
Abstract
Identifying drug transporters and their in vivo significance will help to explain why some central nervous system (CNS) drugs cross the blood-brain barrier (BBB) and reach the brain parenchyma. We characterized the transport of the drug Clonidine at the luminal BBB by in situ mouse brain perfusion. Clonidine influx was saturable, followed by Michaelis–Menten kinetics (Km = 0.62 mmol/L, Vmax = 1.76 nmol/sec per g at pH 7.40), and was insensitive to both sodium and trans-membrane potential. In vivo manipulation of intracellular and/or extracellular pH and Trans-stimulation showed that Clonidine was transported by an H+-coupled antiporter regulated by both proton and Clonidine gradients, and that diphenhydramine was also a substrate. Organic cation transporters (Oct1–3), P-gp, and Bcrp did not alter Clonidine transport at the BBB in knockout mice. Secondary or tertiary amine CNS compounds such as oxycodone, morphine, diacetylmorphine, methylenedioxyamphetamine (MDMA), cocaine, and nicotine inhibited Clonidine transport. However, cationic compounds that interact with choline, Mate, Octn, and Pmat transporters did not. This suggests that Clonidine is transported at the luminal mouse BBB by a new H+-coupled reversible antiporter.
Keywords
Introduction
The blood-brain barrier (BBB) formed by the endothelial cells of the brain capillaries isolates the brain parenchyma from the systemic circulation and regulates the composition of the brain extracellular fluid. Tight junctions between adjacent endothelial cells of the BBB restrict the nonspecific paracellular diffusion of compounds. Additional BBB features allow the control of the transcellular diffusion of compounds like drug transporters involved in influx and/or efflux (Ohtsuki and Terasaki, 2007). It is essential, therefore, to identify the transporters and properties that govern their functions at the BBB and to understand how some drugs can reach the brain parenchyma and why others do not. Studies on the transport properties at the BBB should indicate why many cationic compounds that are active in the central nervous system (CNS) reach the brain parenchyma.
Systems that transport organic cations, such as organic cation transporters (OCT1-3) (SLC22A1-3), OCTN1-3 (SLC22A4-5, 21), PMAT (SLC29A4), and MATE1-2 (SLC47A1-2) and that interact with tetraethylammonium (TEA), have been identified in various cells in vitro. However, studies on the transport of secondary or tertiary amine drugs, such as methylenedioxymethamphetamine (MDMA, ecstasy), oxycodone, and Clonidine, led to the discovery of an H+ antiporter that is not inhibited by TEA. Its functional properties differ from those of any of the already identified organic cation transporters (Fischer et al, 2006; Okura et al, 2008; Kuwayama et al, 2008). However, as the molecular nature of the transporter involved has not been identified, it is commonly known as the ‘tertiary or secondary amine/H+’ antiporter.
Many in vitro studies on Clonidine, an organic cation that acts on the CNS, have shown that neurones (Fischer et al, 2007), porcine brain endothelial cells (Huwyler et al, 1997), keratinocytes, placental cells, and enterocytes (Fischer et al, 2006; Grafe et al, 2004; Müller et al, 2004) produce a clonidine/H+ antiporter. Thus, Clonidine is a useful probe for characterizing this amines/H+ antiporter in vivo.
This study was carried out to characterize Clonidine transport at the mouse luminal BBB and to obtain more information on drug interactions and inhibition of this transport system. We measured the transport of [3H]clonidine at the luminal membrane of the endothelial cells that form the mouse BBB by in situ brain perfusion. This method can accurately determine BBB permeability and it is possible to control the composition of the vascular perfusate for a short time (Cattelotte et al, 2008; Smith, 1996; Takasato et al, 1984). We used exposure to various chemicals and ions, together with studies on transporter knockout (KO) mice, to identify the properties of Clonidine transport at the mouse BBB. Our results suggest that Clonidine transport across the luminal BBB involves a distinct amines/H+ antiporter that interacts with organic compounds that have secondary or tertiary amine moieties, such as diphenhydramine (DPH), and certain addictive drugs. The features of this amines/H+ reversible antiporter at the mouse BBB highlight its importance for facilitating the entry of its substrates into the brain. It is a new permeability factor that can be used to identify drugs that might act on the CNS.
Materials and methods
Animals
Adult 30–40 g, 7- to 11-week-old male Swiss and FVB mice were obtained from Janvier (Genest, France). The KO mouse strains, Oct1, 2(−/-), Oct3(−/-), and mdr1a, mdr1b, bcrp(−/-), were bred in-house from progenitors obtained from the laboratory of Dr Alfred H Schinkel (The Netherlands Cancer Institute, The Netherlands). These KO strains were derived from FVB mice. The mice were housed in a controlled environment (22°C ± 3°C; 55% ± 10% relative humidity) and a 12-h dark-light cycle, with access to food and tap water ad libitum. All experimental procedures complied with ethical rules of the French Ministry of Agriculture for experimentation with laboratory animals (law 87–848).
Drugs and Chemicals
[3H]Clonidine (69 to 72 Ci/mmol) and [14C]sucrose (565 mCi/mmol) were purchased from Perkin-Elmer Life Sciences (Paris, France). Acetazolamide (ACTZ), agmatine, ammonium chloride, carnitine, Cimetidine, cocaine, codeine, para-chloroamphetamine, corticoster-one, desipramine, dimethylamiloride (DMA), dopamine, histamine, methadone, nalbuphine, nicotine, phloretin, procainamide, quinidine, serotonin, TEA, verapamil, xylazine were purchased from Sigma (St Quentin, France). Tramadol was a gift from Ethypharm (Châteauneuf-Thymerais, France). Choline chloride, Clonidine, guanidine, HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), lithium chloride, and valinomycin were Fluka products (St Quentin, France). Dimethylsulfoxide (DMSO) and DPH were obtained from Inresa (Strasbourg, France). Milnacipran was a gift from Pierre Fabre (Bordeaux, France) and GF120918 was a gift from GlaxoSmithKline (Research Triangle Park, NC, USA). Morphine, diacetylmorphine, and oxycodone were purchased from Francopia (Gentilly, France). MDMA was synthesized (purity >99%) in the laboratory of Professor Galons (Faculté de Pharmacie, Université Paris Descartes, Paris, France).
In Situ Brain Perfusion
Surgical procedure and perfusion: The transport of [3H]clonidine into the mouse brain was measured by in situ brain perfusion (Cattelotte et al, 2008). Mice were usually anesthetized by injection with ketamine–xylazine (140–8 mg/kg, intraperitoneally), but they were anesthetized with halothane (Belamont, Neuilly-sur-Scine, France) in some experiments. The right external carotid was ligated rostral to the occipital artery at the level of the bifurcation of the common carotid. The right common carotid was ligated and a catheter (polyethylene tubing 0.30 mm, i.d. × 0.70 mm) was inserted above the ligation. The syringe containing the perfusion liquid was placed in an infusion pump and connected to the catheter. Before perfusion, the thorax was opened and the heart was cut. Perfusion was started immediately at a flow rate of 2.5 mL/min. Each mouse was perfused with [3H]clonidine (0.3 μCi/mL) and [14C]sucrose (0.1 μCi/mL) as a vascular marker. Perfusion was terminated after 120 secs by decapitation, unless otherwise specified. The brain was removed from the skull and dissected out on a freezer pack. The right cerebral hemisphere and two aliquots of perfusion fluid taken from the end of the catheter after it had been removed from the artery lumen were placed in tared vials and weighed. Samples were digested in 2 mL of Solvable (Perkin-Elmer Life Sciences) at 50°C, cooled to room temperature and mixed with 9 mL of Ultima Gold XR (Perkin-Elmer Life Sciences). Dual label counting was carried out in a Packard Tri-Carb 1900TR (Perkin-Elmer Life Sciences).
