient with aSAH. In this case, the spreading depolarization propagates from opto-electrode 5 (blue) to 3 (red) (propagation rate: 3.1 mm/min). The high-frequency activity is already suppressed by a preceding CSD at electrode 4 (trace 9), when depression of activity is induced by spreading depolarization at electrode 5 (trace 10). Traces 11 and 12: Typical inverse hemodynamic response to spreading depolarization as characterized by a severe decrease of regional cerebral blood flow (CBF) in response to the depolarization. Such severe decrease of regional CBF in response to CSD is termed spreading ischemia. The prolonged negative cortical DC shift (compare trace 8) is the defining electrophysiological feature for the inverse hemodynamic response. It indicates that the hypoperfusion is significant enough to produce a mismatch between neuronal energy demand and supply (Dreier et al, 1998, 2009).
Translation of this concept from bench to bedside comprised two steps. First, it was necessary to examine whether spreading depolarizations occur at all in SAH patients; second, does spreading ischemia develop? The first step consisted of a prospective multicenter study assessing incidence and timing of spreading depolarizations and DCI in 18 SAH patients who required aneurysm surgery (Dreier et al, 2006). Spreading depolarizations were recorded with subdural electrode strips placed on cerebral cortex for up to 10 days. Delayed ischemic strokes were verified by serial computed tomography scans or MR imaging. Electrocorticography detected spreading depolarizations in 72% of patients. Delayed cerebral ischemia occurred, time locked to recurrent spreading depolarizations in every single case. In four patients, delayed ischemic strokes developed in the recording area. These strokes were associated with progressive prolongation of the ECoG depression periods to > 60 minutes (Figure 4).
The second step of the translation from bench to bedside targeted spreading ischemia. For this purpose, a prospective, multicenter study was performed in 13 patients with SAH using novel subdural opto-electrode technology for simultaneous laser-Doppler flowmetry and DC-ECoG, combined with measurements of PtiO2 (Dreier et al, 2009). Isolated spreading depolarizations were observed in 12 patients and were associated with either physiological, absent, or inverse regional CBF responses. Whereas the normal hemodynamic response was associated with tissue hyperoxia, the inverse response caused tissue hypoxia. Clusters of prolonged spreading depolarizations with persistent depressions were recorded in five patients close to structural brain damage as observed by neuroimaging. Such clusters were associated with spreading ischemias that were significantly longer (duration of up to 144 minutes) than those coupled to isolated spreading depolarizations. Consistently, Bosche et al (2010) reported that progressively hypoxic responses of PtiO2 can develop during clusters of spreading depolarizations.
Intracerebral Hemorrhage
Spontaneous ICH constitutes 15% to 20% of all strokes and has the highest mortality within stroke: around 40%. Most survivors are disabled. Primary ICH accounts for around 80% of cases and is due to ruptured arteries/arterioles in hypertensive or angiopathic patients. Secondary ICH is caused by bleeding in tumors or congenital malformations or is due to coagulopathy or anticoagulant treatment. According to the International Surgical Trial in Intracerebral Haemorrhage (STICH) study (Mendelow et al, 2005), evacuation of the hematoma is mainly indicated if the bleeding is expanding, or to treat the primary cause. Accordingly, operations on ICH patients have declined (Kirkman et al, 2008), and only a limited number of patients have been recruited to the COSBID study (Strong et al, 2002; Fabricius et al, 2006). Most of these patients survived at 6 months, and they probably represent a relatively benign group within the ICH population. Nevertheless, CSDs have been recorded in >60% of ICH patients. In two normotensive young males, hematomas due to arterio-venous malformations were accompanied by multiple episodes of CSD, whereas in two older females with hematomas due to hypertension, no CSD episodes were recorded for 64 to 72 hours (Fabricius et al, 2006). These cases possibly reflect the experimental finding of an inverse correlation between blood pressure and severity of CSDs (Sukhotinsky et al, 2010).
The presence of an ischemic penumbra around intracerebral hematomas in man remains a controversial issue (Siddique et al, 2002; Herweh et al, 2009). In our preliminary data, we found that the mean duration of the ECoG depression during CSD was comparatively short in ICH patients (Figure 5), and PIDs—a putative electrophysiological signature of the penumbra—were rarely recorded (2% of events). This is in accordance with the view that ICH is usually not associated with a large ischemic penumbra.