Perfusion fluid: Perfusion was attained using Krebs carbonate-buffered physiological saline (mmol/L): 128.0 NaCl, 24.0 NaHCO3, 4.2 KCl, 2.4 NaH2PO4, 1.5 CaCl2, 0.9 MgSO4, and 9.0
The ionic composition of the Krebs carbonate perfusate could be changed to remove all carbonate, Cl−, Na+, or K+. In some experiments, carbonate in the Krebs perfusion fluid was replaced by sodium chloride. In chloride-free perfusion fluid, chloride was replaced by gluconate and nitrate. Sodium was replaced by potassium chloride in the ‘K+’ perfusion fluid (K+ 154 mmol/L), by lithium chloride in the ‘Li+’ perfusion fluid (Li+ 128 mmol/L, K+ 26 mmol/L), by mannitol in the ‘mannitol’ perfusion fluid (mannitol 256 mmol/L, K+ 26 mmol/L, Cl− 3 mmol/L), and by choline chloride in the ‘choline’ perfusion fluid (choline 128 mmol/L, K+ 26 mmol/L). In the K+-free buffer, K+ was replaced with sodium salts (Na+ 132.2 mmol/L). The perfusion fluid that controlled the extracellular vascular pH (pHe) was always adjusted using a digital pH meter (± 0.05 pH units) before adding [3H]clonidine and [14C]sucrose. The perfusate was then placed in the syringe, which was connected to the mouse catheter for immediate perfusion.
[ 3 H]Clonidine transport: The initial rate of [3H]clonidine (9 nmol/L) transport was measured under trans-influx zero conditions by perfusion for 120 secs unless otherwise specified. Cis-inhibition was used in some experiments, by perfusing [3H] Clonidine together with an unlabeled compound. All the solutions with unlabeled organic compounds for cis-inhibition were made on the day of the experiment. Valinomycin, quinidine, and phloretin were dissolved in DMSO. The final DMSO concentration in the perfusate never exceeded 0.5% (v/v) and did not affect the integrity of the BBB and the [3H] Clonidine transport (data not shown).
Trans-stimulation was performed using two syringes, each in its own infusion pump, and connected to the carotid catheter via a four-way valve. This enabled us to switch from one to the other syringe for the appropriate time. The brain was first ‘loaded’ by perfusion with [3H]clonidine (0.3 μCi/mL; ~9 nmol/L) in Krebs carbonate buffer (pHe 7.40) for 120 secs. The second perfusion was with or without DPH (10 mmol/L) (pHe 6.40) for 60 secs.
Apparent brain distributional volume and initial brain transport parameters: All calculations were carried out as described earlier (Smith, 1996; Takasato et al, 1984). The brain vascular volume was estimated using the [14C]sucrose distribution volume (Vv; μL/g), as sucrose diffuses across the BBB very slowly:
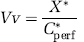
where X* (d.p.m./g) is the [14C]sucrose amount measured in the right brain hemisphere and Cperf* (d.p.m./μL) the [14C]sucrose concentration in the perfusion fluid. If the Vv was above the normal value (>20 μL/g; Cattelotte et al, 2008), the mouse was discarded from the study.
The apparent brain distribution volume (Vbrain, μL/g) was calculated from [3H]Clonidine in the right brain hemisphere parenchyma as follows:
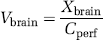
where Xbrain (d.p.m./g) is the calculated amount of [3H] Clonidine in the right cerebral parenchyma and Cperf (d.p.m./μL) the [3H]clonidine tracer concentration in the perfusion fluid. Brain tissue total [3H]clonidine radioactivity was corrected for vascular volume using vascular space subtraction as follows:
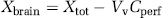
where Xtot (d.p.m./g) is the total quantity of [3H]clonidine measured in the brain tissue (parenchyma and cerebrovas-culature capillary bed). The amount of [3H]clonidine in the vascular space (VvCperf) was calculated and subtracted from the total [3H]clonidine (Xtot) (Equation (3)). This yielded the amount of [3H]clonidine outside the vascular brain space (Xbrain) and permitted the calculation of Vbrain according to Equation (2) and the initial brain transport expressed as Kin (μL/sec per g) calculated from
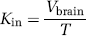
where T is the [3H]clonidine perfusion time (secs).
The brain flux (Jin; nmol/sec per g) is obtained by
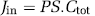
where Ctot is the total Clonidine concentration in the perfusate; PS (permeability–surface area product; μL/sec per g) was calculated from Kin using the Crone–Renkin equation
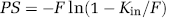
where F is vascular flow (μL/sec per g). Equation (5) can be reduced for a flow-independent solute (Kin/F <0.3) to obtain Equation (6).
The Jin (nmol/sec per g) was calculated according to this relationship for a flow-independent compound as
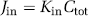
and
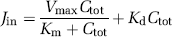
where Vmax (nmol/sec per g) is the maximal velocity of transport, Km (mmol/L) the apparent Michaelis–Menten transport parameter, and Kd (μL/sec per g) is an unsaturable component. The data were fitted to Equation (6) using nonlinear regression analysis.
Data Analysis
Transport parameters (Kin, Vblain) are given as means ± s.d. from 4 to 7 mice, unless specified otherwise. Student's unpaired t-test was used to identify significant differences between groups. The tests were all two tailed and statistical significance was set at P < 0.05. Transport parameters (Km, Vmax, Kd) were estimated by fitting Clonidine brain flux versus Clonidine concentration data using Equation (6) and nonlinear regression using the WinNonlin software (Pharsight, Mountain View, CA, USA). The errors associated with these parameters are asymptotic standard errors returned by the nonlinear regression routine and are a measure of the best fit value.
Results
Physical Integrity of the Blood-Brain Barrier
An intact mouse BBB is a prerequisite for measuring [3H]clonidine brain transport parameters, and this must be assessed in every mouse individually. Although the [14C]sucrose Vv is not shown for each mouse, this was always measured and was below 20 μL/g, in agreement with the normal distribution Vv for [14C]sucrose (Cattelotte et al, 2008). The [14C]sucrose volumes (data not shown), and hence the integrity of the BBB, were not altered by the modified perfusion fluid or by various organic compounds added to the perfusion fluid to study [3H]clonidine transport.
Time Course of [3H]Clonidine Transport
The first set of perfusion experiments explored the time course of [3H]clonidine transport (Figure 1). The [3H]clonidine (~9 nmol/L) Vbrain increased linearly with the perfusion time, suggesting that the transport rate remained unchanged over this time. The initial linear rate is said to be ‘unidirectional,’ so that transport parameters measured for this initial linear phase are qualitatively and quantitatively similar over this time. In this phase, the luminal membrane of the BBB represents the single kinetics interface that affects the extravascular [3H]clonidine ‘brain’ distribution. An appropriate perfusion time should ensure that at least 40% of the tracer remains outside the vascular space (Smith, 1996). This requirement was met after perfusion for 2 secs, but we generally used 120 secs in single-time experiments to ensure the accumulation of sufficient tracer and a 3H/14C ratio above 3. This was also sufficient time for biochemical changes (e.g., pH) to take place in the endothelial cells. The hypothetical effect of the cationic anesthetic mixture (ketamine and xylazine) on the evaluation of [3H]clonidine brain transport (Figure 1; Vbrain 732 ± 51 μL/g at 120 secs; Kin ~6.1 μL/g per sec) was assessed using mice anesthetized with halothane (Vbrain 745 ± 60, μL/g; 120 secs; n = 5). The [3H]clonidine Vbrain obtained using the two systems of anesthesia was not different; hence, ketamine/xylazine anesthesia appeared to have no effect on [3H]clonidine brain transport.
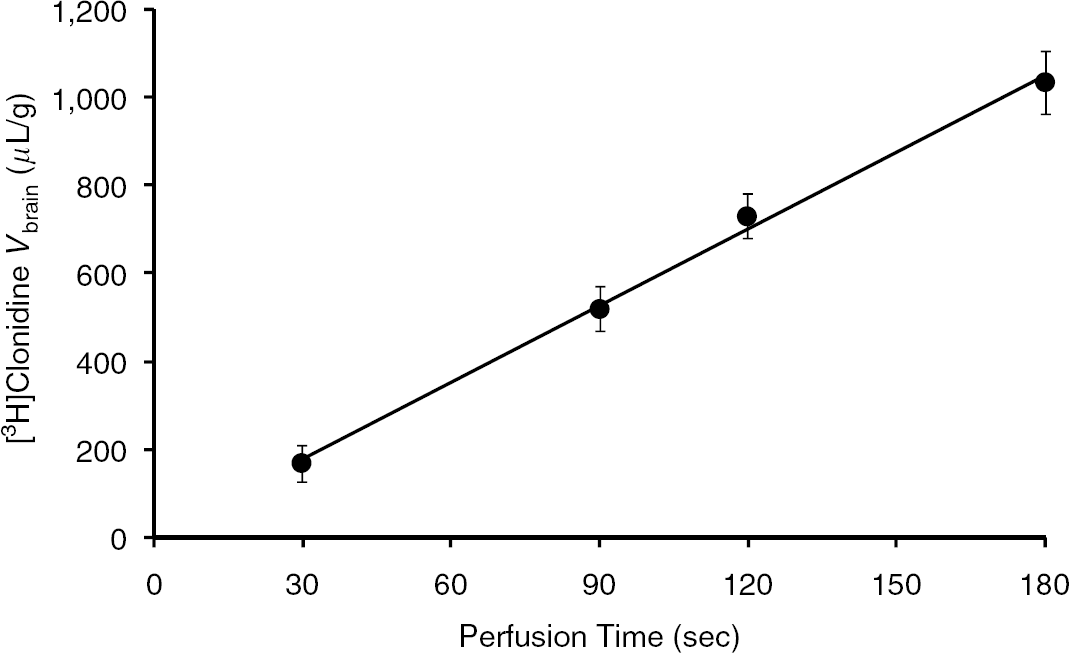
Time course of [3H]clonidine uptake by the right brain hemisphere (solid line) of Swiss mice, expressed as apparent distribution volume (Vbrain, μL/g) determined by in situ brain perfusion for 30, 90, 120, and 180 secs. Regressions analysis of the individual data yields r2 = 0.97. Data are means ± s.d. of 4 to 5 animals.
Clonidine Brain Flux at Multiple Concentrations
The brain influx (Jin) of [3H]clonidine (~9 nmol/L) was measured after a 120-sec perfusion with Clonidine concentrations of 9 nmol/L to 10 mmol/L, obtained by adding unradiolabeled Clonidine to the Krebs carbonate buffer (pHe 7.40). The plot of the Clonidine flux against the total Clonidine concentration yielded an apparent Km of 0.62 ± 0.02 mmol/L and a Vmax of 1.76 ± 0.03 nmol/sec per g (Figure 2). The best nonlinear regression plot was adequate with a Hill coefficient of 1. The Clonidine unsaturated apparent component (Kd; pHe 7.40) certainly reflected passive diffusion. Nonlinear regression analysis gave a Kd (0.008 μL/sec per g) that represents ~0.1% of the total brain Clonidine flux. An Eadie–Hofstee transformation plot (Jin versus Jin/Ctot) gave a straight line (r2 = 0.94; figure not shown), suggesting that one saturable system is involved in Clonidine transport at the luminal side of the BBB.
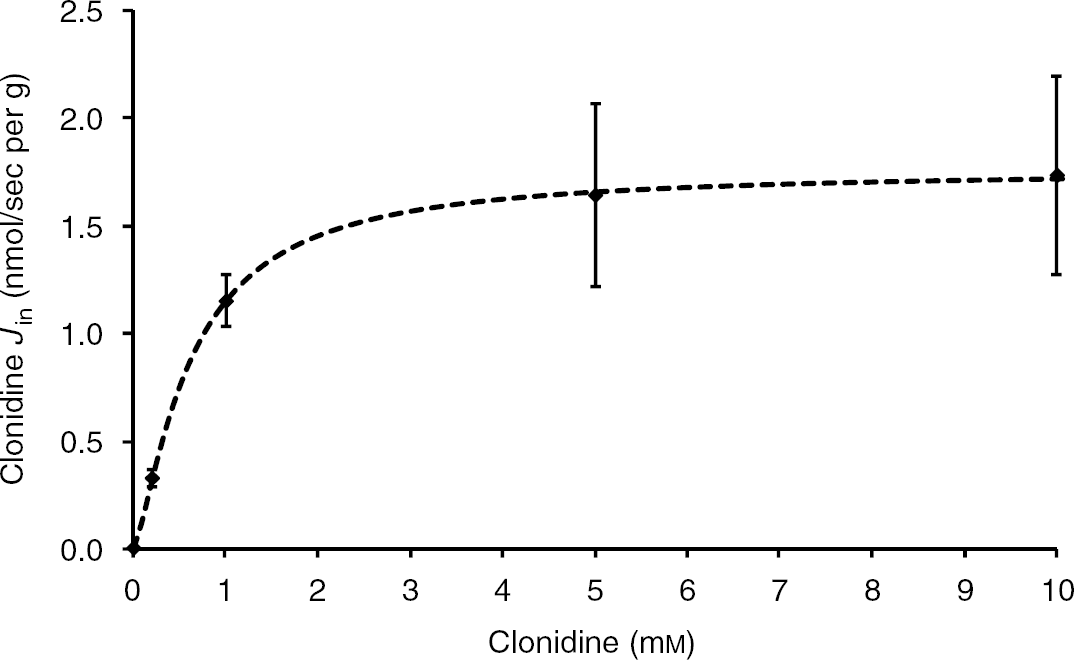
Clonidine brain flux (Jin; nmol/sec per g) measured in the right brain hemisphere of Swiss mice and fitted to the Clonidine concentration in the Krebs carbonate perfusion fluid at pHe 7.40. Data are means ± s.d. of 5 to 7 animals. The dotted line is the data fitted to the Michaelis–Menten equation by nonlinear least-square regression. The estimated parameters for the brain are Km of 0.62 ± 0.02 mmol/L, Vmax of 1.76 ± 0.03 nmol/sec per g.
Effects of Extracellular and Intracellular pH on [3H]Clonidine Transport at the Blood-Brain Barrier
Alteration of the Extracellular Vascular pHe: The Krebs carbonate perfusion fluid pHe was set to yield pH 5.40, 6.40, 7.40, and 8.40. The [3H]clonidine transport (Kin) increased significantly nearly 300% between pHe 5.40 and 6.40, and between pHe 6.40 and 7.40, and by ~150% between pHe 7.40 and 8.40 (Figure 3). Hence, the brain transport of [3H]clonidine is strongly pHe dependent, and is decreased by acidification of the extracellular vascular space. As Clonidine contains one basic nitrogen (pKa 8.20 in water), the amounts of the uncharged and cationic species depend on pHe and can be easily calculated in water, but not in blood, which promotes greater amine dissociation and ionization. Clonidine is ~99% cationic at pH 5.40 and 6.40, 86% cationic at pH 7.40, and 39% cationic at pH 8.40. The pH dependency of Clonidine transport into the brain could be due to passive diffusion of the uncharged molecule, which permeates more freely according to the pH partition theory. However, the increase in Clonidine transport between pH 5.40 and 6.40 cannot be due to a change in the amount of uncharged Clonidine (<1%) or by a change in the passive diffusion rate. Moreover, although the amount of uncharged Clonidine at pH 7.40 is only ~14% more than at pH 6.40, the brain transport of [3H]clonidine increased by ~300% from pH 6.40 to 7.40.
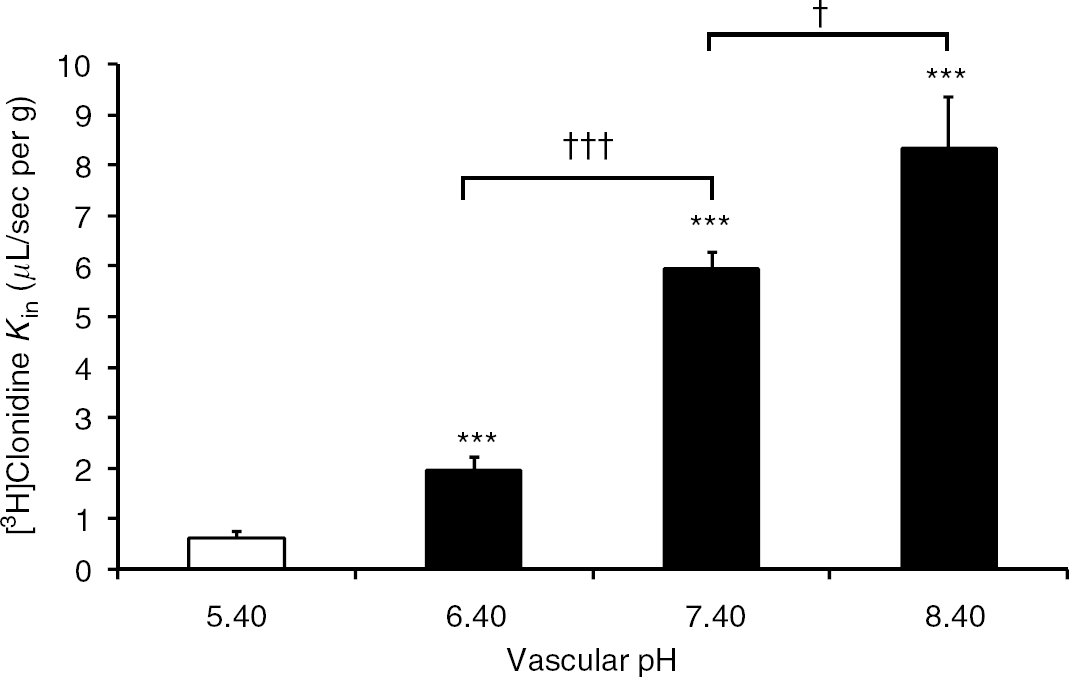
Effect of Krebs bicarbonate perfusion fluid at a pHe of 5.40, 6.40, 7.40, or 8.40 on the [3H]clonidine brain transport (Kin; μL/sec per g) measured by in situ mouse brain perfusion for 120 secs. Data are means ± s.d. of 5 to 7 animals. ***P < 0.001 comparing with the control group (pHe 5.40). †P < 0.05, †††P < 0.001 comparing pHe 7.40 with pHe 6.40 or 8.40.