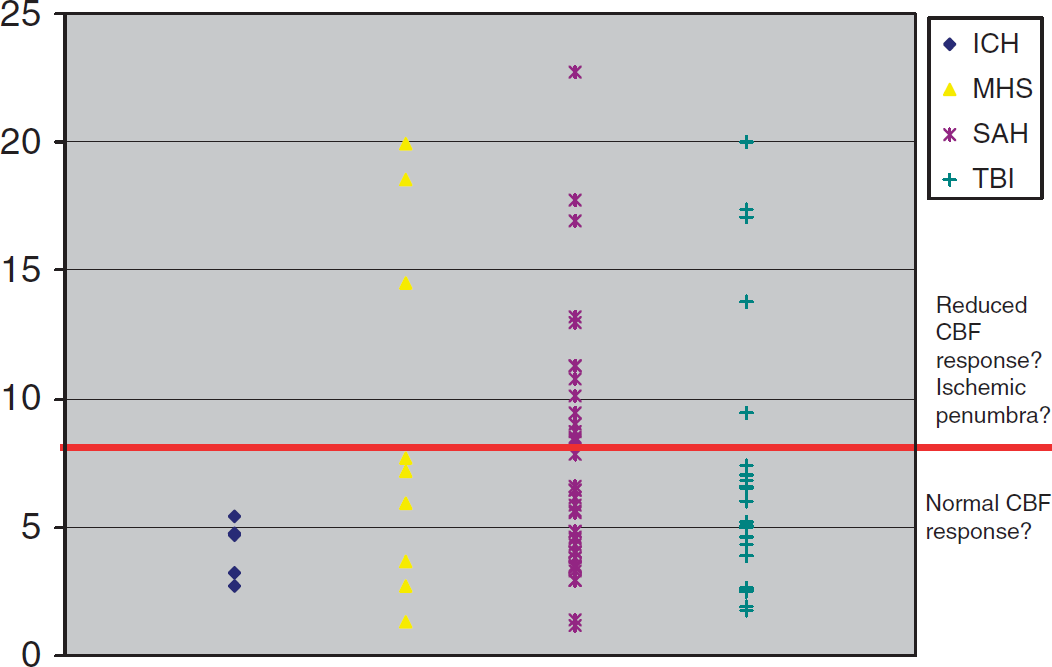
Duration (in minutes) of recovery time of cortical electrical silence induced by depolarization in different diseases. The duration of the depression of cortical activity reflects the ability of the tissue to recover after cortical spreading depression (CSD) and thereby the perfusion and metabolic state at the point of measurement. The clinical deterioration of patients we have monitored is often accompanied by clusters of CSD, which show a gradual increase of recovery time. Therefore, assessment of recovery time may be an early warning sign that secondary damage is under development. So when is duration prolonged? In this figure, the mean duration of depression for all CSD episodes from 87 patients (intracerebral hemorrhage (ICH): 5, malignant hemispheric stroke (MHS): 13, subarachnoid hemorrhage (SAH): 43, traumatic brain injury (TBI): 26) is displayed in minutes. The figure suggests a bimodal distribution with a cluster below ∼8 minutes, probably reflecting a fast recovery and tolerable perfusion conditions, whereas the more widespread group above ∼8 minutes may reflect a reduced blood flow response indicating penumbral conditions. Interestingly, the small group of patients with ICH all showed a fast recovery, possibly because a widespread penumbra seems to be a rare feature in this condition.
Aggravation of brain injury after ICH has been associated with rebleeding, mass effect (Xi et al, 2006), and with brain edema, which develops in a stepwise manner: a transient initial peri-hematoma edema is associated with transient reduction in blood flow in the same area (Herweh et al, 2009), and at 24 to 48 hours thrombin-dependent edema and disruption of the blood–brain barrier develop (Thiex and Tsirka, 2007). Finally, lysis of the red blood cells leads to deposits of degradation products in the parenchyma and precipitates an inflammatory reaction (Xi et al, 2006; Thiex and Tsirka, 2007; Wang and Dore, 2007). As CSD is associated with edema formation and neuroinflammation, one might speculate whether the recruitment of cortical areas into the lesion in ICH patients is in part due to repeated CSD episodes. Interestingly, the addition of MK801 to rtPA injected for local fibrinolytic therapy into intracerebral hematomas in swine prevented the expansion of the peri-hematoma edema within 10 days of observation (Thiex et al, 2007). It is not known if this effect was caused by MK801 preventing CSD (Lauritzen and Hansen, 1992). Intracerebral hemorrhage induced in swine provoked multifocal, frequent CSDs in all animals (Mun-Bryce et al, 2001). The observation of multiple CSDs in human ICH should inspire more experimental work to investigate whether the occurrence of CSDs is associated with changes in lesion volume.