Alkalinization of the Brain Endothelial Cells: An increase in the intracellular pH (pHi) leads to the inversion of the proton gradient that favors the movement of protons into cells. The BBB endothelial cells were made alkaline by an acute ‘pulse’ of NH4C1. Adding NH4C1 to the extracellular medium leads to an equilibrium between NH3 and NH4+, but only NH3 readily diffuses through the cell membrane. This NH3 then consumes protons within the cells to form a new equilibrium with NH4+, causing a rapid increase in pHi and consequently inverses the proton gradient. Mouse brains were perfused with Krebs carbonate perfusion fluid (pHe 7.40) containing NH4C1 (30 mmol/L) and [3H]clonidine for 120 secs. This increase in pHi significantly reduced the brain transport of [3H]clonidine (Figure 4A).
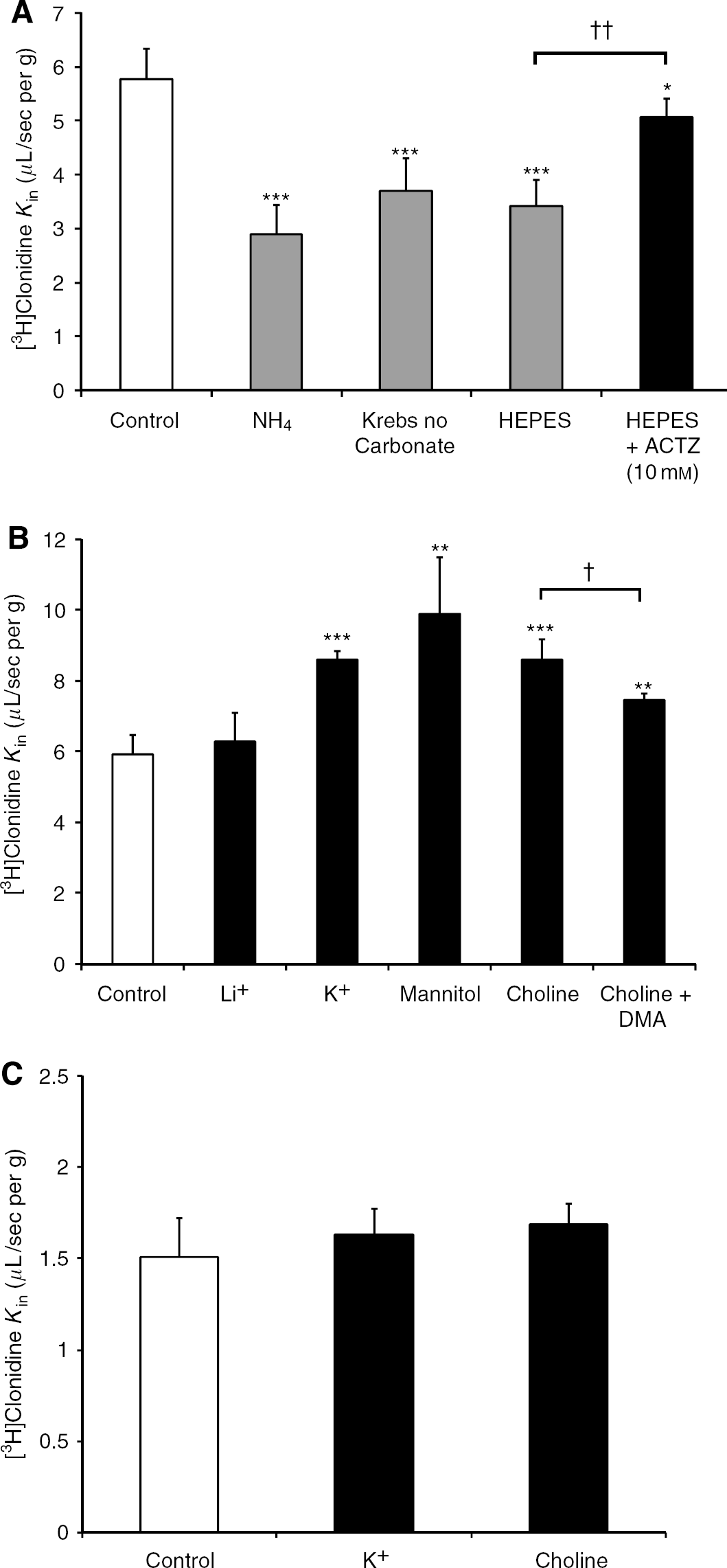
Effects of vascular perfusion fluids that could interfere with the regulation of the intracellular pHi at the BBB. The [3H]clonidine mouse brain transport (Kin; μL/sec per g) was measured by in situ brain perfusion for 120 secs. In all panels, the control group was always perfused with regular Krebs carbonate buffer. The perfusion fluids were at pHe 7.40 (
We also assessed the rapid, transient increase in pHi by replacing the carbonate buffer (blood) by carbonate-free buffer. The carbonate-free extracellular medium causes the intracellular CO2 to diffuse out of endothelial cells to establish a new equilibrium between the intracellular and extracellular media. The loss of intracellular CO2 results in HCO3−and H+ being converted into CO2 by carbonic anhydrase (CA), which then diffuses toward the vascular space (Ennis et al, 1996; Taylor et al, 2006). Perfusion with the bicarbonate-free buffer thus led to an abrupt, rapid increase in pHi that required CA within the cells and at the luminal membrane of the BBB (Taylor et al, 2006). The circulating blood was replaced by HEPES or Krebs carbonate-free buffer for in situ brain perfusion with [3H]clonidine for 120 secs. The brain [3H]clonidine transport after perfusion with carbonate-free perfusion fluid (pHe 7.40) was significantly lower than the transport by brains perfused with Krebs carbonate fluid (pHe 7.40; Figure 4A). This ‘carbonate/no carbonate’ change was dramatically reduced when ACTZ (10 mmol/L), a permeating CA inhibitor, was added to the carbonate-free HEPES (pHe 7.40; Figure 4A). [3Clonidine brain transport measured in the Krebs carbonate buffer (pHe 7.40) containing different amounts of HEPES (5 to 25 mmol/L) was not different from the transport measured in Krebs carbonate HEPES-free buffer, suggesting that HEPES had no direct effect on [3H]clonidine transport (data not shown).
Acidification of the Brain Endothelial Cells: Decreasing the pHi alters the strength of the proton gradient. Removing Na+ from the extracellular medium rapidly reduces the pHi of the BBB by increasing the acid load (Hsu et al, 1996). We acidified the BBB by removing the vascular Na+ to study the effect of endothelial cell acidification on [3H]clonidine transport. We replaced the Na+ in the Krebs carbonate buffer with choline, lithium, mannitol, or 154 mmol/L K+. We perfused the mouse brains with [3H]clonidine in these various Na+ -free carbonate buffers for 120 secs at pHe 7.40 (Figure 4B) and 6.40 (Figure 4C) and compared the transport parameters with those obtained with regular Krebs carbonate fluid (control; pHe 7.40 or 6.40). [3H]Clonidine brain transport (Kin) was significantly greater than control in all diverse Na+ -free buffers except lithium buffer (pHe 7.40; Figure 4B). Transport in the choline and K+ (154 mmol/L) Na+-free buffers was not statistically different from the regular Krebs carbonate at pHe 6.40 (control; pHe 6.40; Figure 4C). The ‘mannitol’ carbonate perfusion fluid lacked Na+ and contained only 3 mmol/L Cl−, unlike the other Na+-free buffers (lithium, choline, high ‘K+’), which contained 130 mmol/L Cl−.