Traumatic Brain Injury
Patients with TBI may require neurosurgery either for evacuation of intraparenchymal or acute subdural hematoma, or for decompression of a swollen hemisphere. The spontaneous occurrence of depolarizations in some 50% to 60% of such cases has now been shown by independent investigators using different monitoring techniques (Mayevsky et al, 1996; Strong et al, 2002) and also at seven different centers within the COSBID consortium (Fabricius et al, 2006; Hartings et al, 2009). Patterns of CSD vary widely, from sparse single events in some patients to temporal clusters of events repeating every 20 to 30 minutes for ≥12 hours in others, and sometimes recurring after 5 to 7 days (Figure 6). The prevalence and heterogeneity of this clinically ‘silent’ activity raise fresh questions for TBI research and potentially shed light on current and past enigmas.
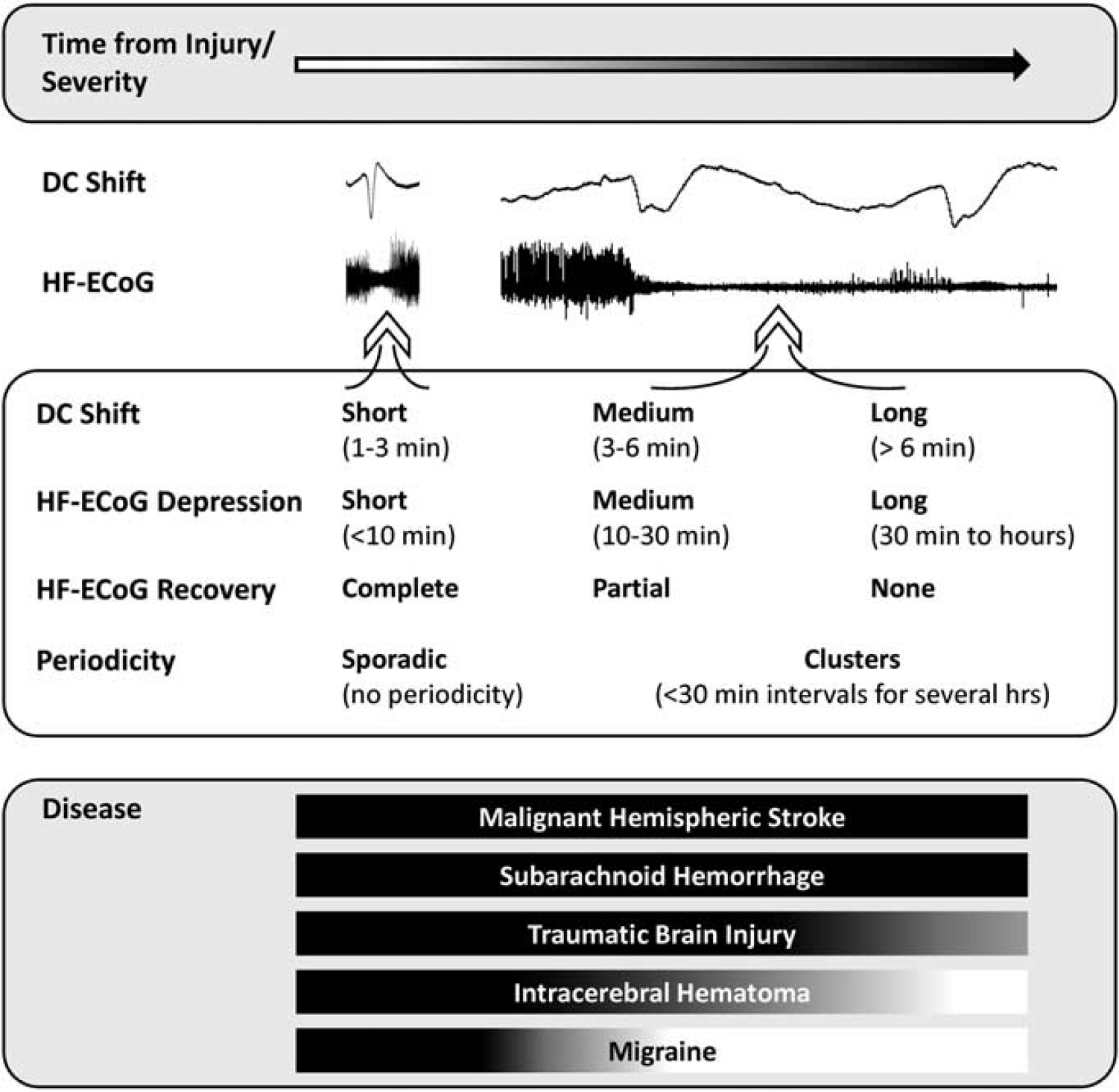
Graded nature of cortical spreading depolarizations and their occurrence in disease states. Various electrocorticogram (ECoG) measures reflect the putative pathologic severity of cortical spreading depressions (CSDs) observed in the human brain, including (1) the duration of the DC shift, (2) the duration of depression of high-frequency spontaneous activity (HF-ECoG), (3) the extent of recovery of HF-ECoG after depression, and (4) the periodicity of recurrent CSD events. At one end of the spectrum, putatively benign CSDs appear sporadically with short-lasting DC shifts and depression periods and complete recovery of HF-ECoG. At the other end, depolarization durations (DC shift) are prolonged, with persistent HF-ECoG depression and no or incomplete recovery, and events recur repetitively in clusters. This latter type shares features in common with depolarizations observed in various experimental models where (1) perfusion deficits delay membrane repolarization and recovery of HF-ECoG and (2) CSD is proven to cause pannecrosis of affected cortex. The bottom panel shows the prevalence of each type of CSD in different diseases, based on current evidence, with the full spectrum observed in malignant hemispheric stroke (acute progressive infarction) but only short-lasting events believed to occur in migraine (no acute cortical damage). In some patients, the severity of CSD may increase progressively after injury through hours or days of recurring events.
The nearly complete breakdown of ion homeostasis caused by CSD, with consequent intracellular Ca2+ surge and release of excitatory amino acids, suggests that CSD may contribute to excitotoxicity, a primary mechanism of neuronal damage after TBI (Obrenovitch and Urenjak, 1997). CSD may contribute to the nonischemic metabolic crisis in TBI elucidated by Vespa et al: using cerebral microdialysis, they found that persistently low glucose (<0.2 mmol/L) was common in severe TBI, with unknown etiology in 72%, and was correlated with poor outcome. Lactate–pyruvate ratio was also raised without other evidence of ischemia (Vespa et al, 2003). CSD may underlie such microdialysis markers of disturbed metabolism (Parkin et al, 2005; Hartings et al, 2008), as PIDs cause cumulative depletion of glucose and accumulation of lactate (Hopwood et al, 2005). CSD is also associated with increased glucose utilization in experimental TBI (Sunami et al, 1989) and, when recurrent, progressively depletes glucose even in the healthy brain (Hashemi et al, 2009). Stepwise depletion of brain glucose in association with individual depolarizations has now been shown in patients with acute ischemic or traumatic brain injury (Feuerstein et al, 2010). Finally, data from Hernandez et al (2006) can be interpreted as indicating that established risk factors (at admission) for adverse outcome from TBI account for only 30% of outcome variance: could CSD contribute to some of the other 70%? Interim analysis of COSBID data suggest that the occurrence of CSD is associated with worse 6-month outcome and is a stronger predictor than the established covariates such as age and admission GCS—at least in patients requiring craniotomy. In these patients, the presence of CSD was not associated with the occurrence of parenchymal lesions, but rather varied with the degree of traumatic SAH.