The anion exchanger HCO3−/Cl−, also called the AE transporter, restores the pHi at the BBB (Nicola et al, 2008). However, a lack of chloride in the extracellular medium leads to the immediate and sustained acidification of the brain endothelial cells by a molecular mechanism that is presently unknown (Hsu et al, 1996; Nicola et al, 2008). When we replaced the chloride in the Krebs carbonate buffer (pHe 7.40) with gluconate and nitrate the [3H]clonidine transport, Kin (7.60 ± 0.51 μL/sec per g; n = 4; P < 0.05) was significantly (1.3-fold) greater than that for control mice perfused with Krebs carbonate fluid (5.95 ± 0.52 μL/sec per g), as might be expected after a decrease in the BBB pHi. The Cl−/H+ antiporter (CLC) may be involved in pHi acidification after chloride removal, but this requires dedicated confirmatory studies (Nicola et al, 2008; Picollo and Pusch, 2005).
Influence of Sodium on [3H]Clonidine Brain Transport
It can be difficult to assess the influence of sodium on transport across the BBB, as acute exposure to Na+-free perfusion fluid could alter the pHi via Na+-dependent transporters involved in regulating the pHi, such as the Na+ /H+ antiporter (NHE) and the Na+/HCO3− cotransporter (NBC). We perfused brains with [3H]clonidine plus 1 mmol/L DMA, an NHE inhibitor, in the choline buffer and found that the increased [3H]clonidine transport was significantly reduced (Figure 4B). Sodium does not seem to be required for [3H]clonidine transport, as suggested by the lack of statistic difference between [3H]clonidine transport measured in the Li+ buffer and in the Krebs carbonate buffer (Figure 4B). However, this conclusion cannot be definitive as Li+ can sometimes take the place of Na+ in carrier transport process. Then, we perfused brains with [3H]clonidine in regular Krebs carbonate (control; pHe 6.40) and in carbonate Na+-free buffer containing choline or K+ at pHe 6.40 (Figure 4C) to distinguish the rapid removal of vascular Na+ from a decrease in pHi. The data indicate that Na+ is not essential for [3H]clonidine transport across the BBB (Figure 4C).
Effects of Changes in the Trans-membrane and Membrane Dipole Potentials on [3H]Clonidine Brain Transport
We used perfusion with K+-free or high K+ fluids with or without valinomycin (10 μmol/L) to assess the effect of the trans-membrane potential on [3H]clonidine transport. Neither K+-free perfusion fluid nor Krebs carbonate buffer containing valinomycin altered [3H]clonidine transport. A high K+ content dissipates the trans-membrane potential gradient, and this could affect simple diffusion and/or carrier-mediated transport components, hence decreasing the transport of cationic compounds into depolarized cells. High K+ perfusate (154 mmol/L) resulted in significantly enhanced [3H]clonidine transport (Figures 4B and 5), but Na+ -free perfusion fluid could interfere with pHi regulation. The rate of [3H]clonidine transport in this perfusion fluid was not different from that obtained with the ‘choline’ or ‘mannitol’ perfusion fluids; they contained no Na+ but had the regular K+ content (4.2 mmol/L; Figure 4B). We excluded the effect of cell acidification due to a lack of Na+ by inverting the proton gradient. Mouse brains were perfused with [3H]clonidine in regular Krebs carbonate (control; K+ 4.2 mmol/L) and high K+ (154 mmol/L) carbonate buffers at pHe 6.40 (Figure 4C). We found no effect of trans-membrane depolarization on [3H]clonidine transport (Figure 4C).
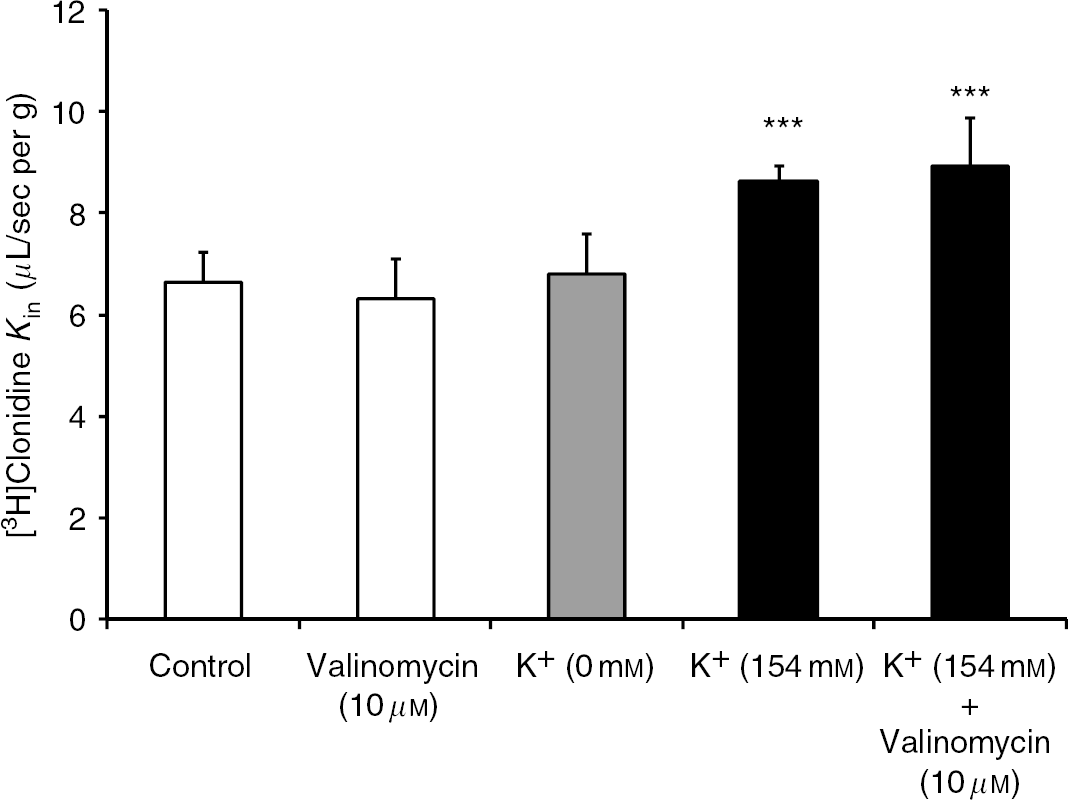
[3H]Clonidine brain transport (Kin; μL/sec per g) measured by in situ mouse brain perfusion for 120 secs after perfusion (pHe 7.40) with regular Krebs carbonate fluid without (control) or with valinomycin (10 μmol/L), Krebs carbonate fluid without K+ (0 mmol/L), and high K+ (154 mmol/L) Na+-free Krebs carbonate fluid with or without valinomycin (10 μmol/L). Data are means ± s.d. of 5 to 7 animals. ***P < 0.001 compared with the control group.
The effect of the dipole membrane potential on [3H]clonidine transport at the BBB was assessed by adding phloretin (0.5 mmol/L) to the perfusion fluid (pHe 7.40). The [3H]clonidine transport (Kin) with phloretin (7.65 ± 0.29 μL/sec per g; n = 4) was significantly greater (1.27 times) than that in control Swiss mice without phloretin (6.02 ± 0.31 μL/sec per g; n = 5; P < 0.001). The same experiment performed with saturating Clonidine (10 mmol/L) showed that [3H]clonidine transport was significantly (1.5-fold) greater when phloretin (0.5 mmol/L) was present in the perfusate than in control Swiss mice perfused with Clonidine (10 mmol/L) without phloretin (data not shown).
Effects of P-gp, Bcrp, and Oct1, 2, 3 on [3H]Clonidine Brain Transport
The brains of mdr1a, 1b, bcrp(−/-) triple KO mice and FVB control mice were perfused with [3H]clonidine. The influence of P-gp (mdr1a) and Bcrp (Abcg2) on [3H]clonidine transport was also assessed by including the P-gp and Bcrp inhibitor, GF120918 (2 μmol/L), in the perfusate. The rates of [3H]clonidine transport (Kin) in control mice without GF120918 (5.24 ± 0.66 μL/sec per g; n = 4), with GF120918 (4.75 ± 0.29 μL/sec per g; n = 4) and in triple KO mdr1a, mdr1b, bcrp(−/-) mice (4.50 ± 0.19 μ/sec per g; n = 4) were not statistically different. The rates of [3H]clonidine transport (Kin) in control mice (6.25 ± 0.75 μL/sec per g; n = 5), in double KO Oct1, 2(−/-) mice (6.11 ± 0.71 μL/sec per g; n = 5), and in Oct3(−/-) mice (6.40 ± 0.28 μL/sec per g; n = 5) were not statistically different.