Experimental data on the effects of CSD in TBI are equivocal, and should be interpreted with the caution that animal models seldom replicate the complexity of primary and secondary injury of severe TBI in humans. In most rodent models of cerebral contusion, only a single CSD is triggered by the initial mechanical insult, which is followed by few or no subsequent CSD waves in the ensuing hours (Ozawa et al, 1991; Nilsson et al, 1993). To determine whether CSD contributes to the maturation of cortical contusions in normally perfused and oxygenated animals, Baumgarten et al (2008) applied an approach used earlier by others in experimental stroke: they elicited additional CSDs, remote and independent from the primary injury, which then invaded the evolving contusion. There was no effect of these additional CSDs on injury volume at 24 hours, although secondary insults were specifically (and in one sense, commendably) minimized in this study. Different results were obtained in an in vitro cortical slice model of trauma, where a single wave of CSD caused by traumatic impact increased the volume of cortical damage compared with conditions under which the same cortical impact did not induce CSD (Church and Andrew, 2005).
In contrast to these cerebral contusion models, the pathophysiology of human TBI is highly complex and heterogeneous. Primary injury may consist of any combination of parenchymal contusion, ICH, SAH, extraparenchymal hematoma, and diffuse axonal injury (Saatman et al, 2008), and is often critically compounded by secondary insults including hypotension, hypoxia, fever, and brain edema leading to elevated intracranial pressure and secondary global ischemia. These factors raise the likelihood of metabolic insufficiencies leading to membrane failure and elevated [K+]o and may explain the higher incidence of CSD in human TBI. Indeed, Trabold et al (2006) tested the effect of hypotension experimentally by compounding cortical cold lesions with transient hypotension: this triggered a second CSD and was associated with a significant increase in lesion volume compared with controls. Rogatsky et al (2003) similarly simulated severe TBI with secondary deficits. Using fluid percussion injury in rats, different TBI severities resulted in substantial ICP elevations that significantly correlated with the number of CSDs. Interestingly, the greatest injury severity was associated with 44% mortality, and 50% of these rats exhibited continuous repetitive cycles of CSD. The authors concluded that ‘the rising number of CSD cycles under condition of an ICP level ≥20 mm Hg may show, with high probability, the unfavorable development of TBI, caused by growing secondary hypoxic insult.’
Thus, these studies are consistent with secondary insults as risk factors for developing CSD and further raise the possibility that CSD is a focal, cortical mechanism by which global or systemic insults adversely affect outcome. Clinical observations are congruent with this possibility, as CSD was found to be significantly associated with hyperthermia, hypotension, and elevated ICP (Hartings et al, 2009). Figure 7, for instance, shows the clinical translation of the results of Rogatsky et al (2003): occurrence of repetitive cycles of CSD in a patient with ICP sustained >20 mm Hg. Further observational studies are needed to determine whether other factors, such as medications and variations in plasma osmolality, electrolytes, and glucose, are also associated with changes in the frequency, properties, and impact of CSD after TBI (Hopwood et al, 2005; Sakowitz et al, 2009).
Repetitive cycles of cortical spreading depression (CSD) during sustained elevated ICP in an elderly patient with delayed deterioration associated with a severe right temporo-parietal traumatic contusion. Top and middle traces show the low-frequency (LF-electrocorticogram (ECoG); low-pass filtered at 0.05 Hz) and high-frequency (HF-ECoG; high-pass filtered at 0.5 Hz) components of recordings from three bipolar channels of a linear subdural electrode strip. Arrows highlight individual CSDs, evidenced as propagating, large amplitude slow potential changes causing the suppression of HF-ECoG. Fourteen CSDs occurred in the 8-hour epoch shown; overall, a total of 105 CSDs were recorded in this patient during 70 hours of ECoG monitoring. Throughout this period, ICP remained elevated >20 mm Hg and often persisted at 30 to 40 mm Hg. The patient died 5 days after injury on withdrawal of ventilator support, in compliance with her living will.
Discussion
Identification of Cortical Spreading Depression in Human Brain Cerebral Cortex: Why Only Now?
This review focuses on the possible implications for clinical management of the demonstration and characterization of CSD in the injured human brain, in the light of relevant aspects of the experimental pathophysiology. For many years, CSD was used experimentally to study the functional organization of the brain and behavior (Bures et al, 1984), cerebral ion homeostasis (Hansen, 1985), and blood flow and energy metabolism (Mayevsky and Chance, 1974). In addition, leading clinicians regarded CSD as irrelevant for human brain disorders including migraine based on the lack of evidence of CSD in the ECoG obtained in patients during epilepsy surgery (Gloor, 1986). Noninvasive registration of CSD for example by conventional EEG technique is difficult due to the brief duration of the depolarization wave and EEG silence that moves slowly across the cortical surface and involves a cortical volume that is too small to be detected (Lauritzen, 2001). In the clinical centers where MEG equipment has been available, the results that have emerged have been ambiguous (Bowyer et al, 2001). In comparison, examination of migraine patients using a variety of brain imaging techniques has shown changes in perfusion and has been more successful in identifying CSD, and ongoing research is focused on the possibility of detecting CSD with related techniques in patients with acutely injured brain cortex as described in this review.