Effect of Organic Compounds and CNS Drugs on [3H]Clonidine Brain Transport
Effects of cold organic compounds on [3H]clonidine transport were examined by cis-inhibition. Mice were perfused with Krebs carbonate buffer (pHe 7.40) for 120 secs (Table 1). Most of the chemicals used are known to affect transporters involved in handling organic cations. The profile of [3H]clonidine transport inhibition could help to identify the function of the transporters involved. The quaternary aliphatic ammonium cations, choline and TEA, did not significantly affect [3H]clonidine transport; neither did Cimetidine nor the aliphatic zwitterion quaternary ammonium, carnitine. The tertiary amine, DPH, significantly inhibited [3H]clonidine transport. However, primary monocationic amines, such as dopamine, histamine, serotonin, guanidine, and polycationic amines, such as agmatine, were all without effect. Some CNS drugs inhibited [3H]clonidine brain transport. All the CNS drugs that inhibited [3H]clonidine transport had a single cationic charge and a tertiary amine moiety, except the secondary amines para-chloroamphetamine and MDMA.
Effects of organic cations and CNS cationic drugs on the [3H]clonidine mouse brain transport
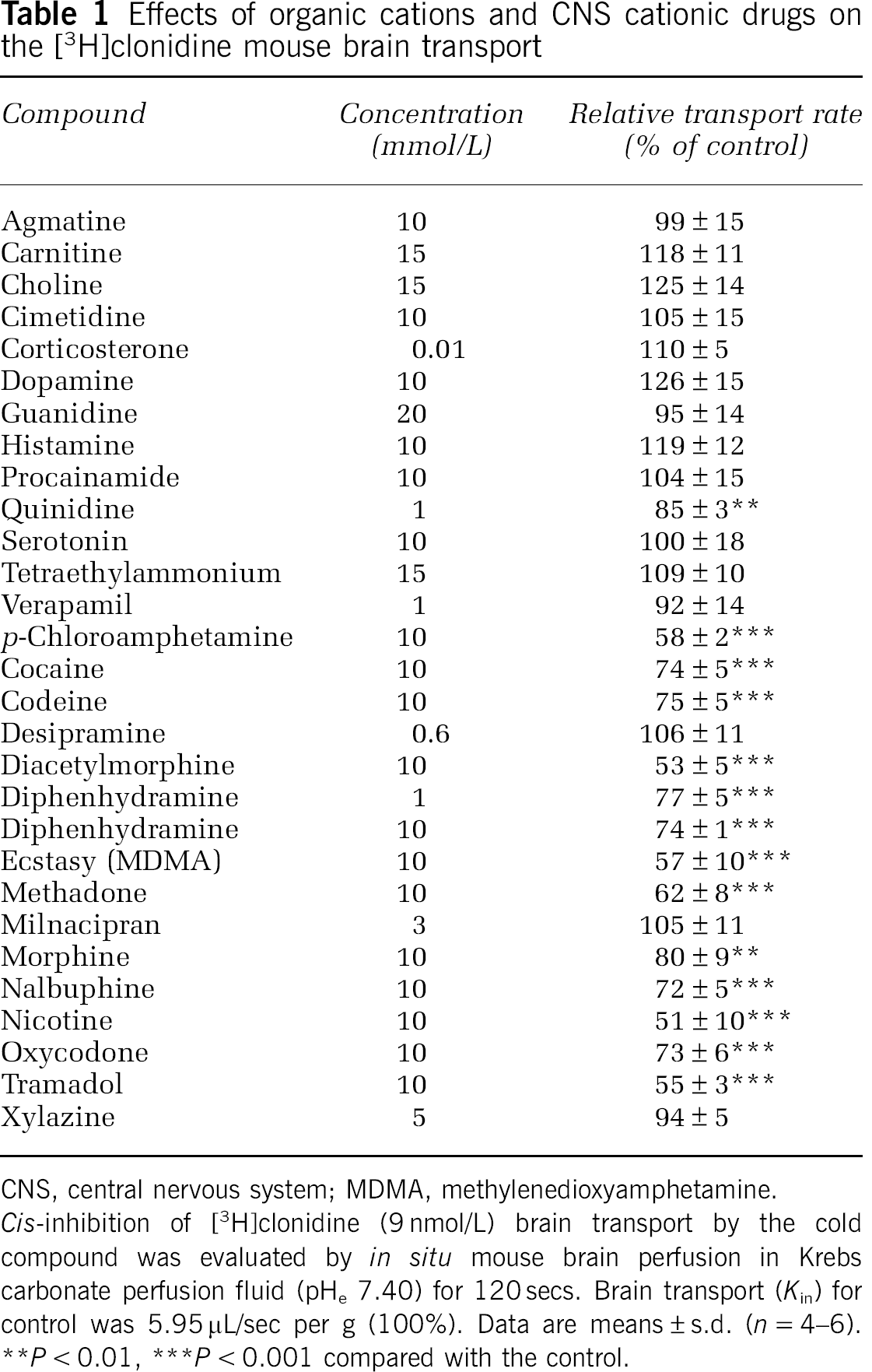
CNS, central nervous system; MDMA, methylenedioxyamphetamine.
Cis-inhibition of [3H]clonidine (9 nmol/L) brain transport by the cold compound was evaluated by in situ mouse brain perfusion in Krebs carbonate perfusion fluid (pHe 7.40) for 120 secs. Brain transport (Kin) for control was 5.95 μL/sec per g (100%). Data are means ± s.d. (n = 4–6).
P < 0.01
P < 0.001 compared with the control.
Effect of Trans-stimulation by Diphenhydramine on [3H]Clonidine Transport
We used trans-stimulation to determine whether DPH was a substrate (Figure 6). Mouse brains were first loaded with [3H]clonidine by in situ perfusion for 120 secs (control). We then used washout (WO) perfusion to perfuse with the addition of DPH (10 mmol/L; WO + DPH) at pHe 6.40 or without DPH. The trans-efflux of [3H]clonidine was significantly stimulated by DPH exposure, suggesting that DPH shares the same transporter. The observed trans-stimulation also suggests that the transporter can transport Clonidine in both directions.
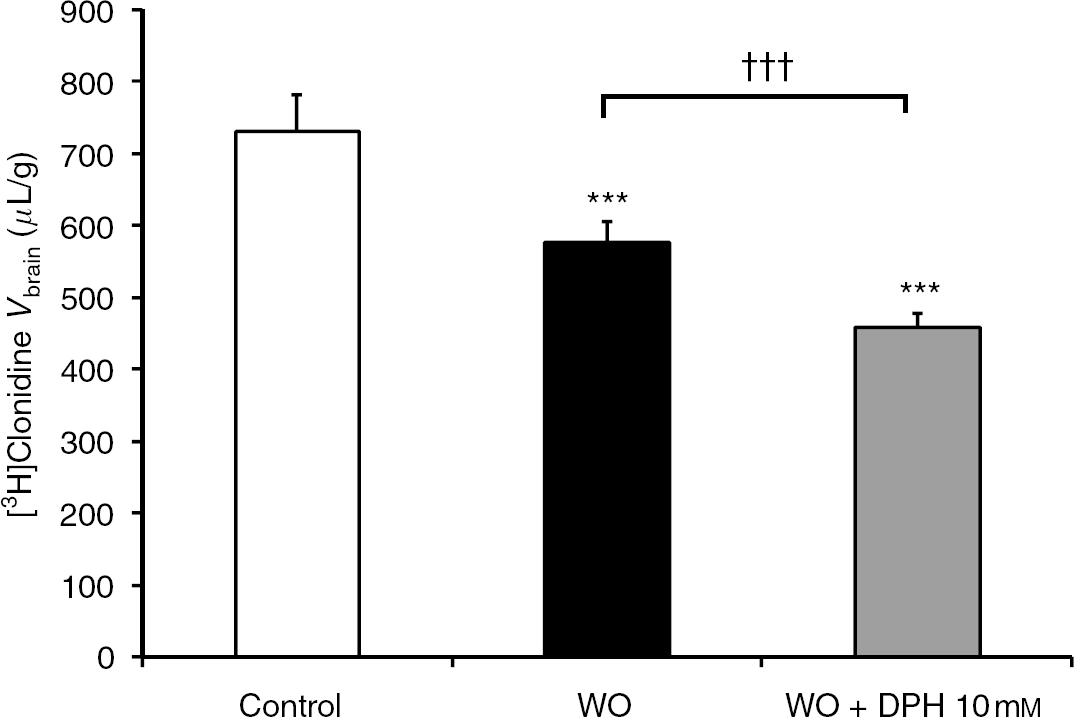
[3H]Clonidine brain distribution volume (Vbrain; μL/g) measured by in situ mouse brain perfusion for 120 secs in Krebs carbonate buffer (pHe 7.40) alone (control) or followed by a second 60 secs washout (WO) perfusion. The second perfusion was with Krebs carbonate fluid (pHe 6.40) without (WO) or with (WO + DPH) diphenhydramine (DPH; 10 mmol/L). Data are means ± s.d. of 5 to 7 animals. ***P < 0.001 compared with the control group. †††P < 0.001 comparing WO with or without DPH (10 mmol/L).