Clearly, the invasive nature of the methodology used in the patient studies reported here represents a limitation in broadening the indication for CSD studies in the intensive care unit (Strong et al, 2002). Nonetheless, in these patients there is a need to monitor not only a single CSD as an indication of brain injury, but to monitor cortical function and the evolution of CSD over prolonged time periods. Current ECoG methods allow this, providing information on variations in occurrence, duration, and recovery rate of the CSD that offers a sensitive, graded assessment of the functional state and viability of the cerebral cortex from penumbra to normal cortical tissue (Figures 5 and 6). The first achievement of the COSBID study has been to accomplish this in a large patient sample, providing the opportunity to assess the impact of CSD and its variants on secondary brain damage and in consequence patient outcome (Figure 6). Less invasive ECoG approaches may allow the translation and application of this experience to a broader range of patients in the future (Waziri et al, 2009).
How Will Patients Benefit from This?
It is not unreasonable to suggest that the emerging evidence for occurrence of CSDs and especially PIDs in patients with acute brain injury is leading to a paradigm shift in our understanding of the pathophysiology of acute brain injury in humans. The obvious benefit is to allow us to begin to define appropriate treatment for patients with tissue-at-risk in whom the occurrence of the depolarization waves might pose an important threat to the tissue. Clearly, there is still much to be clarified until CSD has found its place among the complicated series of events, which lead from head trauma, intracranial hemorrhage, and focal ischemia to established brain damage. Importantly, association does not prove causality, and this will only be confirmed with a successful interventional study targeted at CSDs or at vasoreactivity to them. Most immediately, we need to examine (1) the extent to which CSDs that are apparently less harmful (hyperemic and quick repolarization) are associated with bad or good outcome and (2) the hypothesis that PIDs predict adverse outcome independent of classical risk factors. This latter concept has the stronger experimental support: currently accumulating clinical data point in this direction but will require further analysis and peer review. In the case of TBI, if the hypothesis is sustained, the incidence of CSDs of different patterns becomes a potential early surrogate endpoint for a prospective clinical trial in patients shown to be experiencing CSDs, although demonstration of improved outcome would still be essential. In malignant hemispheric stroke, the issues are different in that PIDs are essentially always present and an inherent component of the disease process: for reasons given below, a clinical trial targeting vasoreactivity might offer a reasonable prospect of success. However, when considering potential treatments designed to mitigate depolarizations, we also need to take some account of the body of experimental literature that describes potential beneficial effects of CSD. For example, preconditioning the rat brain with CSD for 48 hours induces ischemic tolerance after an interval of days, consequently reducing the infarcted lesion size in the acute phase after focal cerebral ischemia (Yanamoto et al, 2004). In addition, induction of CSD for 48 hours increases the density of mitotic cells, divided cells, and new neurons in the subventricular zone in the following days by an NMDA receptor-dependent mechanism (Yanamoto et al, 2005). Further studies will need to address whether CSD episodes in the human cortex may be neuroprotective.
In the meantime, clinical management in the neurocritical care unit can focus on control of factors that are known to increase incidence and duration of CSDs: systemic hypotension, pyrexia, hypoxia, and unduly low plasma glucose. A definition of what is euglycemia for a patient with brain injury remains to be agreed, but values in the range 8 to 9.5 mmol/L (144 to 171 mg/dL) are associated in experimental stroke with a low incidence of PIDs (Hopwood et al, 2005). This range lies below that which is more clearly associated with adverse clinical outcome from ischemic stroke (Gray et al, 2007).