Discussion
Some features of the BBB restrict the entry of cationic drugs into the brain parenchyma. One of these is the ATP-binding cassette (ABC) efflux transporters, P-gp and Abcg2. However, these ABC transporters did not affect the BBB access of Clonidine (Mw 230), in agreement with in vitro reports and physicochemical observations that substrates of P-gp must have molecular weight of over 280 Da (e.g., morphine) (Mahar Doan et al, 2002). However, the evidence for a saturable Clonidine brain influx highlights the critical role of an unidentified cation transporter at the luminal mouse BBB. The apparent BBB Michaelis-Menten (Km) parameter for Clonidine transport into the mouse brain (pHe 7.40) agrees well with in vitro values for porcine BBB microvessel endothelial cells (1.3 mmol/L; Huwyler et al, 1997) and human SH-SY5Y neuronal cells (0.7 mmol/L; Fischer et al, 2007). These data provide the background for in vivo characterization of this transport system and the need to evaluate the influence of the driving force factors such as ions.
Modifying the vascular proton concentration altered the Clonidine brain transport in a way that suggests the presence of a clonidine/H+ exchanger rather than a change in passive diffusion, according to the pH partition theory. The unsaturated component (Kd; pHe 7.40) confirms that the passive diffusion of Clonidine is negligible, as it is in other cells (Fischer et al, 2007; Grafe et al, 2004) and again argues for the presence of a sensitive pH carrier-mediated process. However, this pH dependency of Clonidine influx could be due to an outwardly proton-gradient-dependent antiporter or due to a change in the ionization state of the transporter at the vascular side, which could affect Clonidine linkage and/or its transport. We tested these possibilities in experiments in which the pHi and/or the pHe was altered so as to change the proton gradient acting between vascular space and BBB endothelial cells.
Several mechanisms are responsible for keeping the pHi of the endothelial cells forming the BBB more acidic than the surrounding medium under basal condition (Taylor et al, 2006). In vitro experiments have shown that a change in the extracellular medium can lead first to an abrupt, sustained change in pHi. Later-acting cell mechanisms then attempt to restore the basal pHi as illustrated by in vitro BBB experiments (Hsu et al, 1996; Nicola et al, 2008; Taylor et al, 2006). We therefore adapted the in situ brain perfusion procedures to perform acute pHi changes at the mouse BBB, as these alterations were more predictable, and take place on the same time scale as the brain perfusion time. Switching the perfusion fluid from one equilibrated with a carbonate buffer (e.g., the blood) to a carbonate-free-buffered fluid leads to an abrupt increase in the pHi. This inversion of the proton gradient is effective at the BBB because the brain endothelial cells have both cytosolic and luminal membrane-bound CA, which catalyze the rapid change in the pHi and transformation of CO2 in the vascular bed to HCO3−, which diffuses poorly (Ghandour et al, 1992; Taylor et al, 2006). Our ACTZ inhibition experiment indicates that the intracellular alkalinization produced by removing carbonate requires CA, as it does in vitro (Taylor et al, 2006). Similarly, we used only a short-time exposure to NH4C1 to rapidly increase the pHi and consequently reduce Clonidine brain transport. We could not study the delayed acidification that is produced 15 to 20 min after the removal of NH4Cl from the extracellular fluid (Taylor et al, 2006) owing to the limited duration of our experimental procedure.
Sodium and chloride are transported by carriers located at the BBB luminal membrane shown to be involved in the regulation of the brain endothelial cell pHi, such as NHEs, AEs and NBCs (Ennis et al, 1996; Hsu et al, 1996; Taylor et al, 2006). Removal of Na+ or Cl− from the vascular fluid leads to acute BBB acidification, which strengthens the outward proton gradient and enhances [3H]clonidine transport. However, replacing Na+ by Li+ did not alter the [3H]clonidine brain transport. In fact, the NHE and NBC transporters could accept Li+ in place of Na+, but not K+ or some other cations (Amlal et al, 1998; Dunham et al, 2005; Jean et al, 1986; Sciortino and Romero, 1999; Szabó et al, 2000). The vascular pHe was reduced to 6.40 to limit the enhanced outward proton gradient produced by removing the Na+ from the perfusing fluid. This also shows that Na+ is not required for Clonidine transport at the BBB. These changes in pHe and/or pHi show that Clonidine transport is H+-coupled and that both the extracellular and intracellular protons can affect the rate and direction, as suggested by trans-stimulation studies showing both opposite and reversible transport of proton and Clonidine. Reversible transport means that the transporter can rotate either ‘clockwise’ or ‘counter-clockwise’ depending on the magnitude of the proton or substrate gradient. As the physiological proton concentrations (pH 7.40 to 7.20) correspond to ~40 to 60 nmol/L, the amine substrate concentration over the nmol/L range could determine the direction of the transporter, despite an unfavorable proton gradient (and vice versa), as was also suggested by Kuwayama et al (2008).
Depolarization of the plasma trans-membrane potential by dissipating the potassium electrochemical gradient could affect the activity of some transport processes. The activities of the organic cation transporters, OCT1-3 and PMAT, are known to be reduced by trans-membrane depolarization (Koepsell et al, 2007; Engel and Wang, 2005). However, Clonidine transport is insensitive to BBB trans-membrane depolarization. Moreover, the dipole membrane potential, which hinders the passive diffusion of cationic compounds, particularly those with a delocalized charge (Cattelotte et al, 2009; Chiu et al, 1992), slightly restricts the passive permeability of the BBB to Clonidine.
One of our objectives was to determine whether Clonidine transport at the BBB is due to a known organic cation transporter. To do this, we used various chemicals and KO mice for Oct1-3 transporters. We first showed that Clonidine transport was distinct by its resistance to inhibition by TEA, a prototypical substrate/inhibitor of OCT, OCTN, and MATE transporters. This also confirmed our studies with deficient Oct1-3 mice. Only OCTN1, MATE, CTL1, and PMAT transporters are known to be coupled with protons. Clonidine transport is not mediated by Mates, as it is not a substrate of Mate1 and because Cimetidine, TEA, and guanidine, which are potent inhibitors of rodent Mate1 and/or Mate2, are ineffective (Hiasa et al, 2006, 2007; Ohta et al, 2006). Other chemical inhibitions rule out the involvement of the choline transporters, PMAT, and carnitine transporters, OCTN2-3 (Inazu et al, 2005; Wu et al, 1999). Moreover, PMAT is sensitive to the trans-membrane potential and is inhibited by dopamine, histamine, and Cimetidine (Engel et al, 2004; Engel and Wang, 2005; Xia et al, 2007). Thus, these experiments suggest that Clonidine transport at the mouse luminal BBB is mediated by a new organic cation/H+ antiporter, as suggested in other cells (Fischer et al, 2006, 2007). As summarized in Figure 7, there must be the same or a similar transporter system able to transport Clonidine at the abluminal BBB to permit its access to the brain parenchyma.
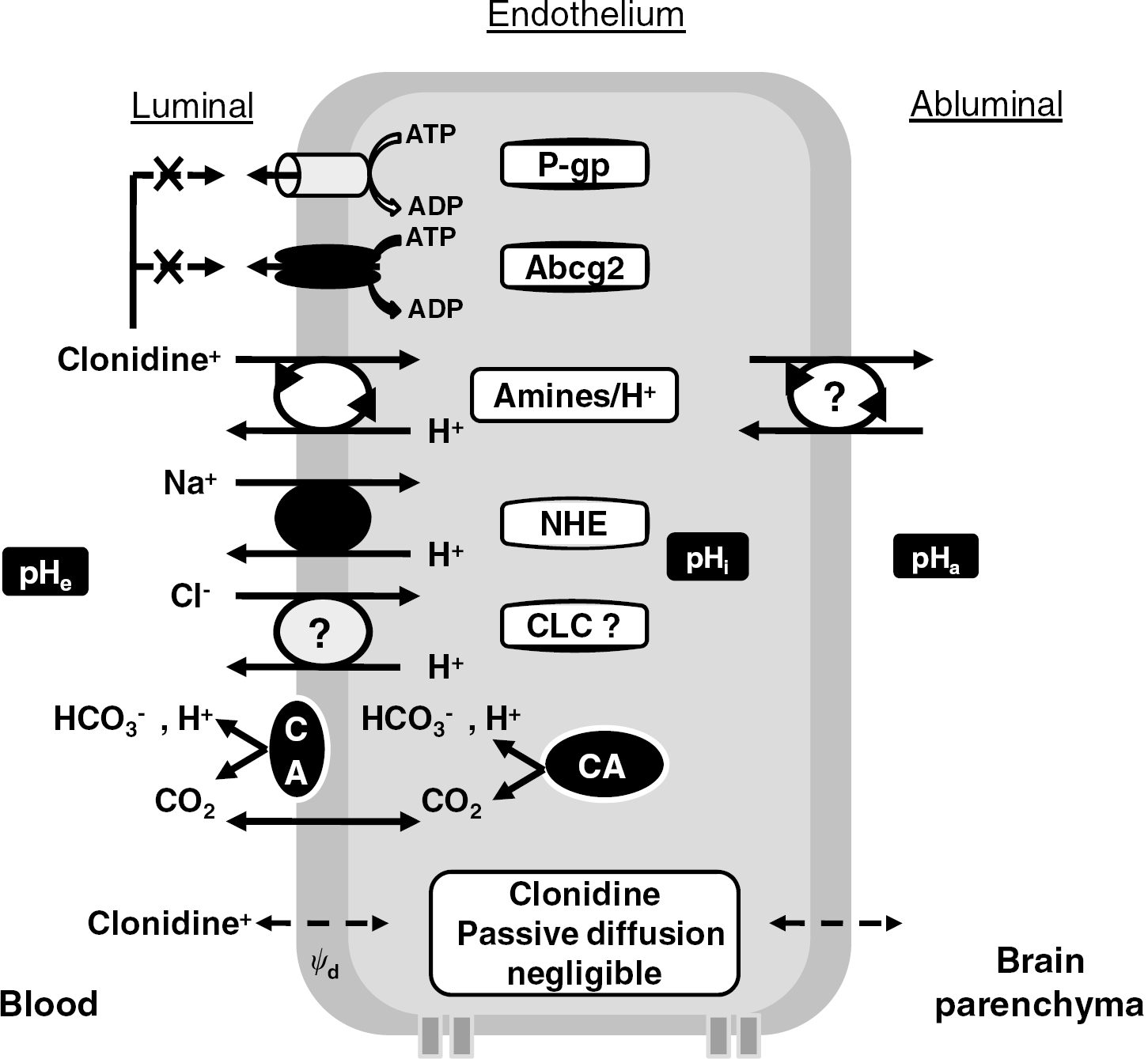
Putative pathways and mechanisms for the transport of Clonidine at the mouse BBB. The passive diffusion of Clonidine is negligible and, unlike the trans-membrane plasma potential (Δψp), the dipole membrane potential (ψd) significantly hinders its crossing of the bilayer. The Clonidine transport is not controlled by two major ABC efflux transporters: P-gp (mdr1a) and Abcg2 (Bcrp). However, Clonidine is a substrate of an ‘amines/H+’ antiporter that acts reversibly at least at the luminal BBB (clockwise for influx) depending on the concentration gradient. Only the NHE and CLC transporters and the enzyme CA that were manipulated by ionic alterations or chemical inhibitions are shown in this diagram, although other transporters are known to be involved in the control of pHi. Similarly, organic cation transporters such as Octs, Pmat, or Mates are not shown, as their presence at the mouse BBB in vivo is presently unknown.
In vivo investigations by the groups of Pardridge and Oldendorf showed the possible involvement of an H+ antiporter at the rat BBB. It could facilitate the entry of some amine drugs, such as propranolol, amphetamine, and nicotine, into the brain (Oldendorf et al, 1979; Pardridge et al, 1984; Pardridge and Connor, 1973). However, these earlier studies provided no information on the molecular and/or functional identity of this transporter, as evidence regarding this was obtained only recently (Koepsell et al, 2007). The carrier-mediated transport of DPH, which could involve the Clonidine BBB transporter, was suggested to be present at the BBB and to work as an H+ antiporter in Caco-2 cells (Au-Yeung et al, 2006; Goldberg et al, 1987; Mizuuchi et al, 2000). An H+ antiporter for oxycodone was recently found in immortalized rat brain endothelial cells, as was a similar antiporter for MDMA in Caco-2 cells (Kuwayama et al, 2008; Okura et al, 2008). These studies show features very similar to this Clonidine transporter, and MDMA transport is, similar to that of Clonidine, trans-stimulated by DPH and reversible (Kuwayama et al, 2008). These results strongly suggest that DPH, oxycodone, and MDMA are also substrates of this clonidine/H+ antiporter. Although morphine inhibits the transport of Clonidine, its substrate capacity for an influx BBB carrier process could not be suggested (Cisternino et al, 2004). Moreover, morphine is poorly transported at the rodent's BBB, even after the P-gp efflux is inhibited (Cisternino et al, 2004; Oldendorf et al, 1972). These observations suggest that morphine is probably not an efficient substrate of this amines/H+ antiporter, unlike oxycodone (Okura et al, 2008). Although little differences exist in the molecular structure between morphine, codeine, and heroin (diacetylmorphine), it was postulated that the great differences in the brain uptake of these cationic drugs are due to their passive permeability at the BBB (Oldendorf et al, 1972). Although a wide range of CNS drug interactions is illustrated in our study, a general feature of the compounds that interfere with clonidine transport is that they have an aliphatic secondary or tertiary amine moiety linked through a chain containing 1 to 4 atoms (carbon and/or heteroatoms) to a planar aromatic structure (phenyl, pyridine). Some have aryl, alkyl, halogen, dioxol, and hydroxyl groups on the aromatic ring, although hydroxyl seems to hamper interaction with the transporter. Data on more inhibitors/substrates are now required to assess the interacting site using quantitative structure activity relationship (QSAR) analysis.
Two main factors are currently considered to distinguish drugs that enter the CNS from those that do not: P-gp transport and passive permeability. There have been many attempts to predict these effects by a variety of approaches such as computational QSAR methods (Clark, 2003; Mahar Doan et al, 2002). One difficulty lies in establishing physicochemical descriptors of drugs based on compounds believed to cross the BBB by passive diffusion, but that in fact do not. These studies show that a positive charge due to secondary or tertiary amine moiety, a feature of many drugs that enter the CNS, can be considered to favor crossing the BBB (Clark, 2003). This, of course, needs now to be confirmed, especially as some secondary/tertiary amines could be transported by carrier-mediated processes at the BBB.
Clonidine and DPH share a common carrier-mediated, reversible transport process that involves an H+-coupled antiporter at the luminal side of the mouse BBB. This carrier-mediated process is a new factor determining the brain permeability of drugs. It could involve other CNS drugs such as some opiates and amphetamines. This transporter appeared to be a promising new brain drug delivery system to enhance the brain distribution of substrate/vectors. It also raised the potential for a new pharmacother-apeutic target that needs the development of drug inhibitors to limit the brain access of addictive drugs and possibly help abstinence.
Footnotes
Acknowledgements
We thank Dr Alfred H Schinkel for supplying all the KO mouse strains. We thank Dr Joseph W Polli for generously supplying GF120918, and Professor Hervé Galons for generously supplying MDMA. We thank Dr Anne-Lise Roy for help in the search for chemical structures and Dr Owen Parkes for editing the English text.
The authors declare no conflict of interest.