Future Directions of Research
Identifying the signaling pathways regulating the human brain's susceptibility to develop CSD may provide opportunities for manipulating those pathways therapeutically. Antiepileptic drugs may prevent CSD and migraine (Marrannes et al, 1988; Martins-Ferreira et al, 2000; Ayata, 2009), but in patients with acutely injured brain cortex, the effects of antiepileptic drugs are unpredictable and patients may develop both seizures and CSD in the presence of high doses of antiepileptic drugs that normally decrease neuronal excitability (Fabricius et al, 2008). Further studies are needed to examine whether a change in the choice of antiepileptic drug (phenytoin is most commonly used) can be proven effective in decreasing cortical susceptibility to CSD initiation. Once the CSD is triggered, the propagation of the depolarization wave depends on preserved function of NMDA receptors in normal brain tissue, which is accompanied by huge transmembrane Na+ and Ca2+ currents (Marrannes et al, 1988; Lauritzen and Hansen, 1992). Accordingly, NMDA receptor blockade abolishes CSD very effectively in normal brain tissue. In injured brain, nerve cells and blood cells rupture and release K+ (and other substances), which rises in the extracellular fluid. This is accompanied by a relative insensitivity of injured cortical tissue to NMDA receptor blockade (Petzold et al, 2005), which potentially may limit the extent to which NMDA receptor blockade can be used therapeutically in patients with CSD. In patients, the preliminary data are ambiguous. On the one hand, the CSD episodes were discontinued in two patients with multiple CSDs that were treated with ketamine, a weak NMDA receptor antagonist (Sakowitz et al, 2009). On the other hand, a cluster of CSD occurred in another patient despite the presence of ketamine (Dreier et al, 2009). The effect of NMDA receptor antagonists deserves further study.
In addition, the existence of multiple pathways regulating CBF during and after CSD may allow therapeutic modification of the hyperemia during the depolarization wave itself, and of the persistent oligemia or ischemia after CSD episodes in different brain areas and conditions, including the ischemic penumbra. The decrease in CBF that accompanies CSD may be produced by excessive generation of vasoconstrictors released from neurons (e.g., NPY) as well as astrocytes (e.g., thromboxane), or decreased release of vasodilators (e.g., NO), which could be ameliorated by use of appropriate drugs. It has already been shown that the impact of CSD on blood vessels in terms of vasoconstriction and reduced reactivity can be ameliorated by high doses of L-arginine, the substrate for nitric oxide synthase (Fabricius et al, 1995; Scheckenbach et al, 2006). Drugs that enhance the production or effect of NO may prove useful in treating patients with CSD. Specifically, the so-called spreading ischemia coupled to the cortical depolarization phase instead of spreading hyperemia may be an interesting target for treatment. Thus, NO donors or nimodipine, an L-type calcium antagonist, caused spreading ischemia to revert to almost normal spreading hyperemia with accelerated recovery of the tissue from the depolarization in rats (Dreier et al, 1998, 2001). A problem of such approaches is that systemic treatment with those vasodilators lowers systemic blood pressure, which then seems to promote the inverse hemodynamic response (Dreier et al, 2002; Sukhotinsky et al, 2010). Thus, it will be important to learn more about the process of the inverse hemodynamic response to develop more selective strategies. There is experimental evidence that potassium, released by spreading depolarization, is involved as a vasoconstrictor inducing spreading ischemia (Windmuller et al, 2005), and the vascular cells termed pericytes may contribute to capillary constrictions in normal brain tissue (Peppiatt et al, 2006) as well as in ischemic tissue (Yemisci et al, 2009). Pericyte-mediated constriction may constitute the mechanism for the ‘no-reflow’ phenomenon after cerebral ischemia, and the decrease of CBF after CSD. The use of new detection techniques at the bedside, either invasively by using laser-Doppler probes embedded in the electrode strip (Dreier et al, 2009), or noninvasively using transcranial Doppler or near-infrared-spectroscopy for rapid assessment of CBF (Cheng et al, 2002; Zweifel et al, 2010) may allow for real-time modification of therapies based on immediate observation of beneficial and adverse drug effects. The fact that CSD can be monitored and inhibited by a variety of therapies provides hope for future clinical trial design: if CSD inhibition is a primary mechanism of protection, monitoring of ECoG or of a (validated) surrogate could be used to identify for inclusion only those patients who might benefit.
Footnotes
Acknowledgements
The authors apologize to those whose work we have not been able to cite for reasons of space. AJS thanks the Wellcome Trust and HeadFirst for their support during the period covered by this review.
The authors declare no conflict of interest.